

Abstract
Clostridium difficile is the major cause of antibiotic-associated diarrhea and pseudomembranous colitis in healthcare settings. However, the host factors involved in the intestinal inflammatory response and pathogenesis of C. difficile infection (CDI) are largely unknown. Here we investigated the role of leukotrienes (LTs), a group of pro-inflammatory lipid mediators, in CDI. Notably, the neutrophil chemoattractant LTB4, but not cysteinyl (cys) LTs, was induced in the intestine of C57BL/6 mice infected with either C. difficile strain VPI 10463 or strain 630. Genetic or pharmacological ablation of LT production did not ameliorate C. difficile colitis or clinical signs of disease in infected mice. Histological analysis demonstrated that intestinal neutrophilic inflammation, edema and tissue damage in mice during acute and severe CDI were not modulated in the absence of LTs. In addition, CDI induced a burst of cytokines in the intestine of infected mice in a LT-independent manner. Serum levels of anti-toxin A immunoglobulin (Ig) G levels were also not modulated by endogenous LTs. Collectively, our results do not support a role for LTs in modulating host susceptibility to CDI in mice.
Keywords: Clostridium difficile, leukotriene, eicosanoid, nosocomial infection
1. INTRODUCTION
The Gram-positive, anaerobic, spore-forming bacillus Clostridium difficile is the major cause of intestinal disease associated with antibiotic therapy [1]. Clinical manifestations of C. difficile infection (CDI) range from mild diarrhea to more severe manifestations such as pseudomembranous colitis, toxic megacolon and death [2]. In the 21st Century, CDI has emerged in nosocomial and community settings [3, 4], while its overall incidence and severity have increased [5, 6]. As a result, CDI constitutes a significant public health problem. Because antibiotic therapy for CDI has not completely eliminated this threat, there is a pressing need for interventions that will reduce disease severity in vulnerable populations.
Upon entering of the gastrointestinal tract, C. difficile germinates, outgrows and produces toxins, including toxin A (TcdA) and toxin B (TcdB), which trigger clinical manifestations of CDI [7]. C. difficile toxins have cytotoxic effects leading to disruption of the intestinal epithelium and subsequent mucosal inflammation [8]. Bacterial intoxication of the host results in the release of pro-inflammatory cytokines and chemokines that incite a robust inflammatory response characterized by neutrophil infiltration, which contributes to collateral tissue damage [9, 10]. The complex autocrine and paracrine signaling mechanisms that mediate the recruitment of neutrophils and the development of symptomatic colitis are incompletely characterized. Understanding the molecular drivers of acute inflammation is an important strategy for developing novel, host-centered therapies that can be used in concert with antibiotic or probiotic approaches to CDI therapy and/or prevention.
Among pro-inflammatory factors induced by C. difficile toxins are the leukotrienes (LTs) [11, 12]. The LTs are lipid mediators derived from the polyunsaturated cell membrane fatty acid arachidonic acid (AA), and they are produced mainly by myeloid cells. In response to many inflammatory cell stimuli, AA is liberated from cell membranes. It is then enzymatically converted to LTs through the rapid and sequential action of 5-lipoxygenase (5-LO) and either leukotriene A4 hydrolase (LTA4H) or leukotriene C4 synthase (LTC4S), generating LTB4 and cysteinyl (cys)LTs (LTC4, LTD4 and LTE4), respectively [13]. Upon binding to their specific receptors, LTs exert their functions in inducing inflammation mainly by recruitment and activation of cells to the site of injury [14]. LTB4, for example, is one of the most potent neutrophil chemokines [15].
The LTs have been implicated in the pathogenesis of several human gastrointestinal inflammatory conditions including ulcerative colitis and Crohn’s disease [16–18]. Furthermore, it was also shown that exogenously-applied LTB4 elicited ileitis in rats that closely resembled TcdA-induced inflammation [11]. Specifically, LTs were associated with intestinal neutrophilic inflammation [11, 19] and also with fluid accumulation in rat ileum stimulated with TcdA [11]. That stated, the role of LTs in CDI pathogenesis has not been studied using animal models of infection caused by live C. difficile. Given the implication of LTs in inflammatory intestinal disorders and in inflammation induced by TcdA, we sought to test the hypothesis that LTs are important mediators of disease severity and tissue inflammation in CDI. If true, this would be significant because there are already a number of pharmacological tools for modulating LT synthesis and signaling that could be further studied for a role in managing CDI.
Using a single antibiotic to induce susceptibility to CDI, we demonstrated that LTs are strongly induced during mild or severe CDI in mice. However, using pharmacological or genetic approaches (including 5-LO−/− mice lacking the gene encoding the 5-LO enzyme), we were unable to support our hypothesis. The models used in this work suggest that LTs neither govern colonization resistance against C. difficile nor the severity of disease. Furthermore, the tissue damage, cell influx and cytokine production induced by CDI in the intestine appear to be independent of LT production. Thus, our data suggest that C. difficile-induced colitis is independent of LTs.
2. MATERIALS AND METHODS
2.1. Animals and housing
C57BL/6 mice were obtained from a breeding colony at The University of Michigan Medical School that was previously established using mice purchased from Jackson Laboratories (Bar Harbor, ME). 5-LO deficient mice (5-LO−/−) on a C57BL/6 genetic background were purchased from Jackson Laboratories. To equilibrate their microbiota, 5-LO−/− and wild-type C57BL/6 (WT) mice were co-housed for 2 weeks prior to cefoperazone administration. Mice were housed with autoclaved water, food and bedding and all experiments and manipulations were performed in a biosafety level 2 laminar flow hood. Experimental procedures were conducted with the approval of the University Committee on Use and Care of Animals of the University of Michigan (protocol 3553) and animals were housed in an AAALAC-accredited facility.
