

Abstract
The parasitoid wasp genus Nasonia has rapidly become a genetic model system for developmental and evolutionary biology. The release of its genome sequence led to the development of high-resolution genomic tools, for both interspecific and intraspecific research, which has resulted in great advances in understanding Nasonia biology. To further advance the utility of Nasonia vitripennis as a genetic model system and to be able to fully exploit the advantages of its fully sequenced and annotated genome, we developed a genetically variable and well-characterized experimental population. In this study, we describe the establishment of the genetically diverse HVRx laboratory population from strains collected from the field in the Netherlands. We established a maintenance method that retains genetic variation over generations of culturing in the laboratory. As a characterization of its genetic composition, we provide data on the standing genetic variation and estimate the effective population size (Ne) by microsatellite analysis. A genome-wide description of polymorphism is provided through pooled resequencing, which yielded 417 331 high-quality SNPs spanning all five Nasonia chromosomes. The HVRx population and its characterization are freely available as a community resource for investigators seeking to elucidate the genetic basis of complex trait variation using the Nasonia model system.
Keywords: effective population size, genetic variation, laboratory strain, parasitoid wasp, pooled resequencing, single-nucleotide polymorphism (SNP)
Introduction
The parasitoid wasp genus Nasonia has rapidly become a genetic model system for developmental and evolutionary biology (Desplan & Beukeboom 2003; Werren & Loehlin 2009; Brown 2010; Godfray 2010; Muers 2010). As a genetic model, it has inherent advantages that rival even the current model insect systems with a much longer history and tradition. Its haplodiploid reproduction offers advantages of haploid genetics (e.g. lack of genetic dominance) in a complex eukaryotic system (Pultz et al. 2000). Several other important characteristics (easy husbandry, visible mutation markers, four interfertile species, short-generation time and large family sizes, long-term storage after diapause induction) have laid the foundations for the large-scale study of Nasonia. However, the most important step in the development of Nasonia as a genetic model has been the availability of its genome sequence (Werren et al. 2010), which largely expanded the understanding of basic biological processes in this species complex.
Of all the members within the Nasonia genus, Nasonia vitripennis is the best studied species, for which a wealth of information has been available on life-history and behavioural traits (e.g. Wylie 1966; Legner & Gerling 1967; Whiting 1967; Smith & Pimentel 1969; Davies 1975; Werren 1980, 1983a,b; King 1993; Rivers & Denlinger 1995; Drapeau & Werren 1999; Gadau et al. 1999; Bordenstein et al. 2001; Rivero & West 2002, 2005; Beukeboom & van den Assem 2004; Shuker & West 2004; Lalonde 2005; Shuker et al. 2005, 2007a,b; Bordenstein & Werren 2007; Pannebakker et al. 2008; Gadau & Wolschin 2009; Geuverink et al. 2009; Werren & Loehlin 2009; Raychoudhury et al. 2010a; Desjardins et al. 2010; Rivers et al. 2012; Niehuis et al. 2013; Gibson et al. 2013; Brucker & Bordenstein 2013). The release of its genome sequence led to the development of high-resolution genomic tools, for both interspecific (Niehuis et al. 2010; Loehlin et al. 2010a; Loehlin & Werren 2012; Desjardins et al. 2013) and intraspecific (Beukeboom et al. 2010; Pannebakker et al. 2010; Wang et al. 2013) research. The last years have seen a large number of studies that used the Nasonia genome sequence, reporting scientific breakthroughs in fundamental and applied biology (Loehlin et al. 2010b; Pannebakker et al. 2010, 2011; Verhulst et al. 2010; Werren et al. 2010; Lynch et al. 2011; Koevoets et al. 2012; Loehlin & Werren 2012; Brucker & Bordenstein 2013; Gibson et al. 2013; Niehuis et al. 2013; Paolucci et al. 2013). However, to further advance the utility of N. vitripennis as a genetic model system and to be able to fully exploit the advantages of its fully sequenced and annotated genome, a genetically variable and well-characterized experimental population, which is available for use in research worldwide, is highly required (e.g. Burke et al. 2010; Huang et al. 2012; Svenson et al. 2012).
In this study, we describe the establishment of the genetically diverse outbred HVRx laboratory population from strains that were collected from birds nest boxes and carrion baits in the Netherlands. Furthermore, we determine its (population) genetic characteristics and establish a breeding method that retains genetic variation over generations. To characterize the HVRx population, we describe the standing genetic variation and effective population size by microsatellite analysis. Pooled resequencing further yielded 417 331 high-quality SNPs, which are used for a genome-wide, high-resolution characterization of the population polymorphism. The HVRx population and its characterization are freely available as a community resource for investigators seeking to elucidate the genetic basis of complex trait variation using the Nasonia model system, for instance as a stock for association mapping in experimental evolution studies.
Materials and methods
HVRx population establishment and maintenance
In 2001, five Nasonia vitripennis strains were collected from different localities within 20 km radius from longitude 5.3 E and latitude 52.1 N in the Netherlands: Elspeet (strain ‘B5’), Bussum (strain ‘Bussum’) and the Hoge Veluwe (strains ‘HV55’, ‘HV287’ and ‘HV295’). Strains from the Hoge Veluwe were collected from bird nest boxes, while those from Elspeet and Bussum were collected from baits. Baits consisted of mesh screen bags, containing 25 laboratory hosts (Calliphora vicina) and a piece of liver. Each field strain was initiated by collecting all emerging female wasps from a nest box or bait and providing these with hosts. The number of founding females was not known, but host patches are usually colonized by multiple foundresses (Grillenberger et al. 2009b). After four generations of culturing (hosting about 100 females), ten mated females were randomly collected from each strain and individually provided with two hosts. From the offspring of each mated female, three virgin females and one male were isolated and mixed to initiate the HV population. Thus, a total 30 virgin females and 10 males from each strain were mixed in a 250-mL culture bottle and allowed to mate randomly. After 24 h, 200 hosts were added and the wasps were placed at 25 °C, under 16:8 light:dark conditions. After emergence, the HV population was split into two replicate populations (HV1 and HV2), by randomly isolating 100 mated females for each replicate and providing them with 200 hosts per replicate. Both replicates were maintained at 15 °C, 16:8 light:dark conditions.
To maximize genetic diversity, we merged the HV1 and HV2 replicates to form one single HVRx population after approximately 36 generations of separate mass culturing. From both replicates, we hosted 80 mated females on 200 hosts, and the offspring was allowed to mate randomly before dividing the wasps over four mass culture tubes (70 × 20 mm) each containing 50 fly hosts for oviposition. After 1 week, the parasitized hosts were distributed over four new mass culture tubes. After emergence, approximately 30 mated females from each mass culture tube were transferred to new mass culture tubes to initiate the next generation (Fig. 1). This breeding procedure ensures the maintenance of a large outbred population by allowing random mating between wasps of different tubes and was designed to preserve genetic diversity across generations. The HVRx population was maintained on C. vicina pupae as hosts, at 25 °C, 16: 8 light:dark conditions.
