

Abstract
According to contemporary epidemiological and experimental evidence, we propose a novel classification of cancers based on pathogenesis instead of classifications based on histological appearance of cancer. This new scheme first defines cancers as either 1.inborn errors of development or 2.sporadic ones, and then sub-defines the former into 1A. inborn inherited errors of development, being those due to mutations contributed by one or both parents’ gametes to the developing conceptus, and 1B. inborn induced errors of development when the malformations and/or cancers are due to environmental carcinogenic exposure during pregnancy. It is anticipated that the origin of an increasing number of so-called sporadic cancers will turn out to be linked to the inborn induced errors of development group.
Keywords: Carcinogenesis, Pathobiology, Metastasis, Neoplasia, Mutation, Tissue organization field, Cancer theories
Introduction
Human cancers have been classified according to diverse criteria primarily to serve the needs of the medical profession to diagnose, stage, prognosticate and treat the disease. aFor these pragmatic purposes, pathological classifications of cancer are based on organ of origin of the tumor and predominant cell type (epithelial or mesenchymal), with a long list of sub-classifications including (but not limited to) whether the tumor is benign or malignant (e.g., adenoma vs. carcinoma; fibroma vs. sarcoma), encapsulated or invasive, whether it contains components of different cell types or tissues (e.g., “desmoplastic adenocarcinoma”), and whether it displays low or high proliferation rates (mitotic indices). In recent years, a major effort has been devoted to characterize tumors based on their molecular features, particularly referring to genetic mutations and/or patterns of gene expression. Prognostic and therapeutic advances based on molecular features included, for example, identifying the estrogen and progesterone receptor and HER2/Neu status of breast cancers, or the mutational status of gastric stromal sarcomas and response to Gleevec [1]. The correlation between genomic somatic mutations in cells of “sporadic” cancers with a specific targeted therapy has been interpreted as being causal. However, according to recent pronouncements by the very proponents of such theoretical links, successes have been disappointing [2–5]. Admittedly, cancers are complex biological phenomena whose full understanding remains incomplete [3,6].
Regardless of whether carcinogenesis represents either a cell-based [3] or a tissue-based [6,7] phenomenon, from an etiopathogenic perspective, two main types of cancers are apparent: they can either be inherited or “sporadic”. Inherited cancers refer to those cancers that have an obvious or suspected link to germ-line mutations present in chromosomes that are passed on from one generation to the next. Sporadic cancers refer to those cancers that are assumed to lack an obvious inherited component; instead, it has been proposed that they are the result of the life-long accumulation of spontaneous or induced mutations in a single “normal” cell. Again, this classification does not address whether the cancer process is initiated within a cell as Boveri [8], Nowell [9] and most others have favored since the 20th century, or instead, at the tissue level [10].
Theories of carcinogenesis and metastases
There are divergent opinions regarding the level of biological organization at which cancer originates. The somatic mutation theory of carcinogenesis (SMT) defines cancer as a cell-based disease [8,9,11]. Its fundamental premise is that cancer is due to the accumulation of spontaneous or induced somatic mutations and/or chromosomal aberrations that alter the control of proliferation in a single cell that eventually will generate a tumor [8,12,13]. Boveri called this cell “the cancer cell” [8] and later it was renamed “the renegade cell” by Weinberg [14]. In this context, cancer becomes a clonal disease [9]. A seldom mentioned additional premise associated to SMT has been that quiescence rather than proliferation is the default state of cells in multicellular organisms [15–17]. Adoption of this latter premise implies that cells need to be stimulated directly in order to proliferate; following this rationale, since the 1950s, “growth factors” and, since the 1980s, oncogenes have been proposed as stimulators of cell proliferation [15,18].