2.2. Preparation of C. difficile spores
C. difficile spores from strains VPI 10463 (ATCC 43255) and 630 (ATCC BAA-1382) were prepared as follows. An overnight C. difficile culture in Columbia Broth was inoculated in 40 mL of Clospore media [20] and incubated anaerobically at 37°C. After 5 to 7 days, spores were washed at least 3 times in cold sterile water at 3200 rpm for 20 min. The spore pellets were resuspended in 1 mL of sterile water and stored at 4°C. Prior to infection, an aliquot of stock was heated at 65°C for 20 min to kill vegetative cells and viable spores enumerated by plating for colony forming units (CFU) on taurocholate-cycloserine-cefoxitin-frutose agar (TCCFA) plates.
2.3. Administration of antibiotic and experimental procedures
A murine model of antibiotic associated CDI established by Young lab [21] was used for this work. C57BL/6 female mice at age 5–8 weeks were given 0.5 mg/mL of cefoperazone (MP Biomedicals, Solon, OH) in sterile drinking water for 5 days. A fresh supply of cefoperazone in drinking water was replaced every 2 days during the period of antibiotic administration. After antibiotic, mice were allowed to consume normal sterile drinking water for 2 days and were orally gavaged with C. difficile of either VPI 10463 (100 spores) or 630 strain (1 × 104 spores). Control mice were mock infected with sterile PBS. Animals infected with VPI 10463 were monitored every 6 hours while animals infected with 630 were monitored daily for signs of disease including diarrhea, hunched posture and weight loss. The colonization status of C. difficile-infected animals was monitored by bacterial enumeration in anaerobic culture on TCCFA (Anaerobe Systems, Morgan Hill, CA). Mice were euthanized by CO2 asphyxiation after 6, 24 and 36 hours of the VPI 10463 infection or after 2, 4 and 8 days of the 630 infection and then cecal snips were harvested and were snap frozen in liquid nitrogen and stored at −80°C for further analysis. Survival curves reflected humane endpoints for euthanasia, which were considered when mice showed 20% of weight loss or if mice became moribund. Mice were euthanized by CO2 inhalation.
2.4. In vivo MK886 treatment
For pharmacological inhibition of LTs, VPI 10463 infected mice were orally gavaged with 10 mg/kg of MK886 in PBS (Cayman Chemical Co, Ann Arbor, MI) or vehicle control (ethanol similarly diluted in PBS) given 1 hour prior to infection and 24 hours later. The dose of MK886 used in these studies was based on previously published mouse studies [22, 23] and significantly reduced endogenous LT production in our model (data not shown).
2.5. Measurement of leukotrienes and cytokines
A specific enzyme immunoassay (Enzo Life Science, Farmingdale, NY) was used to quantify LTB4 and cysLTs in cecal homogenates per the manufacturer’s instructions. Cecal tissue stored at −80°C was homogenized in 1mL of PBS containing 1 mmol/L EDTA and 10 μmol/L indomethacin for 20 sec on ice and then centrifuged at 13,000 rpm at 4°C. The supernatant was suspended in 4mL of ethanol on ice, vortexed, and sample was spun at 2000 rpm for 10 min at 4°C for protein precipitation. Supernatant was dried under nitrogen bath and reconstituted in 500 μL of assay buffer prior to performing the assay. Quantification of cytokines was performed by the University of Michigan Cancer Center Immunology Core. Cecal tissue stored −80°C was homogenized in 1mL of PBS and tested for cytokines using ELISA Kits (R&D Systems, Minneapolis, MN). The levels of LTs and cytokines were expressed per gram of tissue.
2.6. Histopathological analysis
Ceca were harvested and fixed by immersion in 10% formalin, and then transferred to 70% ethanol. Tissue was processed, paraffin embedded, sectioned, and stained with H&E by McClinchey Histology Lab Inc.. The histopathological scores were assigned in a blinded fashion by a board-certified pathologist (ILB). Each section was evaluated using a semi-quantitative method and scored histologically as 0–4, as described previously [24]. The following criteria were used to define a histological scoring system. Edema: 0, no edema; 1, mild focal or multifocal edema with minimal (<2× normal width) submucosal expansion; 2, moderate multifocal edema with moderate (2× to 3× normal width) submucosal expansion; 3, severe multifocal to coalescing edema with severe (>3× normal width) submucosal expansion; 4, same as score 3 with diffuse submucosal expansion. Neutrophilic inflammation: 0, no inflammation; 1, minimal multifocal neutrophilic inflammation; 2, moderate multifocal neutrophilic inflammation (greater submucosal involvement); 3, severe multifocal to coalescing neutrophilic inflammation (greater submucosal involvement, with or without mural involvement); 4, same as score 3 with abscesses or extensive mural involvement. Epithelial damage: 0, no epithelial changes; 1, mild multifocal superficial epithelial damage (vacuolation, increased apoptosis and villus tip attenuation/necrosis); 2, moderate multifocal superficial epithelial damage (vacuolation, increased apoptosis, villus tip attenuation/necrosis); 3, severe multifocal to coalescing mucosal damage, with or without pseudomembrane (intraluminal neutrophils and sloughed epithelium in fibrinous matrix); and 4, same as score 3 with significant pseudomembrane or epithelial ulceration. Representative photomicrographs were taken using an Olympus DP72 12.5 megapixel digital camera mounted to an Olympus BX45 light microscope and using the software provided by the manufacturer (cellSens Standard 1.7.1, Olympus Corporation). Photo processing and composite plate construction was performed in Adobe Photoshop CS2, version 9.0. Photo processing was confined to global adjustments of brightness, contrast, sharpness, and image size that did not materially alter the interpretation of the image. Correction of peripheral lens distortion was performed if needed for low magnification photos.