Fig 1.
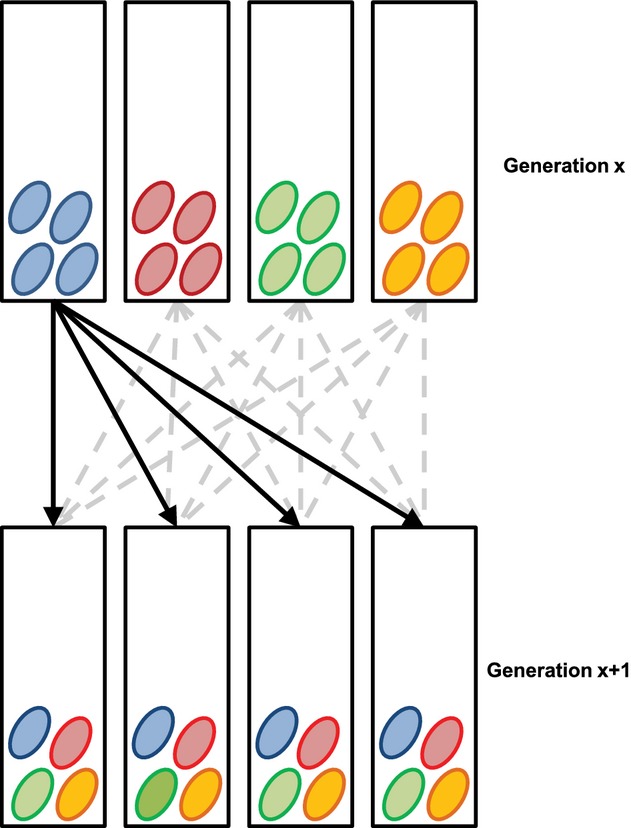
Nasonia vitripennis HVRx outbred laboratory population maintenance schedule. In each of four mass culture tubes, 40 mated female N. vitripennis wasps of generation x are provided with 50 Calliphora spp. hosts. After oviposition, the parasitized hosts are redistributed over four clean mass culture tubes, to ensure optimal mixing of the wasps over all four culture tubes and to allow mating between all wasps emerging within generation x + 1.
Microsatellite analysis
To determine and monitor the genetic diversity of the HVRx population over time, we isolated DNA from females from the HV1 (n = 14) and HV2 (n = 14) strains, and from the HVRx population in generations 1 (n = 12), 5 (n = 24), 10 (n = 24) and 32 (n = 24), using a standard high-salt-chloroform protocol (Maniatis et al. 1982). A set of 40 microsatellite markers, evenly distributed over the five chromosomes of N. vitripennis and combined into six multiplex sets, was used to determine genetic variation (Beukeboom et al. 2010; Pannebakker et al. 2010; Koevoets et al. 2012). Details of these microsatellite markers are listed in Table S1 (Supporting information). Microsatellite markers were amplified using the Qiagen multiplex PCR kit (Qiagen, Hilden, Germany) according to the manufacturer's recommendations (PCR profile: 15 min at 95 °C, followed by 30 cycles of 30 s at 94 °C, 1.5 min at annealing temperature and 1 min at 72 °C, followed by 45 min at 72 °C). Amplification was in 5 μL volumes using Applied Biosystems Veriti or Applied Biosystems 9700 thermocyclers (Applied Biosystems, Foster City, CA, USA). Fragments were diluted 400 times, separated on a Applied Biosystems 3730 DNA Analyzer and analysed using GeneMapper v4.0 (Applied Biosystems). Population genetic statistics (allelic richness R, heterozygosity HE, FST) were calculated using fstat 2.9.3 (Goudet 2001). Statistical analyses were carried out using r 2.14.1 (R Development Core Team 2012).
Theoretical estimate of effective population size
To evaluate the expected effective population size (Ne) achieved by our set-up, we simulated a population propagated under the four-tube breeding schedule that was used for maintenance of the HVRx population, as well as a population of identical census size but propagated as a single large one (i.e. in a single large tube). We modelled the sampling of a single biallelic locus in a haplodiploid system with 30 (four-replicates) or 120 (single-replicate) mated females per tube, to match the approximate number of mated females transferred each generation. For each replicate, mating between diploid females and haploid males was simulated, with females producing offspring from a single male selected with replacement from a vector of males of length rNf, where Nf is the number of females and r is the observed male over female ratio. For the multiple-replicate scheme, mated females from different tubes were mixed in equal proportions into new tubes (following Fig. 1).
For each simulation, we calculated Ft, the allelic differentiation with respect to the founding population, as: 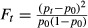 (Nicholson et al. 2002), where pt is the allele frequency in generation t and p0 is the frequency in the founding population. Ne was then estimated by solving the following nonlinear equation:
(Nicholson et al. 2002), where pt is the allele frequency in generation t and p0 is the frequency in the founding population. Ne was then estimated by solving the following nonlinear equation: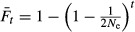 , where
, where 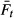 was obtained by averaging Ft over 100 000 independent simulations. This simulated estimate of Ne was compared with the classic theoretical prediction of inbreeding effective size in a haplodiploid populations derived by Wright (1933) as
was obtained by averaging Ft over 100 000 independent simulations. This simulated estimate of Ne was compared with the classic theoretical prediction of inbreeding effective size in a haplodiploid populations derived by Wright (1933) as
where Nf and Nm are the number of females and the number of males, respectively.
Empirical estimation of effective population size
We used approximate Bayesian computation (ABC, Tavare et al. 1997; Marjoram et al. 2003) to obtain estimates of the posterior distribution of realized Ne over the past 32 generations from the microsatellite data. We estimated the following summary statistics from the data in generation 5, 10 and 32: number of alleles, expected heterozygosity and Ft (as defined above and averaged over all loci and alleles). This resulted in a vector of length 9, O, with each element Oij containing the observed value for summary statistic i in generation j. We then simulated 10 000 000 instances of multilocus genotypes sampled from a diploid Wright–Fisher population subject to drift over 32 generations, with proposal values of Ne drawn from a uniform distribution between 0 and 1000. Starting allele frequencies were set to match those observed in the base population. In each independent simulation, the same summary statistics as above were measured in generation 5, 10 and 32, yielding a vector S of simulated summary statistics, with each element Sij corresponding to a value in Oij. Following Pritchard et al. (1999), we only accepted iterations for which | Sij – Oij |/Oij < ε for each element of Sij and Oij. The threshold value ε was set to 0.11 to achieve an acceptance rate of 0.001.