From the second half of the 20th century onward, SMT gained standing due to staggering advances in genetics and molecular biology, and the de-emphasizing of physiology and developmental biology in cancer pathogenesis. The initial claim that cancers were due to a single mutation in a cell in culture conditions [19], referred to as malignant transformation, was followed by claims that the number of somatic mutations responsible for such a transformation in human tumors increased from single digits to the thousands [20,21]. Eventually, the SMT morphed from its original simplicity into the 6 “hallmarks of cancer” in 2000 [11], with 2 more being added a decade later [22].
In contrast to the SMT, the tissue organization field theory of carcinogenesis (TOFT) posits that cancer is i) a tissue-based disease, and that, explicitly, ii) proliferation is the default state of all cells [10]. That cancer is due to a pathological interaction between tissues is not a new claim; this was predicated by German pathologists during the second half of the 19th century [23,24]. The merits of both SMT and TOFT to explain carcinogenesis have been debated elsewhere based on experimental, clinical and theoretical grounds [6,7,25].
In the last decade, given the increasing lacks of fit between the premises of SMT and the evidence gathered from the huge amount of data generated by novel technical improvements in gene sequencing, a merging of the cell-based SMT and tissue-based components has been proposed as an add-on to the original SMT [11,18,26–28]. Based on grounds that SMT and TOFT are centered on a) different levels of biological organization (cell for SMT, tissue for TOFT) and b) opposite premises regarding the proliferative default state (quiescence for SMT and proliferation for TOFT), we and others have argued against accepting this compromise [6,7,10,29,30]. Moreover, in addition to evidence challenging the need for somatic mutations to significantly participate in the carcinogenic process [31], experimental and clinically-based evidence has documented that solid tumor carcinogenesis can occur in the absence of somatic mutations [32,33]. Also, equally robust experimental and clinical evidence shows that stromal alterations lead to neoplasia of the parenchyma as exemplified in leukemia [34–36].
As a result of the lacks of fit referred to above, another variant of SMT has been proposed, namely, the cancer stem cell (CSC) theory of carcinogenesis. Despite aggressive efforts directed at identifying normal stem cells and cancer stem cells, these entities remain as operational and rather elusive concepts [37–39]. In addition, the stem cell niche appears to be made up of epithelial cells plus the adjacent stroma; under this perspective, stemness is likely to be conferred by the niche and not by an autonomous epithelial cell-based property [37,40]. Thus, the CSC theory would represent another example of the type of “compromise” theory (SMT plus a tissue-based component) referred to above.
A pathogenetic classification of cancers
In Figure 1, we outline our proposed novel etiopathogenic classification of cancers. It is comprised of two main groups of tumors, namely, 1. Inborn errors of development. Within this group, we identify 2 sub-groups of neoplasms. First, there are 1A. Inborninheritederrors of development - the result of a process initiated by a germ-line mutation(s) in the genome of one or both gametes (sperm and/or ovum). The mutated genome of the resulting zygote will endow all the cells in the morphogenetic fields of the developing organism with such a genomic mutation(s). The altered expression of these mutated genes could take place either during organogenesis, tissue remodeling and/or repair. This provides a temporal-dependent dimension to the carcinogenic process. Examples of this variety of inborn error of development include retinoblastoma, Gorlin syndrome, xeroderma pigmentosa, BRCA-1 and-2 neoplasia, and many others (for a comprehensive listing and description of these tumors see [41,42]).
Figure 1.