2.7. Anti-toxin A/B IgG measurement
Levels of IgG specific to C. difficile toxin A and toxin B were measured in serum from infected mice. 96-well EIA plates (Corning Incorporated, Tewksbury, MA) were coated with 100μl of either purified C. difficile toxin A or toxin B (List Laboratories, Campbell, CA) at 5μg/mL in 0.05M sodium bicarbonate buffer pH 9.6 (Sigma, Saint Louis, MO) overnight at 4°C. Following the coating, the plates were washed six times. The first three washes used PBS-Tween consisting of Phosphate buffer saline (PBS) (Gibco by Life Technologies, Paisley, Scotland, UK) with 0.05% Tween-20 (Fisher Scientific, Fair Lawn, NJ). The second three washes were performed with PBS alone. Following the washes the plates were blocked with blocking buffer consisting of 2% Bovine Serum Albumin (Sigma, Saint Louis, MO) in PBS-Tween for 1 hour at 37°C. Serum was diluted to 1:400 in blocking buffer and incubated for 1 hour at 37°C. Each sample was run in duplicate. Following six washes, three with PBS-Tween and three with PBS, a 1:500 preparation of goat anti-mouse IgG HRP secondary antibody (Southern Biotech, Birmingham, AL) was added to the wells and incubated for 1 hour at 37°C to detect bound antibody. Each plate had both a negative control consisting of toxin coated wells treated with the secondary antibody and substrate as well as a positive control consisting of toxin coated wells reacted with either mouse anti-toxin A (clone TCC8) or mouse anti-toxin B (clone 2CV) monoclonal antibody diluted 1:1000 in blocking buffer (tgcBiomics, Mainz, Germany). The plates were washed six times (three times with PBS-Tween and three times with PBS) and 150μL of ABTS (2,2′azinbis [3-ethylebenzthiazoline-6-sulfonic acid] diammonium salt) substrate (Peirce Biotechnology, Rockford, IL) was added to each well and incubated at room temperature for 30 min. Following the incubation, the color-generation reaction was stopped by the addition of 100μL 1% Sodium dodecyl sulfate (Sigma, Saint Louis, MO) to each well. The optical density at 410 nm was recorded on a VersaMax plate reader (Molecular Devices, Sunnyvale, CA). The absorbance for each sample was corrected by subtracting the OD410 of the negative control wells from the OD410 measured in the sample wells.
2.8. Statistical analyses
Statistical analyses were performed using GraphPad Prism software (La Jolla, CA). Data are presented as means ± SEM of values determined from the indicated number of samples. Differences were analyzed with one-way ANOVA followed by Bonferroni post-test for multiple comparisons or by using Student t test as appropriate. Mann-Whitney U test was used to assess the differences in categorical histological scores. Kaplan-Meier plots were used to analyze survival in the challenged mice. A P value ≤ 0.05 was considered significant.
3. RESULTS
3.1. LTB4 is increased during C. difficile colitis in cefoperazone-treated mice
Antibiotic-exposed mice have proven a useful tool for studying C. difficile pathogenesis [25]. Cefoperazone treatment was recently described to alter the intestinal microbiome and metabolome to a highly susceptible state for CDI in mice [26, 27]. The timeline of infection is described in Figure 1A. Because a putative role for LTs in governing host responses to CDI could depend on the severity of disease induced by infection, we used two well-characterized strains of C. difficile that are known to induce either mild or severe infection in cefoperazone-treated mice. C. difficille strain 630 was compared in this manner with strain VPI 10463 [21]. The strain VPI is a higher toxin producer in vivo when compared to strain 630, as observed in cefoperazone-treated mice [21]. Mice infected orally with 1 × 104 spores of 630 strain developed a mild disease characterized by a gradual and slight weight loss (<10% of baseline body weight) which peaked between days 4 and 6 after infection (Fig. 1B). This infection was associated with a mild inflammatory response in the intestine analyzed at day 4 post inoculation (Fig. 1C). In contrast, mice infected with 1 × 102 spores of strain VPI 10463 developed a much more severe disease, with rapid severe weight loss (≥ 20% of initial weight body) (Fig. 1F), and an acute intestinal inflammatory response characterized by intense neutrophilic inflammation, edema and epithelial damage only 32 hours post-infection (Fig. 1G). In addition, mice infected with VPI developed clinical signs of disease such as diarrhea and hunched posture, necessitating euthanasia within 34 to 40 hours after infection. Using these two models of CDI severity we evaluated the LT profile in the cecal tissues of infected mice. Cecal LTB4 levels were significantly increased at days 4 and 8 after 630 infection (Fig. 1D) and by 32 hours after VPI 10463 infection (Fig. 1H). However, no statistical difference was observed in intestinal levels of cysLTs between mice infected with 630 (Fig. 1E) or VPI 10463 (Fig. 1I) compared to uninfected animals.
Figure 1. LTB4 production is induced in C. difficile infection.

Experimental timeline for C. difficile infection (A). C57BL/6 mice were treated for 5 days with cefoperazone in drinking water followed by 2 days of recovery period in regular drinking water and then orally infected with 104 spores of strain 630 (left panels) or 102 spores of strain VPI 10463 (right panels). The 630-infected mice develop a mild disease with slight weight loss (B), and mild intestinal inflammation and damage (C), while VPI 10463-infected mice develop a more severe disease showing a rapid weight loss (F) and intense, edematous intestinal inflammation and damage (G). Animals infected with either 630 or VPI 10463 strain showed increased intestinal level of LTB4 (D, H) but not cysLT (E, I) compared to uninfected animals. Leukotrienes were measured in cecal homogenates by ELISA at the indicated time points. Data are presented as representative images of H&E stained cecum sections or as means ±SEM. Size bars in histological images = 200 μm. Asterisks indicate the cecal lumen and arrows indicate submucosal edema. Statistical comparisons were made using one-way ANOVA followed by Bonferroni’s multiple comparison tests. * P < 0.05 (n = 12 for weight loss and n = 4–5 for ELISA).