Genome resequencing and analysis
To resequence the HVRx base population, DNA from 20 pooled females from generation four was extracted and purified using Qiagen DNeasy columns (Qiagen). A 101-bp unpaired and a 76-bp paired-end library were constructed and subsequently sequenced by the University Medical Centre Groningen (UMCG) Genome Analysis Facility according to standard Illumina protocols. The unpaired library was run on a single lane and the paired-end library on five lanes of an Illumina Genome Analyzer II (Illumina, San Diego, CA, USA). Sequence data were provided to NasoniaBase (Munoz-Torres et al. 2011) and submitted to the NCBI Short Read Archive with accession no. SRP022050.
Reads were aligned to the v2.0 of the N. vitripennis genome (Werren et al. 2010) using Mosaik Aligner v1.1.0021 (https://code.google.com/p/mosaik-aligner/). SNPs were filtered from the HVRx assembly using Varscan.v2.2.8 (Koboldt et al. 2012), according to the following rules: minimum consensus quality: 20; minimum count of minor allele: 2, minimum coverage: 8. Reads with mapping qualities less than 20 were discarded. SNP data were provided to NasoniaBase (Munoz-Torres et al. 2011) and deposited in Dryad (Dryad Entry doi:10.5061/dryad.f2330).
Mean SNP densities were calculated in nonoverlapping 100 kb windows, with a minimum mean coverage of 8. Genome-wide diversity analysis was performed using the popoolation pipeline (Kofler et al. 2011). We estimated nucleotide diversity π in nonoverlapping 100 kb windows, using the same parameters augmented with a maximum coverage of 1 000 000, a fraction of the window that has a coverage between the minimum and maximum coverage of 0.6. Additional statistical analyses were carried out using r 2.14.1 (R Development Core Team 2012).
Results
After being maintained in the laboratory for approximately 36 generations, the HV1 and HV2 populations had genetically diverged (FST = 0.26, P < 0.05, Table1). The mean heterozygosity was not statistically different between both replicates (mean HE ± SE HV1 = 0.53 ±0.03, HV2 = 0.46 ± 0.05, t74 = 1.41, P = 0.163, Table2), which is reflected in the heterozygosity per locus for HV1 and HV2 (Table S2, Supporting information). The mean allelic richness did show differences between both replicate populations (mean R ± SE HV1 = 3.08 ± 0.18, HV2 = 2.52 ± 0.14, t78 = 2.50, P = 0.015, Table2).
Table 1.
Genetic differentiation in the Nasonia vitripennis HVRx outbred laboratory population. Pairwise FST values between HV1 and HV2 founder populations and generations 1, 5, 10 and 32 of the HVRx population over 40 microsatellite loci combined
| HV1 | HV2 | HVRx-G1 | HVRx-G5 | HVRx-G10 | HVRx-G32 | |
|---|---|---|---|---|---|---|
| HV1 | 0.00 | 0.26* | 0.04* | 0.04* | 0.06* | 0.11* |
| HV2 | 0.00 | 0.16* | 0.17* | 0.18* | 0.22* | |
| HVRx-G1 | 0.00 | 0.02 | 0.03* | 0.07* | ||
| HVRx-G5 | 0.00 | 0.03* | 0.06* | |||
| HVRx-G10 | 0.00 | 0.05* | ||||
| HVRx-G32 | 0.00 |
Significant values P < 0.05 after Bonferroni correction following G-statistics (as implemented in Fstat).
Table 2.
Genetic variation in the Nasonia vitripennis HVRx outbred laboratory population
| Expected heterozygosity HE | Allelic richness R | |
|---|---|---|
| HV1 | 0.53 (0.03)a | 3.08 (0.18)ab |
| HV2 | 0.46 (0.04)ab | 2.52 (0.14)a |
| HVRx-G1 | 0.58 (0.04)a | 3.52 (0.22)b |
| HVRx-G5 | 0.60 (0.03)ac | 3.45 (0.20)b |
| HVRx-G10 | 0.56 (0.03)a | 3.40 (0.18)b |
| HVRx-G32 | 0.56 (0.03)a | 3.34 (0.21)b |
Table shows the mean expected heterozygosity HE and the mean allelic richness R. Standard errors are in parentheses.
Different lowercase letters indicate significant differences at P < 0.05 for each estimate.
The HV1 and HV2 replicates were merged to start the HVRx population with maximum genetic diversity. We subsequently cultured the HVRx population following a breeding procedure designed to retain genetic diversity over generations (Fig. 1). To confirm the effectiveness of our breeding procedure, we monitored the population genetic characteristics of the HVRx population at generations 1, 5, 10 and 32 after establishment.
Genetic differentiation
Already at generation 1, the HVRx population had significantly diverged from both original populations (FST = 0.04 (HV1) and 0.16 (HV2), P < 0.05, Table1). After generation 5, the HVRx population started to diverge modestly but significantly between subsequent generations (Table1). However, 32 generations of controlled breeding resulted in a FST value of 0.07 between generation 1 and generation 32, which is considerably lower than that of the differentiation between the original HV1 and HV2 populations after 36 generations (Table1).
Genetic variation
Mean heterozygosities (HE ± SE) for HVRx generations 1, 5, 10 and 32 did not change and were 0.58 ± 0.04, 0.60 ± 0.03, 0.56 ± 0.03 and 0.56 ± 0.03, respectively. An overall comparison, including also the founding populations HV1 and HV2, showed that the only significant difference in heterozygosity was between HV2 and HVRx generation 5 (Tukey's HSD critical value = 0.127, Table2). Our results indicate that heterozygosity was effectively maintained over 32 generations. This pattern is also reflected in the per-locus heterozygosity (Table S2, Supporting information).
Mean allelic richness (R ± SE) showed a slight decrease over generations 1, 5, 10 and 32 and was 3.52 ± 0.22, 3.45 ± 0.20, 3.40 ± 0.18 and 3.34 ± 0.21, respectively. An overall comparison, including also the founding populations HV1 and HV2 only, showed significant differences in allelic richness between HV2 and all HVRx generations (One-way aov; F5,234 = 3.87, P = 2.170e-3; Tukey HSD critical value = 0.741, Table2). Overall, our results indicate that our controlled breeding method effectively limits the decrease in allelic richness over 32 generations.
Effective population size
The empirical estimation of effective population size (Ne), based on the diversity parameters above, yielded a posterior mode of 195 and a mean of 236 diploid individuals. The computer simulations of the different breeding schemes yielded nearly identical effective population sizes of Ne = 177 and Ne = 173 for the four-replicate and single-replicate mating scheme, respectively, entirely consistent with the theoretical prediction of Ne = 173 and falling within 0.9 and 0.3 standard deviations of the posterior mean and mode, respectively (Fig. 2).
Fig 2.