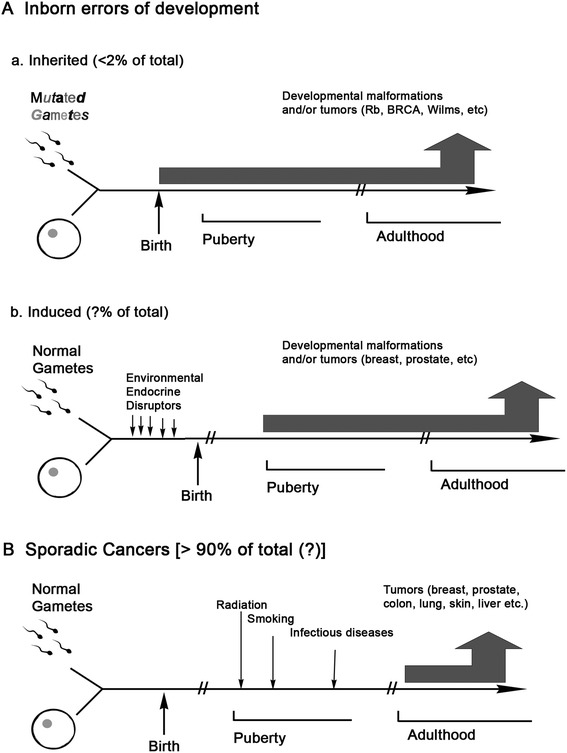
Novel pathogenetic classification of neoplasia. This classification distinguishes 2 main types of neoplasia as inborn errors of development (A) and sporadic cancers (B). A is further classified into inborn errors of development that may be either inherited (a) or induced (b).
Remarkably, King et al. [43] noticed in a cohort of patients carrying BRCA mutations that breast cancer risk by age 50 among carriers born before 1940 was 24%, whereas it was 67% among those born after 1940 [43]. These authors concluded that “non-genetic factors may significantly influence the penetrance even of high-penetrance mutations”. On the one hand, this evidence argues in favor of identifying those non-genetic environmental factors - presumably due to exposure to pollutants that significantly increase the cancer incidence not only of inborn inherited errors of development, but also of sporadic tumors. On the other hand, their observations strengthen the merits for postulating a novel pathogenetic classification of neoplasms that would reflect a significant participation of the environment in carcinogenesis.
Within the group of inborn errors of development, we identify a second subgroup of neoplasms that we named 1B. Inborninducederrors of development (Figure 1). As the name implies, tumors and/or malformations would be due to alterations of the fetal environment, exemplified by exposure to environmental chemicals, such as synthetic hormones (Diethylstilbestrol (DES)) as well as by elevated levels of endogenous hormones [44–46]. Exposure of rodent embryos, fetuses and neonates to environmental estrogens generate premalignant and malignant neoplastic lesions in diverse tissues later in life [47,48]. In humans, exposure of fetuses to DES during the first trimester of pregnancy, correlated with the appearance of vaginal clear cell carcinomas during puberty and early adulthood. A conclusive correlation between exposure to DES and clear cell carcinoma was established because this rare neoplasm appeared in non-DES exposed populations only in post-menopausal women [49]. Animal experiments showing the development of adenosis, pre-neoplastic lesions considered precursors of clear cell carcinomas also suggests a causal link between a synthetic estrogen, DES, and the rare cancer described above [50]. Consistent with the effect of DES exposure in rats, there was an increased incidence of breast carcinomas in the above-mentioned cohort of women when they reached the prevalent age at which breast cancer most commonly occurs [51,52].
Furthermore, reports increasingly indicate that environmental chemicals are important causes in generating tumors. Among them, there are the environmental endocrine disruptors (EED) that are defined as an exogenous chemical, or mixture of chemicals, that interferes with any aspect of hormone action [53]. For example, Bisphenol-A (BPA) has been shown to increase the incidence of hormone-related cancers in rodents [46]. EEDs are suspected to be a factor in human breast and prostate cancers over the last 50 years [47,54]. Because BPA as well as other EEDs are not mutagens, the fact that they induce cancer cannot be adequately explained by the SMT, but is better understood from the TOFT perspective as due to faulty cell-cell and/or tissue-tissue interactions [54,55]. It is in this context that we consider cancer as “development gone awry”.
The World Health Organization’s cancer agency, IARC, has extrapolated industrialization with damaging lifestyle changes as also implying a correlation between direct and/or indirect effects of greater exposure in and out of the workplace (which includes pregnant women) to noxious chemicals and higher cancer incidence [56,57]. Since the 2008 estimates, breast cancer incidence has increased by more than 20% and mortality by 14%. Based on these statistics, IARC subsequently acknowledged the emergence of a “toxic epidemic” of cancers in developing nations [58].