3.2. Endogenously generated LTs do not alter C. difficile infection severity
We next examined the extent to which disease severity during CDI is influenced by endogenous LT production. To address this possibility, we used 5-LO-deficient (5-LO−/−) mice, which lack the enzyme involved in the first committed step in LT biosynthesis. Mice infected with C. difficile strain 630 were monitored over 21 days and the percentage of body weight lost was similar between wild type C57BL/6 (WT) and 5-LO−/− mice (Fig. 2A). Both WT and 5-LO−/− mice reached a peak of weight loss by day 4 after infection followed by gradual weight recovery (Fig. 2A). In concert with weight loss, C. difficile colonization levels in stool from all animals infected with 630 was similar in WT and 5-LO−/− groups when analyzed at day 4 (Fig. 2B). Because severe CDI is characterized by abundant inflammatory responses, which could be influenced by LTs, we next assessed endogenous LTs as modulators of severe infection caused by strain VPI 10463. In addition to using 5-LO−/− mice, we infected mice previously exposed to the LT synthesis inhibitor MK886 or vehicle control. Although VPI 10463 induced rapid weight loss (Fig. 2C, E) and lethality (Fig. 2D, F) in cefoperazone-treated mice, there were no differences in these settings when LTs were pharmacologically (Fig 2C, D) or genetically inhibited (Fig 2E, F), compared to their respective mock-treated or wild type controls. Additionally, culture of cecal contents showed similar levels of C. difficile colonization in WT and 5-LO−/− mice challenged with VPI 10463 after 36 hours of challenge (data not shown).
Figure 2. Abrogation of leukotriene synthesis does not alter disease severity and colonization level in the mouse model of CDI.

Mice lacking the 5-lipoxygenase enzyme (5-LO−/−) and wild type (WT) mice infected with 104 spores of 630 strain (upper panels) exhibited a similar percentage of weight loss during the course of disease (A) and showed no difference in C. difficile colonization levels, as determined in fecal contents 4 days late of infection (B). Animals infected with 102 spores of VPI 10463 strain (middle and bottlom panels) exhibited similar percentages of weight loss (C) and survival (D) when orally treated with 10 mg/kg of the 5-LO inhibitor MK886. 5-LO−/− mice showed no difference in weight loss (E) and survival rate (F) compared to WT. Data are expressed as means ±SEM. Statistical comparisons were made using one-way ANOVA followed by Bonferroni’s multiple comparison tests. Kaplan-Meier plots were used to analyze survival. (n = 8).
3.3. LTs were not associated with intestinal inflammation in C. difficile-infected mice
It is apparent from the present data (Fig. 3) and data from previous study [21], that the histologic changes resulting from CDI colitis in the cefoperazone mouse model are characterized by damage to the intestinal epithelium, which consists of vacuolar degeneration and increased loss of apical tip enterocytes, intestinal neutrophil infiltration within submucosa and mucosa, and severe edema within the submucosa. To assess the extent to which LTs are a component of the inflammatory response and tissue damage in CDI, we ablated the production of these lipids in mice infected with the severe disease-inducing strain VPI 10463. Representative histopathological specimens of cecum harvested by 36 hours of VPI 10463 infection are shown in Figure 3A. Mice treated with MK886 showed no significant difference in edema (Fig. 3B), neutrophilic inflammation (Fig. 3C) or epithelial damage (Fig. 3D) versus vehicle-treated control mice. Similar results were observed in WT and 5-LO−/− mice infected with VPI 10463, revealing that LTs were not involved in histologic changes during CDI in this mouse system (Fig 3E–G).
Figure 3. Effects of leukotrienes on intestinal inflammation and damage caused by CDI.

Mice were infected with VPI 10463 (102 spores) or mock-infected and cecum was harvested after 36 hours for H&E stain. Representative images of tissue sections from controls (uninfected and infected mice) and leukotriene-ablated (MK886 treated or 5-LO−/−) mice are depicted (A). Infected mice treated with 10 mg/kg of the leukotriene inhibitor MK886 (administered 1 hour before and 24 hour after infection) or vehicle had similar scoring for edema (B), neutrophilic inflammation (C) and epithelial damage (D). Genetic abrogation of leukotriene synthesis in 5-LO−/− mice resulted in no difference in edema (E), neutrophilic inflammation (F) and epithelial damage scoring (G) in cecum from infected mice. Mann-Whitney U post hoc test was used to access significant differences. Histological images are of hematoxylin and eosin-stained sections. Size bars indicate 100 μm and arrows indicate submucosal edema accompanied by neutrophilic inflammation.
3.4. LTs failed to modulate the cytokine burst and the adaptive immune response induced during CDI
Because the host response appears to cause collateral tissue damage during CDI, we next sought to investigate the role of LTs in modulating intestinal cytokine production during severe disease. The VPI 10463 infection triggered high levels of cytokines in the cecum of cefoperazone-treated mice after 36 hours of infection (Fig. 4). However, MK886 treatment (left side of the graphs) was not able to modulate the TNF-α (Fig. 4A), IFN-γ (Fig. 4B), IL-1β (Fig. 4C), KC (Fig. 4D), IL-22 (Fig. 4E) and IL-10 (Fig. 4F) concentrations in the intestine of VPI 10463 infected mice. Moreover, cytokine levels in the intestine were similar in WT and 5-LO−/− deficient mice by 36 hours of VPI 10463 infection (right side of the graphs). We also examined whether the adaptive immune response generated during CDI requires LTs. To address this question, we infected WT and 5-LO−/− mice with 630, a non-lethal strain associated with a mild disease in our model, and measured anti-toxin A IgG and anti-toxin B IgG after 21 days of infection (Fig. 5). However, we did not observe differences in anti-toxin A IgG between both groups of mice. Anti-toxin B IgG showed values below the limit of detection (data not shown). Collectively, these results indicate that LTs do not participate in the host response induced by C. difficile.