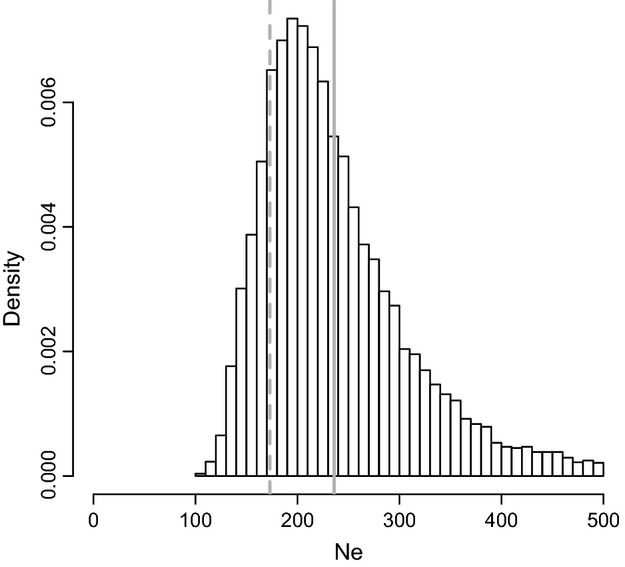
Effective population size Ne in the Nasonia vitripennis HVRx outbred laboratory population. Posterior density of the empirical estimate of Ne based on 40 microsatellite loci, obtained by approximate Bayesian computation. Blue, red and green lines indicate the theoretical prediction based on Wright (1933), the posterior mean and posterior median of the empirical estimate, respectively.
Genomic variation
We found considerable genetic variability in the HVRx population, with a total of 417 331 SNPs distributed over the five chromosomes, or about 1 SNP per 796.7 bp [based on a physical genome size of 332.5 Mb (Gregory, 2013)]. SNP density varied between the chromosomes (one-way aov; F4,1880 = 9.58, P = 1.16e-07, Table3), with the lowest mean density per 100 kb window on chromosome 1. The pattern in SNP density differentiation is retained (one-way aov; F4,1880 = 1105.1, P < 2.2e-16, Table3, Fig. 3) upon adjusting for differences in read depth between the different chromosomes (one-way aov; F4,1880 = 12.268, P = 7.55e-10, Table3, Fig. S1, Supporting information). Interestingly, the highest SNP density per 100 kb window was also found on chromosome 1 at 25.3 Mb showing 438 SNPs (or 39.84 SNPS after adjusting for 10.99X read depth).
Table 3.
Genomic variation per chromosome in the Nasonia vitripennis HVRx outbred laboratory population. Mean SNP density, read depth adjusted SNP density, read depth and nucleotide diversity per chromosome in 100 kb nonoverlapping sliding windows
| Mean SNP density in 100 kb windows (SE) | Mean read depth adjusted SNP density in 100 kb windows (SE) | Mean read depth per 100 kb window (SE) | Mean nucleotide diversity (π) in 100 kb windows (SE) | |
|---|---|---|---|---|
| 1 | 179.57 (5.83)a | 3.96 (0.13)a | 45.05 (0.71)a | 0.0011 (4.00e−05)a |
| 2 | 219.10 (5.74)bc | 4.43 (0.10)bc | 49.32 (0.64)b | 0.0014 (3.54e−05)b |
| 3 | 214.99 (6.44)bd | 4.53 (0.13)bd | 47.95 (0.79)bc | 0.0013 (4.30e−05)b |
| 4 | 197.96 (6.62)acd | 4.21 (0.12)acd | 46.90 (0.70)ac | 0.0013 (4.54e−05)b |
| 5 | 224.59 (6.34)b | 4.37 (0.12)b | 51.07 (0.70)b | 0.0014 (3.85e−05)b |
| All | 202.29 (2.80) | 4.28 (0.06) | 47.84 (0.32) | 0.0013 (1.83e−05) |
Standard errors are in parentheses.
Different lowercase letters indicate significant differences at P < 0.05 for each estimate.
Fig 3.
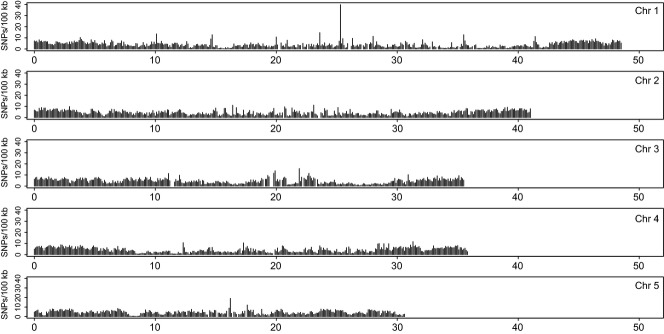
Genome-wide SNP density pattern in the Nasonia vitripennis HVRx outbred laboratory population. Mean SNP density adjusted for read depth in nonoverlapping 100 kb windows, plotted against chromosomal position.
The mean nucleotide diversity per 100 kb window over all chromosomes was 0.13%, showing significant differentiation by chromosome (one-way aov; F4,155 = 9.97, P = 5.76e-08, Table3), with chromosome 1 showing the lowest nucleotide diversity compared with other chromosomes. The distribution of genomic variation over the chromosomes showed similar patterns for both SNP density as for nucleotide diversity. Both measures were higher towards the tips of the chromosomes than the centres (Figs3 and 4). Estimates of nucleotide diversity were lacking in areas where the read depths were low, such as the near the centre of the chromosomes (Fig. 4).
Fig 4.
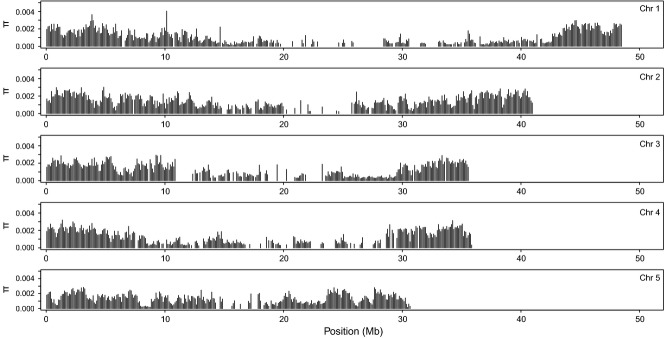
Genome-wide polymorphism pattern in the Nasonia vitripennis HVRx outbred laboratory population. Mean estimates of nucleotide diversity (π) in nonoverlapping 100 kb windows, plotted against chromosomal position.
Discussion
The HVRx laboratory population captures a large amount of genetic variation. With over 400 000 SNPs, it is a valuable resource for investigating the genetic architecture of complex trait variation in the Nasonia genetic model system. The HVRx population provides a genetically variable stock population for experimental evolution experiments, in which the availability of genomic information enables efficient association mapping of complex traits. While genetically diverse stock populations are available for other model species (Drosophila: Burke et al. 2010; Huang et al. 2012; mouse: Svenson et al. 2012; Arabidopsis: Kover et al. 2009), HVRx is the first available for the Hymenoptera genetic model Nasonia.