2. “Sporadic” tumors. Neoplasms due to germ-line mutations have been considered to represent less than 5% of all cancers [42,59], whereas “sporadic” ones represent the vast majority of clinical cancers. The characterization of cancers as “sporadic” extends to any cancer that appears to have no obvious link to germ-line mutations (Figure 1). Their characteristics and properties have been described in great detail in the biomedical literature and will not be dealt with in this article. Our novel classification anticipates, however, that many of what are now considered as “sporadic” cancers will in the future be reclassified as Inborn induced errors of development. Briefly, the term “sporadic” cancers would now be restricted to those cancers that result from carcinogenic exposures after gestation.
Conclusions
A pathogenic classification of cancer is being proposed based on novel insights on experimental carcinogenesis, and from evidence collected through highly sophisticated genome analysis in search of somatic mutations. This classification identifies the presence of 2 major groups of cancers, namely, 1) inborn errors of development, and 2) “sporadic” ones. Based on whether or not genomic mutations are responsible for the emergence of a cancer tissue phenotype, the former group is further classified into 1A) inborn inherited errors of development and 1B) inborn induced errors of development. This classification better reflects current views about how cancers develop, and anticipates that the incidence of “sporadic” cancers now being diagnosed will diminish by being reclassified as our type 1B.
Endnote
aFor the sake of simplicity, we will use the nouns cancers, neoplasms and tumors interchangeably.
Acknowledgements
This research was supported by The Avon Foundation grant #02-2013-025, as well as by the National Institute of Environmental Health Sciences, Award Numbers R01ES08314, U01ES020888. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
Footnotes
Competing interests
The authors declare they have no competing interests.
Authors’ contributions
CS, BD and AMS conceived and participated in the design, coordination and drafting of the manuscript. All authors read and approved the final manuscript.
Contributor Information
Carlos Sonnenschein, Email: carlos.sonnenschein@tufts.edu.
Barbara Davis, Email: bdavistoxpath@gmail.com.
Ana M Soto, Email: ana.soto@tufts.edu.
References
- 1.Jackson SE, Chester JD: Personalised cancer medicine.Int J Cancer 2014. Ahead of print doi:10.1002/ijc.28940. [DOI] [PubMed]
- 2.Hanahan D. Rethinking the war on cancer. Lancet. 2014;383:558–563. doi: 10.1016/S0140-6736(13)62226-6. [DOI] [PubMed] [Google Scholar]
- 3.Weinberg RA. Coming full circle-from endless complexity to simplicity and back again. Cell. 2014;157:267–271. doi: 10.1016/j.cell.2014.03.004. [DOI] [PubMed] [Google Scholar]
- 4.Kaiser J. Profile: Bert Vogelstein. Cancer genetics with an edge. Science. 2012;337:282–284. doi: 10.1126/science.337.6092.282. [DOI] [PubMed] [Google Scholar]
- 5.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sonnenschein C, Soto AM, Rangarajan A, Kulkarni P. Competing views on cancer. J Biosci. 2014;39:1–22. doi: 10.1007/s12038-013-9404-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Soto AM, Sonnenschein C. The tissue organization field theory of cancer: a testable replacement for the somatic mutation theory. Bioessays. 2011;33:332–340. doi: 10.1002/bies.201100025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boveri T. The Origin of Malignant Tumors. Baltimore, MD: Williams & Wilkins; 1929. [Google Scholar]