Figure 4. Leukotrienes are not associated with increased cytokine production in CDI.

Cecal homogenates from mice treated with 10 mg/kg of MK886 (left side) and from 5-LO−/− mice (right side) were used to measure TNF-α (A), IFN-γ (B), IL-1β (C), KC (D), IL-22 (E) and IL-10 (F). Cecal snips were harvested after 36 hours of VPI 10463 infection (102 spores) and ELISA was used to quantify cytokines production in homogenates. Data are expressed as means ±SEM Statistical comparisons were made using one-way ANOVA followed by Bonferroni’s multiple comparison tests. * P < 0.05 (n = 4–8).
Figure 5. Serum IgG response to toxin A in 5-LO−/− mice.
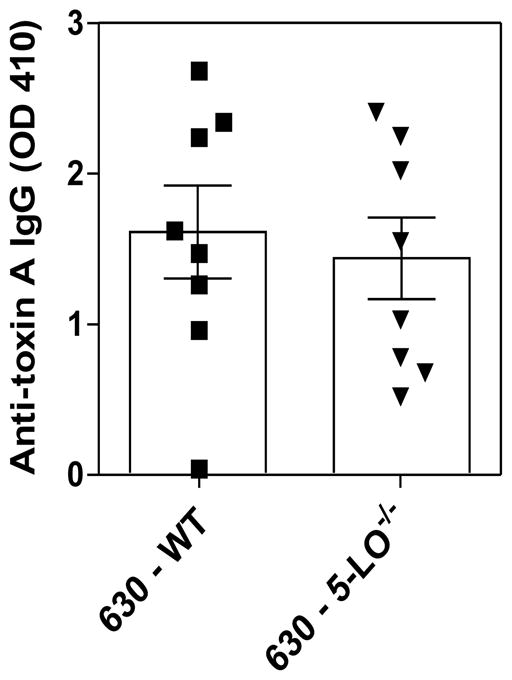
5-LO−/− and wild type mice were i.g. infected with 104 spores of C. difficile (630) and blood collected 21 days after infection. Serum anti-Toxin A IgG responses were determined by ELISA. Data are expressed as means ±SEM. Statistical comparisons were made using unpaired Student t test.
4. DISCUSSION
LTs are potent lipid mediators of inflammation that have been implicated in a several types of colitis, including CDI. The particular capacity for LTB4 to attract neutrophils to sites of inflammation suggested that this eicosanoid might be an important mediator in this infection. Surprisingly, LTs were not found to play a role in governing the outcome or severity of colitis in the CDI mouse models presented here. Despite the fact that LTB4 was highly induced during infection, impairing LT production did not impact physical signs of disease or the histopathological degree of intestinal inflammation.
The inflammatory pathways activated during CDI, and the mechanisms whereby such inflammation translates into tissue damage, are incompletely understood. It has been shown that the large C. difficile toxins lead to inflammasome activation and signaling pathways involving NF-κB, STAT3 and MAP kinase [28–30]. Such toxins also induce monocyte, macrophage and mast-cell activation leading to an intense release of cytokines including IL-1β, TNF-α, IFN-γ and IL-8, which propagate the inflammation and may be responsible in part for histopathological features of CDI [31–33]. Furthermore, neutrophil influx to the epithelium and submucosa is a hallmark of severe CDI [21], but inhibition of neutrophil influx in rabbits did not lead to complete protection [34] and this appeared to reflect the direct injury of epithelial cells by C. difficile toxins [7].
LTs are associated with infections [35, 36] and several inflammatory conditions [37–39], including colitis [40]. For example, LT inhibition has been noted to ameliorate the clinical signs of experimental model of colitis induced by dinitrobenzene sulfonic acid (DNBS) [41]. It was notable in the present study that live C. difficile induced high levels of LTB4 in the intestines of infected mice, but cysLTs were not significantly induced. We chose to measure total cysLTs because of the rapid, sequential metabolism of LTC4 to LTD4 and then to LTE4, which is why we are unable to address whether changes in individual cysLT compounds (i.e., LTC4, LTD4, or LTE4) occurred. It is noteworthy that our results differ from other studies showing that both LTB4 and LTC4 production were induced in rat ileal loops challenged with TcdA [12]. We speculate that these differences reflect differences between true CDI versus intoxication without infection (i.e., using toxin A or B challenge without live pathogens). We hypothesize that the environment during true infection might drive leukotriene activation through one cascade in favor of another (e.g., LTA4 hydrolase leading to LTB4 production instead of LTC4 synthase culminating in LTC4 generation). Future studies might examine differences between true infection vs. toxin challenge, but that is beyond the scope of our work, which was focused on infection per se. The production of LTB4 correlated with clinical signs of disease and inflammation observed in both mild and severe mouse models of CDI employed in this work. Our data are consistent with studies reporting significant increases of LTB4 in animal models of colitis induced by TcdA [11, 42] and in the intestine of CDI patients with ulcerations [43]. Apart from LTB4, prostaglandin E2 [44] and enzymes related to eicosanoid biosynthesis [44, 45] have also been shown to be induced by C. difficile toxin stimulation of the gastrointestinal tissue.