The HVRx population is maintained at a breeding schedule that retains the available genetic variation, evidenced by the fact that over 32 generations, both the heterozygosity HE and the allelic richness R had not changed significantly. While there was greater differentiation between both founding HV replicates (FST = 0.26) than between European and North American Nasonia vitripennis populations (FST = 0.15, Raychoudhury et al. 2010a), our breeding schedule effectively prevented further differentiation over generations (Table1). Using a simple breeding procedure, our set-up results in a simulated population size of N = 177, and an effective population size Ne of 236. This equals to an Ne/N ratio close to 1.5, which is high compared with previously reported estimates for natural and captive populations of other species (Briscoe et al. 1992; Frankham 1995; Simoes et al. 2010). While effective population size is influenced by many factors, such as fluctuations in population size over generations, variation in family size and sex ratio, mating system and selection, work by Simoes et al. (2010) has shown that careful maintenance procedures can efficiently reduce the loss of genetic variability in laboratory populations in organisms that generate a high number of offspring. The results described here for the HVRx population corroborate this finding.
The effective population size is an important parameter for predicting the potential response to selection (Falconer & Mackay 1996). Because the effect of drift is reduced in larger populations, allelic diversity persists longer allowing for greater and faster responses to selection (Weber 2004). While the exact response to selection is determined by intensity of selection, heritability of the trait of interest and effective population size, the empirical estimate of Ne = 236 for HVRx is on upper range used in artificial selection studies (Weber & Diggins 1990; Weber 2004; Zhang & Hill 2005). The population sizes of laboratory populations also received attention in the biological control literature, where the maintenance of genetic variability is an important issue. The population size of the HVRx exceeds the general recommendation to start and maintain natural enemy cultures with an effective population size of Ne > 100 (Roush 1990; Bartlett 1993; Nunney 2003).
The genetic diversity of the HVRx laboratory population is slightly lower than that observed in studies of natural N. vitripennis populations. Evaluating 12 overlapping microsatellite loci, the heterozygosity of the HVRX population after 32 generations (mean 0.52 ± 0.07) is lower than that observed in natural N. vitripennis populations directly upon collection from the field (range of HE: 0.46–0.95 Table S3, Supporting information; Grillenberger et al. 2009a; Raychoudhury et al. 2010b; Paolucci et al. 2013). It is, however, considerably higher than in the highly inbred standard laboratory strain AsymCX (Werren et al. 2010). A similar pattern is observed in the genome-wide nucleotide diversity of the HVRx population (π = 0.0013), which is lower than the levels of synonymous nucleotide diversity (ranging from π = 0.0016 to 0.0026) observed in ∼16 kb sequence from 27 genes in multiple strains of N. vitripennis (Raychoudhury et al. 2010b; Werren et al. 2010). The genome-wide nucleotide diversity of the HVRx population, however, included both synonymous and nonsynonymous polymorphisms, which result in a lower estimate than when just synonymous polymorphisms were used. Overall, the levels of nucleotide diversity in Nasonia are much lower than those observed in natural Drosophila melanogaster populations (e.g. Mackay et al. 2012; Orozco-ter Wengel et al. 2012), which could be explained by founder events, but also by effects more specific to haplodiploids such as the purging of deleterious mutations in haploid males or high levels of inbreeding (Hedrick & Parker 1997).
Analysis of the genomic variation in the HVRx population shows that genomic polymorphism, both in terms of SNP density as well as of nucleotide diversity, is lower in the centre of the chromosomes and higher towards the distal ends. While we have no exact data on the location of the centromeres, the fact that all Nasonia chromosomes are meta- or submetracentric (Gokhman & Westendorff 2000) strongly suggests the centromeres are at or near the centre of the chromosomes. These regions not only show reduced nucleotide diversity in the HVRx population, but also show reduced recombination rates in Nasonia (Niehuis et al. 2010; Desjardins et al. 2013), consistent with the correlation between recombination rate and nucleotide diversity observed in a wide range of organisms (Begun & Aquadro 1992; Nachman 2002). Unfortunately, the nucleotide diversity data are not complete throughout the HVRx genome, due to low read depths at several regions. While we cannot exclude sequence quality issues for these regions, low read depth regions colocate with gaps in the Nvit 2.0 reference sequence (Werren et al. 2010) that make reference mapping impossible in those regions.
The HVRx laboratory population is a genetically diverse N. vitripennis outbred laboratory population that fills a gap between conventional inbred Nasonia laboratory strains and free-living natural populations. It offers higher levels genetic variation than inbred laboratory strains and easier experimental manipulation than free-living populations, by capturing intraspecific genetic variation in a tractable laboratory population that is easily maintained using the provided breeding schedule. While inbred laboratory strains are a valuable tool for studying interspecific differences in Nasonia (e.g. Desjardins et al. 2010; Werren et al. 2010; Koevoets et al. 2012; Loehlin & Werren 2012; Gibson et al. 2013; Niehuis et al. 2013), the intraspecific context is crucial to understand the evolutionary forces underlying adaptation of complex (life-history) traits. The HVRx adds a novel resource for both inter- and intraspecific research in Nasonia, thereby further extending the qualities of the Nasonia genetic model system for developmental and evolutionary research. The combination of a genetically diverse outbred population with full genomic information makes HVRx a solid starting point for studying the genomic changes underlying the evolution of complex traits, for instance as a stock population for experimental evolution in combination with association mapping studies.
Acknowledgments
We thank Albert Kamping and Tim Hollander for their help in setting up the HV and HVRx populations, and Jürgen Gadau for sharing unpublished microsatellite primers with us. The research was supported by the Netherlands Genomics Initiative grants to BAP (NGI Horizon Breakthrough no. 935.19.006 and NGI Zenith no. 935.11.04) and by the Netherlands Organisation for Scientific Research NWO Grant No. ALW 833.02.003 to LWB.
Biography
L.Z. and B.A.P. conceived and performed the research and wrote the paper. S.F. performed the microsatellite analyses. A.H. performed maintenance of HVRx and prepared the material for sequencing. L.W.B. collected the original field strains. J.H., L.Z. and B.A.P. performed statistical analyses.
Data Accessibility
Sequence data were provided to NasoniaBase (http://hymenopteragenome.org/Nasonia/) and submitted to the NCBI Short Read Archive with accession no. SRP022050. SNP data were provided to NasoniaBase and deposited in Dryad (Dryad Entry doi:10.5061/dryad.f2330).
Supporting Information
Additional Supporting Information may be found in the online version of this article
Read depth in the N. vitripennis HVRx outbred laboratory population.
Table S1 Microsatellite marker properties.
Table S2 Expected heterozygosities per microsatellite marker.
Table S3 Heterozygosity in HVRx generation 32 and natural N. vitripennis populations.