- 9.Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:123–128. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 10.Sonnenschein C, Soto AM. The Society of Cells: Cancer and Control of Cell Proliferation. New York: Springer Verlag; 1999. [Google Scholar]
- 11.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/S0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 12.Duesberg P. Chromosomal chaos and cancer. Sci Am. 2007;296:52–59. doi: 10.1038/scientificamerican0507-52. [DOI] [PubMed] [Google Scholar]
- 13.Li L, McCormack AA, Nicholson JM, Fabarius A, Hehlmann R, Sachs RK, Duesberg P. Cancer-causing karyotypes: chromosomal equilbria between destabilizing aneuploidy and stabilizing selection for oncogenic function. Cancer Genet Cytogenet. 2009;188:1–25. doi: 10.1016/j.cancergencyto.2008.08.016. [DOI] [PubMed] [Google Scholar]
- 14.Weinberg RA. One renegade cell: how cancer begins. New York: Basic Books; 1998. [Google Scholar]
- 15.Varmus HE, Weinberg RA. Genes and the Biology of Cancer. New York, NY: Scientific American Library; 1992. [Google Scholar]
- 16.Alberts B, Bray D, Lewis JG, Raff M, Roberts K, Watson JD. Molecular Biology of the Cell. New York, NY: Garland Publishing Inc.; 1994. [Google Scholar]
- 17.Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. Molecular Biology of the Cell. London: Garland Science; 2008. [Google Scholar]
- 18.Weinberg RA. The Biology of Cancer. New York: Taylor & Francis; 2006. [Google Scholar]
- 19.Tabin CJ, Bradley SM, Bargmann CI, Weinberg RA, Papageorge AG, Scolnick EM, Dhar R, Lowy DR, Chang EH. Mechanism of activation of a human oncogene. Nature. 1982;300:143–149. doi: 10.1038/300143a0. [DOI] [PubMed] [Google Scholar]
- 20.Stratton MR. Exploring the genomes of cancer cells: progress and promise. Science. 2011;331:1553–1558. doi: 10.1126/science.1204040. [DOI] [PubMed] [Google Scholar]
- 21.Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458:719–724. doi: 10.1038/nature07943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 23.Ribbert H. Zur Entstehung der Geschwuelste. Duetsche Medizinische Wochenzeitschrift. 1896;30:471–474. doi: 10.1055/s-0029-1204569. [DOI] [Google Scholar]
- 24.Triolo VA. Nineteenth century foundations of cancer research origins of experimental research. Cancer Res. 1964;24:4–27. [PubMed] [Google Scholar]
- 25.Vaux DL. In defense of the somatic mutation theory of cancer. Bioessays. 2011;33:341–343. doi: 10.1002/bies.201100022. [DOI] [PubMed] [Google Scholar]
- 26.Wilkins AS. The enemy within: an epigenetic role of retrotransposons in cancer initiation. Bioessays. 2010;32:856–865. doi: 10.1002/bies.201000008. [DOI] [PubMed] [Google Scholar]
- 27.Laconi E, Sonnenschein C. Cancer development at tissue level. Semin Cancer Biol. 2008;18:303–304. doi: 10.1016/j.semcancer.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 28.Liu H, Radisky DC, Yang D, Xu R, Radisky ES, Bissell MJ, Bishop JM. MYC suppresses cancer metastasis by direct transcriptional silencing of αv and β3 integrin subunits. Nat Cell Biol. 2012;14:567–574. doi: 10.1038/ncb2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bizzarri M, Cucina A. Tumor and the microenvironment: a chance to reframe the paradigm of carcinogenesis. Biomed Res Int. 2014;2014:934038. doi: 10.1155/2014/934038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sonnenschein C, Soto AM. The death of the cancer cell. Cancer Res. 2011;71:4334–4337. doi: 10.1158/0008-5472.CAN-11-0639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maffini MV, Soto AM, Calabro JM, Ucci AA, Sonnenschein C. The stroma as a crucial target in rat mammary gland carcinogenesis. J Cell Sci. 2004;117:1495–1502. doi: 10.1242/jcs.01000. [DOI] [PubMed] [Google Scholar]