To determine the extent to which LTB4 induced by CDI is the cause of associated colitis, we evaluated the capacity of LTs to induce histological alterations in infected mice. It is noteworthy that LTs have been associated with neutrophil influx, intestinal fluid accumulation and tissue damage in isolated rat ileal segments stimulated with TcdA [11, 42]. Also, treatment with the LT-inhibitor drug MK886 limited the TcdB-induced neutrophil migration into the peritoneal cavity of rats [46]. Nevertheless, the current studies found that LT inhibition did not alter neutrophil influx, edema or epithelial damage in the intestine of mice challenged with a high virulent C. difficile strain. Moreover, LTs modulate inflammation and immune responses through the regulation of cytokine production [47, 48], but this work revealed that LT abrogation did not alter the intense production of cytokines triggered by CDI.
Antibody responses against C. difficile antigens are associated with protection in asymptomatic carriers of C. difficile [49] and high antibody production is associated with protection against infection recurrence [50, 51]. Also, passive and active immunization of hamsters [52, 53] and mice [54, 55] has been shown to protect against diarrhea and death. In this light, we explored the possibility that LTs could be associated with adaptive immune responses in CDI. In other models of inflammation, these lipid mediators have been shown to modulate cell migration and activation, including that of T-cells, supporting a role in the development of adaptive immune responses [56–58]. To test this capacity in CDI, we measured anti-toxin A/B antibodies in the serum of normal and LT-deficient mice infected with a non-lethal C. difficile strain (allowing the mice to be followed for 3 weeks post infection). In all mice tested, the concentration of anti-TcdA antibody was similar while the anti-TcdB antibodies were undetectable. This suggests that LTs do no play a role in the development of an antibody response against C. difficile.
While the present work does not implicate LTs in the pathogenesis of CDI, our studies do not rule out this possibility. Our work has several limitations that could be limit the generalizability of our results and conclusions. These experiments were conducted in mice, which does not rule out a possible impact of LTs in human infection. More specifically, these studies were conducted in C57BL/6 mice, and the extent to which the involvement of LTs varies across mouse strains remains to be studied. An additional caveat is that the present work was conducted using two well-characterized strains of C. difficile that reliably produced either a mild form of colitis (strain 630) or a rapidly lethal, severe infection (strain VPI 10463). However, different results might have been obtained had we used primary human clinical isolates of C. difficile and/or strains of the epidemic 027 ribotype. Future work will be needed to determine the impact of strain type of host lipid immunomodulation.
Overall, our findings suggest that LTs are induced in CDI, but the associated colitis is independent of their action. Moreover, our study highlights the complexity of CDI and the importance of using live bacteria to study the pathological mechanisms of CDI-associated diseases.
Acknowledgments
This work was supported by a grant from the NIAID at the NIH (grant number 5U19AI090871-02). BCT was funded by a grant from the CAPES (grant number BEX 9179/11-9). We thank K. Wozniak for support in animal facility and for help with animal harvesting.
Abbreviations
- CDI
Clostridium difficile infection
- TcdA
Clostridium difficile toxin A
- TcdB
Clostridium difficile toxin B
- LT
Leukotriene
- LTB4
Leukotriene B4
- CysLTs
Cysteinyl leukotrienes
- 5-LO
5-lipoxygenase
Footnotes
DISCLOSURES:
The authors have no conflict of interest.
References
- 1.Depestel DD, Aronoff DM. Epidemiology of Clostridium difficile Infection. J Pharm Pract. 2013;26(5):464–75. doi: 10.1177/0897190013499521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bartlett JG, Gerding DN. Clinical recognition and diagnosis of Clostridium difficile infection. Clin Infect Dis. 2008;46(Suppl 1):S12–8. doi: 10.1086/521863. [DOI] [PubMed] [Google Scholar]
- 3.Centers for Disease C and Prevention. Surveillance for community-associated Clostridium difficile--Connecticut, 2006. MMWR Morb Mortal Wkly Rep. 2008;57(13):340–3. [PubMed] [Google Scholar]
- 4.Dumyati G, et al. Community-associated Clostridium difficile infections, Monroe County, New York, USA. Emerg Infect Dis. 2012;18(3):392–400. doi: 10.3201/eid1803.102023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lucado J, Gould C, Elixhauser A. Healthcare Cost and Utilization Project (HCUP) Statistical Briefs. Rockville (MD): 2006. Clostridium Difficile Infections (CDI) in Hospital Stays, 2009: Statistical Brief #124. [PubMed] [Google Scholar]
- 6.Pepin J, et al. Clostridium difficile-associated diarrhea in a region of Quebec from 1991 to 2003: a changing pattern of disease severity. CMAJ. 2004;171(5):466–72. doi: 10.1503/cmaj.1041104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuehne SA, et al. The role of toxin A and toxin B in Clostridium difficile infection. Nature. 2010;467(7316):711–3. doi: 10.1038/nature09397. [DOI] [PubMed] [Google Scholar]
- 8.Koon HW, et al. Human monoclonal antibodies against Clostridium difficile toxins A and B inhibit inflammatory and histologic responses to the toxins in human colon and peripheral blood monocytes. Antimicrob Agents Chemother. 2013;57(7):3214–23. doi: 10.1128/AAC.02633-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pothoulakis C, Lamont JT. Microbes and microbial toxins: paradigms for microbial-mucosal interactions II. The integrated response of the intestine to Clostridium difficile toxins. Am J Physiol Gastrointest Liver Physiol. 2001;280(2):G178–83. doi: 10.1152/ajpgi.2001.280.2.G178. [DOI] [PubMed] [Google Scholar]