References
- Bartlett AC. Maintaining genetic diversity in laboratory colonies of parasites and predators. In: Narang S, Bartlett A, Faust R, editors. Applications of Genetics to Arthropods of Biological Control Significance. Boca Raton, FL: CRC Press; 1993. pp. 133–145. [Google Scholar]
- Begun DJ, Aquadro C. Levels of naturally occurring DNA polymorphism correlate with recombination rates in D. melanogaster. Nature. 1992;356:519–520. doi: 10.1038/356519a0. [DOI] [PubMed] [Google Scholar]
- Beukeboom LW, van den Assem J. A review of Nasonia (Chalcidoidea, Pteromalidae) courtship and mating behaviour, with some additional new observations. Proceedings of the section Experimental and Applied Entomology of the Netherlands Entomological Society. 2004;15:123–132. [Google Scholar]
- Beukeboom LW, Niehuis O, Pannebakker BA, et al. A comparison of recombination frequencies in intraspecific versus interspecific mapping populations of Nasonia. Heredity. 2010;104:302–309. doi: 10.1038/hdy.2009.185. [DOI] [PubMed] [Google Scholar]
- Bordenstein SR, Werren JH. Bidirectional incompatibility among divergent Wolbachia and incompatibility level differences among closely related Wolbachia in Nasonia. Heredity. 2007;99:278–287. doi: 10.1038/sj.hdy.6800994. [DOI] [PubMed] [Google Scholar]
- Bordenstein SRR, O'Hara FPP, Werren JH. Wolbachia-induced incompatibility precedes other hybrid incompatibilities in Nasonia. Nature. 2001;409:707–710. doi: 10.1038/35055543. [DOI] [PubMed] [Google Scholar]
- Briscoe DA, Malpica JM, Robertson A, et al. Rapid loss of genetic variation in large captive populations of Drosophila flies: implications for the genetic management of captive populations. Conservation Biology. 1992;6:416–425. [Google Scholar]
- Brown S. The Nasonia genome sequence: finding gems in the jewel (wasp) box. Insect Molecular Biology. 2010;19:v–vii. doi: 10.1111/j.1365-2583.2009.00989.x. [DOI] [PubMed] [Google Scholar]
- Brucker RM, Bordenstein SR. The hologenomic basis of speciation: gut bacteria cause hybrid lethality in the genus Nasonia. Science. 2013;341:667–669. doi: 10.1126/science.1240659. [DOI] [PubMed] [Google Scholar]
- Burke MK, Dunham JP, Shahrestani P, et al. Genome-wide analysis of a long-term evolution experiment with Drosophila. Nature. 2010;467:587–590. doi: 10.1038/nature09352. [DOI] [PubMed] [Google Scholar]
- Davies I. A study of the effect of diet on the life-span of Nasonia vitripennis (Walk.) (Hymenoptera, Pteromalidae) Journal of Gerontology. 1975;30:294–298. doi: 10.1093/geronj/30.3.294. [DOI] [PubMed] [Google Scholar]
- Desjardins CA, Perfectti F, Bartos JD, Enders LS, Werren JH. The genetic basis of interspecies host preference differences in the model parasitoid Nasonia. Heredity. 2010;104:270–277. doi: 10.1038/hdy.2009.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desjardins CA, Gadau J, Lopez JA, et al. Fine-scale mapping of the Nasonia genome to chromosomes using a high-density genotyping microarray. G3 (Bethesda) 2013;3:205–215. doi: 10.1534/g3.112.004739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desplan C, Beukeboom LW. Nasonia. Current Biology. 2003;13:R860. doi: 10.1016/j.cub.2003.10.042. [DOI] [PubMed] [Google Scholar]
- Drapeau MD, Werren JH. Differences in mating behaviour and sex ratio between three sibling species of Nasonia. Evolutionary Ecology Research. 1999;1:223–234. [Google Scholar]
- Falconer DS, Mackay TFC. Introduction to Quantitative Genetics. Harlow, Essex, UK: Longmans Green; 1996. [Google Scholar]
- Frankham R. Conservation genetics. Annual Review of Genetics. 1995;29:305–327. doi: 10.1146/annurev.ge.29.120195.001513. [DOI] [PubMed] [Google Scholar]
- Gadau J, Wolschin F. Deciphering proteomic signatures of early diapause in Nasonia. PLoS ONE. 2009;4:e6394. doi: 10.1371/journal.pone.0006394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadau J, Page REE, Werren JH, et al. Mapping of hybrid incompatibility loci in Nasonia. Genetics. 1999;153:1731–1741. doi: 10.1093/genetics/153.4.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geuverink E, Gerritsma S, Pannebakker BA, Beukeboom LW. A role for sexual conflict in the evolution of reproductive traits in Nasonia wasps? Animal Biology. 2009;59:417–434. [Google Scholar]
- Gibson JD, Niehuis O, Peirson BRE, Cash EI, Gadau J. Genetic and developmental basis of F2 hybrid breakdown in Nasonia parasitoid wasps. Evolution. 2013;67:2124–2132. doi: 10.1111/evo.12080. [DOI] [PubMed] [Google Scholar]
- Godfray HCJ. Nasonia: a jewel among wasps. Heredity. 2010;104:235–236. doi: 10.1038/hdy.2010.3. [DOI] [PubMed] [Google Scholar]
- Gokhman VE, Westendorff M. The chromosomes of three species of the Nasonia complex (Hymenoptera, Pteromalidae) Beiträge zur Entomologie. 2000;50:193–198. [Google Scholar]
- Goudet J. FSTAT, a program to estimate and test gene diversities and fixation indices. 2001. Available from http://www2.unil.ch/popgen/softwares/fstat.htm.
- Gregory TR. Animal Genome Size Database. 2013. Available from http://www.genomesize.com.