- 32.Mack SC, Witt H, Piro RM, Gu L, Zuyderduyn S, Stutz AM, Wang X, Gallo M, Garzia L, Zayne K, Zhang X, Ramaswamy V, Jager N, Jones DTW, Sill M, Pugh TJ, Ryzhova M, Wani KM, Shih DJ, Head R, Remke M, Bailey SD, Zichner T, Faria CC, Barszczyk M, Stark S, Seker-Cin H, Hutter S, Johann P, Bender S, et al. Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature. 2014;506:445–450. doi: 10.1038/nature13108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Versteeg R. Cancer: tumours outside the mutation box. Nature. 2014;506:438–439. doi: 10.1038/nature13061. [DOI] [PubMed] [Google Scholar]
- 34.Walkley CR, Olsen GH, Dworkin S, Fabb SA, Swann J, McArthur GA, Westmoreland SV, Chambon P, Scadden DT, Purton LE. A microenvironment-induced myeloproliferative syndrome caused by retinoic acid receptor gamma deficiency. Cell. 2007;129:1097–1110. doi: 10.1016/j.cell.2007.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Raaijmakers MH, Mukherjee S, Guo S, Zhang S, Kobayashi T, Schoonmaker JA, Ebert BL, Al-Shahrour F, Hasserjian RP, Scadden EO, Aung Z, Matza M, Merkenschlager M, Lin C, Rommens JM, Scadden DT. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature. 2010;464:852–857. doi: 10.1038/nature08851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kode A, Manavalan JS, Mosialou I, Bhagat G, Rathinam CV, Luo N, Khiabanian H, Lee A, Murty VV, Friedman R, Brum A, Park D, Galili N, Mukherjee S, Teruya-Feldstein J, Raza A, Rabadan R, Berman E, Kousteni S. Leukaemogenesis induced by an activating ?-catenin mutation in osteoblasts. Nature. 2014;506:240–244. doi: 10.1038/nature12883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blanpain C, Fuchs E. Plasticity of epithelial stem cells in tissue regeneration. Science. 2014;344:1242281. doi: 10.1126/science.1242281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kelly PN, Dakic A, Adams JM, Nutt SL, Strasser A. Tumor growth need not be driven by rare cancer stem cells. Science. 2007;317:337. doi: 10.1126/science.1142596. [DOI] [PubMed] [Google Scholar]
- 39.Yoo MH, Hatfield DL. The cancer stem cell theory: is it correct? Mol Cells. 2008;26:514–516. [PMC free article] [PubMed] [Google Scholar]
- 40.Fábián Á, Vereb G, Szöllõsi J. The hitchhikers guide to cancer stem cell theory: markers, pathways and therapy. Cytometry. 2013;83:62–71. doi: 10.1002/cyto.a.22206. [DOI] [PubMed] [Google Scholar]
- 41.Pizzo PA, Poplack DG. Principles and Practice of Pediatric Oncology. Philadelphia: Lippincott, Williams and Williams; 2006. [Google Scholar]
- 42.Garber JE, Offit K. Hereditary cancer predisposition syndromes. J Clin Oncol. 2005;23:276–292. doi: 10.1200/JCO.2005.10.042. [DOI] [PubMed] [Google Scholar]
- 43.King MC, Marks JH, Mandell JB. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science. 2003;302:643–646. doi: 10.1126/science.1088759. [DOI] [PubMed] [Google Scholar]
- 44.Ekbom A, Trichopoulos D, Adami HO, Hsieh CC, Lan SJ. Evidence of prenatal influences on breast cancer risk. Lancet. 1992;340:1015–1018. doi: 10.1016/0140-6736(92)93019-J. [DOI] [PubMed] [Google Scholar]
- 45.Trichopoulos D. Is breast cancer initiated in utero? Epidemiology. 1990;1:95–96. [PubMed] [Google Scholar]