- 10.Madan R, WA Immune responses to Clostridium difficile infection. Trends Mol Med. 2012;18(11):658–66. doi: 10.1016/j.molmed.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McVey DC, Vigna SR. The role of leukotriene B4 in Clostridium difficile toxin A-induced ileitis in rats. Gastroenterology. 2005;128(5):1306–16. doi: 10.1053/j.gastro.2005.03.017. [DOI] [PubMed] [Google Scholar]
- 12.Pothoulakis C, et al. Ketotifen inhibits Clostridium difficile toxin A-induced enteritis in rat ileum. Gastroenterology. 1993;105(3):701–7. doi: 10.1016/0016-5085(93)90886-h. [DOI] [PubMed] [Google Scholar]
- 13.Peters-Golden M, Henderson WR., Jr Leukotrienes. N Engl J Med. 2007;357(18):1841–54. doi: 10.1056/NEJMra071371. [DOI] [PubMed] [Google Scholar]
- 14.Sadik CD, Luster AD. Lipid-cytokine-chemokine cascades orchestrate leukocyte recruitment in inflammation. J Leukoc Biol. 2012;91(2):207–15. doi: 10.1189/jlb.0811402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ford-Hutchinson AW, et al. Leukotriene B, a potent chemokinetic and aggregating substance released from polymorphonuclear leukocytes. Nature. 1980;286(5770):264–5. doi: 10.1038/286264a0. [DOI] [PubMed] [Google Scholar]
- 16.Sharon P, Stenson WF. Enhanced synthesis of leukotriene B4 by colonic mucosa in inflammatory bowel disease. Gastroenterology. 1984;86(3):453–60. [PubMed] [Google Scholar]
- 17.Rask-Madsen J, et al. 5-Lipoxygenase inhibitors for the treatment of inflammatory bowel disease. Agents Actions. 1992;(Spec No):C37–46. [PubMed] [Google Scholar]
- 18.Stanke-Labesque F, et al. Urinary leukotriene E4 excretion: a biomarker of inflammatory bowel disease activity. Inflamm Bowel Dis. 2008;14(6):769–74. doi: 10.1002/ibd.20403. [DOI] [PubMed] [Google Scholar]
- 19.Rocha MF, et al. Clostridium difficile toxin A induces the release of neutrophil chemotactic factors from rat peritoneal macrophages: role of interleukin-1beta, tumor necrosis factor alpha, and leukotrienes. Infect Immun. 1997;65(7):2740–6. doi: 10.1128/iai.65.7.2740-2746.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perez J, Springthorpe VS, Sattar SA. Clospore: a liquid medium for producing high titers of semi-purified spores of Clostridium difficile. J AOAC Int. 2011;94(2):618–26. [PubMed] [Google Scholar]
- 21.Theriot CM, et al. Cefoperazone-treated mice as an experimental platform to assess differential virulence of Clostridium difficile strains. Gut Microbes. 2011;2(6):326–34. doi: 10.4161/gmic.19142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Costa MF, et al. Leukotriene B4 mediates gammadelta T lymphocyte migration in response to diverse stimuli. J Leukoc Biol. 2010;87(2):323–32. doi: 10.1189/jlb.0809563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sforcin JM, et al. Effect of a leukotriene inhibitor (MK886) on nitric oxide and hydrogen peroxide production by macrophages of acutely and chronically stressed mice. J Pharm Pharmacol. 2007;59(9):1249–54. doi: 10.1211/jpp.59.9.0009. [DOI] [PubMed] [Google Scholar]
- 24.Reeves AE, et al. Suppression of Clostridium difficile in the gastrointestinal tracts of germfree mice inoculated with a murine isolate from the family Lachnospiraceae. Infect Immun. 2012;80(11):3786–94. doi: 10.1128/IAI.00647-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lawley TD, Young VB. Murine models to study Clostridium difficile infection and transmission. Anaerobe. 2013;24:94–7. doi: 10.1016/j.anaerobe.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Theriot CM, et al. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat Commun. 2014;5:3114. doi: 10.1038/ncomms4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reeves AE, et al. The interplay between microbiome dynamics and pathogen dynamics in a murine model of Clostridium difficile Infection. Gut Microbes. 2011;2(3):145–58. doi: 10.4161/gmic.2.3.16333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chae S, et al. Epithelial cell I kappa B-kinase beta has an important protective role in Clostridium difficile toxin A-induced mucosal injury. J Immunol. 2006;177(2):1214–20. doi: 10.4049/jimmunol.177.2.1214. [DOI] [PubMed] [Google Scholar]
- 29.Jefferson KK, Smith MF, Jr, Bobak DA. Roles of intracellular calcium and NF-kappa B in the Clostridium difficile toxin A-induced up-regulation and secretion of IL-8 from human monocytes. J Immunol. 1999;163(10):5183–91. [PubMed] [Google Scholar]
- 30.Ng J, et al. Clostridium difficile toxin-induced inflammation and intestinal injury are mediated by the inflammasome. Gastroenterology. 2010;139(2):542–52. 552 e1–3. doi: 10.1053/j.gastro.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 31.de Araujo Junqueira AF, et al. Adenosine deaminase inhibition prevents Clostridium difficile toxin A-induced enteritis in mice. Infect Immun. 2011;79(2):653–62. doi: 10.1128/IAI.01159-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meyer GK, et al. Clostridium difficile toxins A and B directly stimulate human mast cells. Infect Immun. 2007;75(8):3868–76. doi: 10.1128/IAI.00195-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ishida Y, et al. Essential involvement of IFN-gamma in Clostridium difficile toxin A-induced enteritis. J Immunol. 2004;172(5):3018–25. doi: 10.4049/jimmunol.172.5.3018. [DOI] [PubMed] [Google Scholar]