- Grillenberger BK, Gadau J, Bijlsma R, van de Zande L, Beukeboom LW. Female dispersal and isolation-by-distance of Nasonia vitripennis populations in a local mate competition context. Entomologia Experimentalis et Applicata. 2009a;132:147–154. [Google Scholar]
- Grillenberger BK, van de Zande L, Bijlsma R, Gadau J, Beukeboom LW. Reproductive strategies under multiparasitism in natural populations of the parasitoid wasp Nasonia (Hymenoptera) Journal of Evolutionary Biology. 2009b;22:460–470. doi: 10.1111/j.1420-9101.2008.01677.x. [DOI] [PubMed] [Google Scholar]
- Hedrick PW, Parker JD. Evolutionary genetics and genetic variation of haplodiploids and X-linked genes. Annual Review of Ecology and Systematics. 1997;28:55–83. [Google Scholar]
- Huang W, Richards S, Carbone MA, et al. Epistasis dominates the genetic architecture of Drosophila quantitative traits. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:15553–15559. doi: 10.1073/pnas.1213423109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King BH. Flight activity in the parasitoid wasp Nasonia vitripennis (Hymenoptera, Pteromalidae) Journal of Insect Behavior. 1993;6:313–321. [Google Scholar]
- Koboldt DC, Zhang Q, Larson DE, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Research. 2012;22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koevoets T, Niehuis O, van de Zande L, Beukeboom LW. Hybrid incompatibilities in the parasitic wasp genus Nasonia: negative effects of hemizygosity and the identification of transmission ratio distortion loci. Heredity. 2012;108:302–311. doi: 10.1038/hdy.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofler R, Orozco-terWengel P, De Maio N, et al. Kayser M, editor. PoPoolation: a toolbox for population genetic analysis of next generation sequencing data from pooled individuals. PLoS ONE. 2011;6:e15925. doi: 10.1371/journal.pone.0015925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kover PX, Valdar W, Trakalo J, et al. A multiparent advanced generation inter-cross to fine-map quantitative traits in Arabidopsis thaliana. PLoS Genetics. 2009;5:e1000551. doi: 10.1371/journal.pgen.1000551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalonde RG. Egg size variation does not affect offspring performance under intraspecific competition in Nasonia vitripennis, a gregarious parasitoid. Journal of Animal Ecology. 2005;74:630–635. [Google Scholar]
- Legner E, Gerling D. Host-feeding and oviposition on Musca domestica by Spalangia cameroni Nasonia vitripennis, and Muscidifurax raptor (Hymenoptera: Pteromalidae) influences their longevity and fecundity. Annals of the Entomological Society of America. 1967;60:678–691. doi: 10.1093/aesa/60.3.678. [DOI] [PubMed] [Google Scholar]
- Loehlin DW, Werren JH. Evolution of shape by multiple regulatory changes to a growth gene. Science. 2012;335:943–947. doi: 10.1126/science.1215193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loehlin DW, Enders LS, Werren JH. Evolution of sex-specific wing shape at the widerwing locus in four species of Nasonia. Heredity. 2010a;104:260–269. doi: 10.1038/hdy.2009.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loehlin DW, Oliveira DCSG, Edwards R, et al. Non-coding changes cause sex-specific wing size differences between closely related species of Nasonia. PLoS Genetics. 2010b;6:e1000821. doi: 10.1371/journal.pgen.1000821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch JA, Ozüak O, Khila A, et al. The phylogenetic origin of oskar coincided with the origin of maternally provisioned germ plasm and pole cells at the base of the Holometabola. PLoS genetics. 2011;7:e1002029. doi: 10.1371/journal.pgen.1002029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay TFC, Richards S, Stone EA, et al. The Drosophila melanogaster Genetic Reference Panel. Nature. 2012;482:173–178. doi: 10.1038/nature10811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniatis T, Fritsch EF, Sambrook J. Molecular Cloning: A Laboratory Manual. New York: Cold Spring Harbor Laboratory; 1982. [Google Scholar]
- Marjoram P, Molitor J, Plagnol V, Tavare S. Markov chain Monte Carlo without likelihoods. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:15324–15328. doi: 10.1073/pnas.0306899100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muers M. Evo–devo: Nasonia tools up. Nature Reviews Genetics. 2010;11:170–171. doi: 10.1038/nrg2759. [DOI] [PubMed] [Google Scholar]
- Munoz-Torres MC, Reese JT, Childers CP, et al. Hymenoptera Genome Database: integrated community resources for insect species of the order Hymenoptera. Nucleic Acids Research. 2011;39:D658–D662. doi: 10.1093/nar/gkq1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachman M. Variation in recombination rate across the genome: evidence and implications. Current Opinion in Genetics & Development. 2002;12:657–663. doi: 10.1016/s0959-437x(02)00358-1. [DOI] [PubMed] [Google Scholar]
- Nicholson G, Smith AV, Jonsson F, et al. Assessing population differentiation and isolation from single-nucleotide polymorphism data. Journal of the Royal Statistical Society Series B-Statistical Methodology. 2002;64:695–715. [Google Scholar]
- Niehuis O, Gibson JD, Rosenberg MS, et al. Recombination and its impact on the genome of the haplodiploid parasitoid wasp Nasonia. PLoS ONE. 2010;5:e8597. doi: 10.1371/journal.pone.0008597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehuis O, Buellesbach J, Gibson JD, et al. Behavioural and genetic analyses of Nasonia shed light on the evolution of sex pheromones. Nature. 2013;494:7–10. doi: 10.1038/nature11838. [DOI] [PubMed] [Google Scholar]
- Nunney L. Managing captive populations for release: a population genetic perspective. In: van Lenteren JC, editor. Quality Control and Production of Biological Control Agents: Theory and Testing Procedures. Wallingford, UK: CAB International; 2003. pp. 73–97. [Google Scholar]
- Orozco-terWengel P, Kapun M, Nolte V, et al. Adaptation of Drosophila to a novel laboratory environment reveals temporally heterogeneous trajectories of selected alleles. Molecular Ecology. 2012;21:4931–4941. doi: 10.1111/j.1365-294X.2012.05673.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannebakker BA, Halligan DLDL, Reynolds KTT, et al. Effects of spontaneous mutation accumulation on sex ratio traits in a parasitoid wasp. Evolution. 2008;62:1921–1935. doi: 10.1111/j.1558-5646.2008.00434.x. [DOI] [PubMed] [Google Scholar]
- Pannebakker BA, Niehuis O, Hedley A, Gadau J, Shuker DM. The distribution of microsatellites in the Nasonia parasitoid wasp genome. Insect Molecular Biology. 2010;19:91–98. doi: 10.1111/j.1365-2583.2009.00915.x. [DOI] [PubMed] [Google Scholar]
- Pannebakker BA, Watt R, Knott SA, West SA, Shuker DM. The quantitative genetic basis of sex ratio variation in Nasonia vitripennis: a QTL study. Journal of Evolutionary Biology. 2011;24:12–22. doi: 10.1111/j.1420-9101.2010.02129.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolucci S, van de Zande L, Beukeboom LW. Adaptive latitudinal cline of photoperiodic diapause induction in the parasitoid Nasonia vitripennis in Europe. Journal of Evolutionary Biology. 2013;26:705–718. doi: 10.1111/jeb.12113. [DOI] [PubMed] [Google Scholar]