- 46.Soto AM, Sonnenschein C. Environmental causes of cancer: endocrine disruptors as carcinogens. Nat Rev Endocrinol. 2010;6:363–370. doi: 10.1038/nrendo.2010.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Acevedo N, Davis B, Schaeberle CM, Sonnenschein C, Soto AM. Perinatally administered Bisphenol A as a potential mammary gland carcinogen in rats. Environ Health Perspect. 2013;121:1040–1046. doi: 10.1289/ehp.1306734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Murray TJ, Maffini MV, Ucci AA, Sonnenschein C, Soto AM. Induction of mammary gland ductal hyperplasias and carcinoma in situ following fetal Bisphenol A exposure. Reprod Toxicol. 2007;23:383–390. doi: 10.1016/j.reprotox.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Herbst AL, Ulfelder H, Poskanzer DC. Adenocarcinoma of the vagina: association of maternal stilbestrol therapy with tumor appearance in young women. New Engl J Med. 1971;284:878–881. doi: 10.1056/NEJM197104222841604. [DOI] [PubMed] [Google Scholar]
- 50.Robboy SJ, Welch WR, Young RH, Truslow GY, Herbst AL, Scully RE. Topographic relation of cervical ectropion and vaginal adenosis to clear cell adenocarcinoma. Obstet Gynecol. 1982;60:546–551. [PubMed] [Google Scholar]
- 51.Hoover RN, Hyer M, Pfeiffer RM, Adam E, Bond B, Cheville AL, Colton T, Hartge P, Hatch EE, Herbst AL, Karlan BY, Kaufman R, Noller KL, Palmer JR, Robboy SJ, Saal RC, Strohsnitter W, Titus-Ernstoff L, Troisi R. Adverse health outcomes in women exposed in utero to diethylstilbestrol. N Engl J Med. 2011;365:1304–1314. doi: 10.1056/NEJMoa1013961. [DOI] [PubMed] [Google Scholar]
- 52.Palmer JR, Wise LA, Hatch EE, Troisi R, Titus-Ernstoff L, Strohsnitter W, Kaufman R, Herbst AL, Noller KL, Hyer M, Hoover RN. Prenatal diethylstilbestrol exposure and risk of breast cancer. Cancer Epidem Biomar. 2006;15:1509–1514. doi: 10.1158/1055-9965.EPI-06-0109. [DOI] [PubMed] [Google Scholar]
- 53.Zoeller RT, Brown TR, Doan L, Gore AC, Skakkebaek N, Soto AM, Woodruff T, vom Saal FS. Endocrine-dsrupting chemicals and public health protection: a statement of principles from The Endocrine Society. Endocrinology. 2012;153:4097–4110. doi: 10.1210/en.2012-1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Soto AM, Brisken C, Schaeberle CM, Sonnenschein C. Does cancer start in the womb? Altered mammary gland development and predisposition to breast cancer due to in utero exposure to endocrine disruptors. J Mammary Gland Biol Neoplasia. 2013;18:199–208. doi: 10.1007/s10911-013-9293-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wadia PR, Cabaton NJ, Borrero MD, Rubin BS, Sonnenschein C, Shioda T, Soto AM. Low-dose BPA exposure alters the mesenchymal and epithelial transcriptomes of the mouse fetal mammary gland. PLoS One. 2013;8:e63902. doi: 10.1371/journal.pone.0063902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vineis P, Xun W. The emerging epidemic of environmental cancers in developing countries. Ann Oncol. 2009;20:205–212. doi: 10.1093/annonc/mdn596. [DOI] [PubMed] [Google Scholar]
- 57.GLOBOCAN: v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11. 2012. http://globocan.iarc.fr.
- 58.Kessler R. Prevention: air of danger. Nature. 2014;509:S62–S63. doi: 10.1038/509S62a. [DOI] [PubMed] [Google Scholar]
- 59.Gurney JG, Bondy ML. Epidemiology of childhood cancer. In: Pizzo PA, Poplack DG, editors. Principles and Practice of Pediatric Oncology. 5. Philadelphia: Lippincott: Williams &Wilkins; 2006. pp. 1–13. [Google Scholar]