- 34.Kelly CP, et al. Neutrophil recruitment in Clostridium difficile toxin A enteritis in the rabbit. J Clin Invest. 1994;93(3):1257–65. doi: 10.1172/JCI117080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Soares EM, et al. Leukotriene B4 enhances innate immune defense against the puerperal sepsis agent Streptococcus pyogenes. J Immunol. 2013;190(4):1614–22. doi: 10.4049/jimmunol.1202932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morato-Marques M, et al. Leukotrienes target F-actin/cofilin-1 to enhance alveolar macrophage anti-fungal activity. J Biol Chem. 2011;286(33):28902–13. doi: 10.1074/jbc.M111.235309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen M, et al. Neutrophil-derived leukotriene B4 is required for inflammatory arthritis. J Exp Med. 2006;203(4):837–42. doi: 10.1084/jem.20052371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trindade BC, et al. Leukotrienes are upregulated and associated with human T-lymphotropic virus type 1 (HTLV-1)-associated neuroinflammatory disease. PLoS One. 2012;7(12):e51873. doi: 10.1371/journal.pone.0051873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peters-Golden M, et al. Protection from pulmonary fibrosis in leukotriene-deficient mice. Am J Respir Crit Care Med. 2002;165(2):229–35. doi: 10.1164/ajrccm.165.2.2104050. [DOI] [PubMed] [Google Scholar]
- 40.Mazzon E, et al. 5-lipoxygenase modulates the alteration of paracellular barrier function in mice ileum during experimental colitis. Shock. 2006;25(4):377–83. doi: 10.1097/01.shk.0000209530.30564.22. [DOI] [PubMed] [Google Scholar]
- 41.Cuzzocrea S, et al. 5-Lipoxygenase modulates colitis through the regulation of adhesion molecule expression and neutrophil migration. Lab Invest. 2005;85(6):808–22. doi: 10.1038/labinvest.3700276. [DOI] [PubMed] [Google Scholar]
- 42.Burakoff R, et al. Effects of purified Clostridium difficile toxin A on rabbit distal colon. Gastroenterology. 1995;109(2):348–54. doi: 10.1016/0016-5085(95)90320-8. [DOI] [PubMed] [Google Scholar]
- 43.Lauritsen K, et al. In vivo profiles of eicosanoids in ulcerative colitis, Crohn’s colitis, and Clostridium difficile colitis. Gastroenterology. 1988;95(1):11–7. doi: 10.1016/0016-5085(88)90284-3. [DOI] [PubMed] [Google Scholar]
- 44.Alcantara C, et al. Role of inducible cyclooxygenase and prostaglandins in Clostridium difficile toxin A-induced secretion and inflammation in an animal model. J Infect Dis. 2001;184(5):648–52. doi: 10.1086/322799. [DOI] [PubMed] [Google Scholar]
- 45.Lima AA, et al. Role of phospholipase A2 and tyrosine kinase in Clostridium difficile toxin A-induced disruption of epithelial integrity, histologic inflammatory damage and intestinal secretion. J Appl Toxicol. 2008;28(7):849–57. doi: 10.1002/jat.1348. [DOI] [PubMed] [Google Scholar]
- 46.Souza MH, et al. The involvement of macrophage-derived tumour necrosis factor and lipoxygenase products on the neutrophil recruitment induced by Clostridium difficile toxin B. Immunology. 1997;91(2):281–8. doi: 10.1046/j.1365-2567.1997.00243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peres CM, et al. Inhibition of leukotriene biosynthesis abrogates the host control of Mycobacterium tuberculosis. Microbes Infect. 2007;9(4):483–9. doi: 10.1016/j.micinf.2007.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saiwai H, et al. The LTB4-BLT1 axis mediates neutrophil infiltration and secondary injury in experimental spinal cord injury. Am J Pathol. 2010;176(5):2352–66. doi: 10.2353/ajpath.2010.090839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kyne L, et al. Asymptomatic carriage of Clostridium difficile and serum levels of IgG antibody against toxin A. N Engl J Med. 2000;342(6):390–7. doi: 10.1056/NEJM200002103420604. [DOI] [PubMed] [Google Scholar]
- 50.Kyne L, et al. Association between antibody response to toxin A and protection against recurrent Clostridium difficile diarrhoea. Lancet. 2001;357(9251):189–93. doi: 10.1016/S0140-6736(00)03592-3. [DOI] [PubMed] [Google Scholar]
- 51.Leav BA, et al. Serum anti-toxin B antibody correlates with protection from recurrent Clostridium difficile infection (CDI) Vaccine. 2010;28(4):965–9. doi: 10.1016/j.vaccine.2009.10.144. [DOI] [PubMed] [Google Scholar]
- 52.Babcock GJ, et al. Human monoclonal antibodies directed against toxins A and B prevent Clostridium difficile-induced mortality in hamsters. Infect Immun. 2006;74(11):6339–47. doi: 10.1128/IAI.00982-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Giannasca PJ, et al. Serum antitoxin antibodies mediate systemic and mucosal protection from Clostridium difficile disease in hamsters. Infect Immun. 1999;67(2):527–38. doi: 10.1128/iai.67.2.527-538.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Corthier G, et al. Protection against experimental pseudomembranous colitis in gnotobiotic mice by use of monoclonal antibodies against Clostridium difficile toxin A. Infect Immun. 1991;59(3):1192–5. doi: 10.1128/iai.59.3.1192-1195.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gardiner DF, et al. A DNA vaccine targeting the receptor-binding domain of Clostridium difficile toxin A. Vaccine. 2009;27(27):3598–604. doi: 10.1016/j.vaccine.2009.03.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tager AM, et al. Leukotriene B4 receptor BLT1 mediates early effector T cell recruitment. Nat Immunol. 2003;4(10):982–90. doi: 10.1038/ni970. [DOI] [PubMed] [Google Scholar]
- 57.Spanbroek R, et al. 5-lipoxygenase expression in dendritic cells generated from CD34(+) hematopoietic progenitors and in lymphoid organs. Blood. 2000;96(12):3857–65. [PubMed] [Google Scholar]
- 58.Medeiros AI, et al. Leukotrienes are potent adjuvant during fungal infection: effects on memory T cells. J Immunol. 2008;181(12):8544–51. doi: 10.4049/jimmunol.181.12.8544. [DOI] [PubMed] [Google Scholar]