- Pritchard JK, Seielstad MT, Perez-Lezaun A, Feldman MW. Population growth of human Y chromosomes: a study of Y chromosome microsatellites. Molecular Biology and Evolution. 1999;16:1791–1798. doi: 10.1093/oxfordjournals.molbev.a026091. [DOI] [PubMed] [Google Scholar]
- Pultz MA, Zimmerman KK, Alto NM, et al. A genetic screen for zygotic embryonic lethal mutations affecting cuticular morphology in the wasp Nasonia vitripennis. Genetics. 2000;154:1213–1229. doi: 10.1093/genetics/154.3.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2012. Available from http://www.R-project.org/ [Google Scholar]
- Raychoudhury R, Desjardins CA, Buellesbach J, et al. Behavioral and genetic characteristics of a new species of Nasonia. Heredity. 2010a;104:278–288. doi: 10.1038/hdy.2009.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raychoudhury R, Grillenberger BK, Gadau J, et al. Phylogeography of Nasonia vitripennis (Hymenoptera) indicates a mitochondrial–Wolbachia sweep in North America. Heredity. 2010b;104:318–326. doi: 10.1038/hdy.2009.160. [DOI] [PubMed] [Google Scholar]
- Rivero A, West SA. The physiological costs of being small in a parasitic wasp. Evolutionary Ecology Research. 2002;4:407–420. [Google Scholar]
- Rivero A, West SA. The costs and benefits of host feeding in parasitoids. Animal Behaviour. 2005;69:1293–1301. [Google Scholar]
- Rivers DB, Denlinger DL. Fecundity and development of the ectoparasitic wasp Nasonia vitripennis are dependent on host quality. Entomologia Experimentalis et Applicata. 1995;76:15–24. [Google Scholar]
- Rivers DBDB, Kaikis A, Bulanowski D, Wigand T, Brogan R. Oviposition restraint and developmental alterations in the ectoparasitic wasp, Nasonia vitripennis, when utilizing puparia resulting from different size maggot masses of Lucilia illustris Protophormia terraenovae, and Sarcophaga bullata. Journal of Medical Entomology. 2012;49:1124–1136. doi: 10.1603/me11232. [DOI] [PubMed] [Google Scholar]
- Roush R. Genetic variation in natural enemies: critical issues for colonization in biological control. In: Mackauer M, Ehler L, Roland J, editors. Critical Issues in Biological Control. Andover, UK: Intercept; 1990. pp. 263–288. [Google Scholar]
- Shuker DM, West SA. Information constraints and the precision of adaptation: sex ratio manipulation in wasps. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:10363–10367. doi: 10.1073/pnas.0308034101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuker DM, Pen I, Duncan AB, Reece SE, West SA. Sex ratios under asymmetrical local mate competition: theory and a test with parasitoid wasps. The American Naturalist. 2005;166:301–316. doi: 10.1086/432562. [DOI] [PubMed] [Google Scholar]
- Shuker DM, Phillimore AJ, Burton-Chellew MN, Hodge SE, West SA. The quantitative genetic basis of polyandry in the parasitoid wasp, Nasonia vitripennis. Heredity. 2007a;98:69–73. doi: 10.1038/sj.hdy.6800897. [DOI] [PubMed] [Google Scholar]
- Shuker DM, Reece SE, Lee A, et al. Information use in space and time: sex allocation behaviour in the parasitoid wasp Nasonia vitripennis. Animal Behaviour. 2007b;73:971–977. doi: 10.1016/j.anbehav.2006.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoes P, Pascual M, Coelho M, Matos M. Divergent evolution of molecular markers during laboratory adaptation in Drosophila subobscura. Genetica. 2010;138:999–1009. doi: 10.1007/s10709-010-9486-4. [DOI] [PubMed] [Google Scholar]
- Smith GJC, Pimentel D. The effect of two host species on the longevity and fertility of Nasonia vitripennis. Annals of the Entomological Society of America. 1969;62:305–308. [Google Scholar]
- Svenson KL, Gatti DM, Valdar W, et al. High-resolution genetic mapping using the Mouse Diversity outbred population. Genetics. 2012;190:437–447. doi: 10.1534/genetics.111.132597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavare S, Balding D, Griffiths R, Donnelly P. Inferring coalescence times from DNA sequence data. Genetics. 1997;145:505–518. doi: 10.1093/genetics/145.2.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhulst EC, Beukeboom LW, van de Zande L. Maternal control of haplodiploid sex determination in the wasp Nasonia. Science. 2010;328:620–623. doi: 10.1126/science.1185805. [DOI] [PubMed] [Google Scholar]
- Wang X, Wheeler D, Avery A, et al. Function and evolution of DNA methylation in Nasonia vitripennis. PLoS genetics. 2013;9:e1003872. doi: 10.1371/journal.pgen.1003872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber K. Janick J, editor. Population size and long-term selection. Plant Breeding Reviews. 2004;24:249–268. [Google Scholar]
- Weber KE, Diggins LT. Increased selection response in larger populations. II. Selection for ethanol vapor resistance in Drosophila melanogaster at two population sizes. Genetics. 1990;125:585–597. doi: 10.1093/genetics/125.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werren JH. Sex ratio adaptations to local mate competition in a parasitic wasp. Science. 1980;208:1157–1159. doi: 10.1126/science.208.4448.1157. [DOI] [PubMed] [Google Scholar]
- Werren JH. Sex ratio evolution under local mate competition in a parasitic wasp. Evolution. 1983a;37:116–124. doi: 10.1111/j.1558-5646.1983.tb05520.x. [DOI] [PubMed] [Google Scholar]
- Werren JH. Brood size and sex ratio regulation in the parasitic wasp Nasonia vitripennis (Walker) (Hymenoptera: Pteromalidae) Netherlands Journal of Zoology. 1983b;34:123–143. [Google Scholar]
- Werren JH, Loehlin DW. The parasitoid wasp Nasonia: an emerging model system with haploid male genetics. Cold Spring Harbor Protocols. 2009;10 doi: 10.1101/pdb.emo134. doi: 10.1101/pdb.emo134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werren JH, Richards S, Desjardins CA, et al. Functional and evolutionary insights from the genomes of three parasitoid Nasonia species. Science. 2010;327:343–348. doi: 10.1126/science.1178028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiting AR. The biology of the parasitic wasp Mormoniella vitripennis (Walker) Quarterly Review of Biology. 1967;42:333–406. [Google Scholar]
- Wright S. Inbreeding and homozygosis. Proceedings of the National Academy of Sciences of the United States of America. 1933;19:411–420. doi: 10.1073/pnas.19.4.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wylie HG. Survival and reproduction of Nasonia vitripennis (Walk) at different host population densities. The Canadian Entomologist. 1966;98:275–281. [Google Scholar]
- Zhang X-S, Hill WG. Predictions of patterns of response to artificial selection in lines derived from natural populations. Genetics. 2005;169:411–425. doi: 10.1534/genetics.104.032573. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Read depth in the N. vitripennis HVRx outbred laboratory population.
Table S1 Microsatellite marker properties.
Table S2 Expected heterozygosities per microsatellite marker.
Table S3 Heterozygosity in HVRx generation 32 and natural N. vitripennis populations.
Data Availability Statement
Sequence data were provided to NasoniaBase (http://hymenopteragenome.org/Nasonia/) and submitted to the NCBI Short Read Archive with accession no. SRP022050. SNP data were provided to NasoniaBase and deposited in Dryad (Dryad Entry doi:10.5061/dryad.f2330).