

SUMMARY
Mismatch repair (MMR) corrects DNA polymerase errors occurring during genome replication. MMR is critical for genome maintenance, and its loss increases mutation rates several hundredfold. Recent work has shown that the interaction between the mismatch recognition protein MutS and the replication processivity clamp is important for MMR in Bacillus subtilis. To further understand how MMR is coupled to DNA replication, we examined the subcellular localization of MMR and DNA replication proteins fused to green fluorescent protein (GFP) in live cells, following an increase in DNA replication errors. We demonstrate that foci of the essential DNA polymerase DnaE-GFP decreases following mismatch incorporation and that loss of DnaE-GFP foci requires MutS. Furthermore, we show that MutS and MutL bind DnaE in vitro, suggesting that DnaE is coupled to repair. We also found that DnaE-GFP foci decrease in vivo following a DNA damage-independent arrest of DNA synthesis showing that loss of DnaE-GFP foci is caused by perturbations to DNA replication. We propose that MutS directly contacts the DNA replication machinery, causing a dynamic change in the organization of DnaE at the replication fork during MMR. Our results establish a striking and intimate connection between MMR and the replicating DNA polymerase complex in vivo.
Keywords: fluorescence, localization, mismatch repair, MutS, MutL, DnaE
INTRODUCTION
Prokaryotes and eukaryotes have evolved a series of conserved pathways dedicated to DNA repair ensuring genomic preservation [for review (Friedberg et al., 2006)]. One pathway that shares exquisite conservation across all domains of life is DNA mismatch repair (MMR) [for review (Eisen, 1998; Kunkel and Erie, 2005; Modrich, 2006; Schofield and Hsieh, 2003)]. MMR contributes to a variety of cellular pathways, including anti-recombination in Escherichia coli (Rayssiguier et al., 1989) and DNA damage checkpoint activation in eukaryotes (Hickman and Samson, 1999, 2004; Yoshioka et al., 2006). The most well-studied role for MMR is in the correction of DNA replication errors, manifested as mismatches, insertions, and deletion loops [for review (Kunkel and Erie, 2005; Li, 2008; Schofield and Hsieh, 2003)]. In both bacteria and eukaryotes, deletion of the highly conserved MMR genes mutS and mutL results in a several hundred fold increase in mutation frequency (Cox et al., 1972; Fishel et al., 1993; Ginetti et al., 1996; Hamilton et al., 1995). In humans, germline MMR defects predispose people to several different cancers, including hereditary nonpolyposis colorectal cancer (i.e. Lynch syndrome), Turcot syndrome, and several other sporadic cancers (Hamilton et al., 1995; Nystrom-Lahti et al., 2002; Peltomaki, 2005). Furthermore, in pathogenic bacteria loss of MMR contributes to an increase in the occurrence of multidrug resistant strains found in hospital settings [e.g. (Chopra et al., 2003; Denamur et al., 2002)]. These studies underscore the critical importance of MMR to several aspects of human health.
The Gram-negative bacterium E. coli has been the most well understood bacterial MMR system to date. In E. coli, the sensor protein MutS recognizes a mismatch and initiates MMR by recruiting the linker protein MutL (Schofield et al., 2001). MutL coordinates the action of the nicking endonuclease MutH (Ahrends et al., 2006; Hall and Matson, 1999), which senses the methylation state of the DNA at d(GATC) sequences and subsequently nicks the unmethylated strand representing the nascent DNA. MutL and MutH together coordinate the loading of the DNA helicase, UvrD at the incised nick (Hall et al., 1998; Mechanic et al., 2000). UvrD, fueled by ATP hydrolysis, separates the mismatch-bearing strand for degradation by one of several exonucleases (Viswanathan et al., 2001). Once the mismatch containing strand is degraded, DNA pol III holoenzyme is recruited through an unknown mechanism to resynthesize the gapped region (Lahue et al., 1989). Although many of the steps of E. coli MMR are conserved, DNA methylation and endonuclease cleavage by MutH are mostly restricted to E. coli and its very close relatives (Culligan et al., 2000; Eisen, 1998; Eisen and Hanawalt, 1999). It is well accepted that most bacteria and all eukaryotes lack the methylation-directed repair pathway characteristic of E. coli.
It has been shown in E. coli that subunits of DNA polymerase III holoenzyme (pol III) interact with MutS and MutL (Li et al., 2008; Lopez de Saro and O’Donnell, 2001; Lopez de Saro et al., 2006; Pluciennik et al., 2009). MutS and MutL bind to the DNA replication processivity factor, β clamp, and several components of the clamp loader complex (i.e. γ complex) (Li et al., 2008; Lopez de Saro and O’Donnell, 2001; Lopez de Saro et al., 2006; Pluciennik et al., 2009). MutL binding to β clamp is dependent on single-stranded DNA (Lopez de Saro et al., 2006) and mutation of the β clamp binding site on MutL increases the spontaneous mutation frequency, supporting a functional role for interaction between these two proteins during MMR in vivo (Lopez de Saro et al., 2006). Other work shows that E. coli MutL binds to clamp loader proteins γ, δ, and δ′ (Li et al., 2008). In addition, MutS and MutL have been shown to bind to pol III core and the γ complex (Pluciennik et al., 2009). Within pol III core, MutS and MutL interact with the catalytic subunit α (DnaE) (Pluciennik et al., 2009). Thus, in E. coli, MutS and MutL bind to multiple subunits of DNA polymerase III suggesting that these pathways might be physically linked in vivo. Although each of these binding events appears relatively strong, it is not clear how interaction between MMR proteins and components of DNA polymerase III contribute to the mismatch correction pathway in vitro or in vivo.
Cell biological experiments in the Gram-positive bacterium Bacillus subtilis have shown that foci of MutS fused to yellow fluorescent protein (YFP) colocalize or overlap with the replisome protein DnaX fused to cyan fluorescent protein (CFP) in ~48% of cells in response to the mismatch-inducing base-analog 2-aminopurine (2-AP) (Smith et al., 2001). The authors concluded that mismatches are detected at the site of DNA synthesis and that MutS may move away from the replisome following repair complex assembly and continued DNA synthesis (Smith et al., 2001). These results support the hypothesis that mismatch detection is coupled to the DNA replication machinery in B. subtilis and that the site of DNA synthesis is the preferred location for repair (Smith et al., 2001). Experiments using MutS and MutL fused to green fluorescent protein (GFP) have shown that MutS-GFP and MutL-GFP requires ongoing DNA replication for organization into foci in vivo (Smith et al., 2001). Moreover, it was shown that MutS interacts with β clamp in B. subtilis, and that this interaction is critical for MutS-GFP focus formation and efficient MMR in vivo (Dupes et al., 2010; Simmons et al., 2008a). These observations further support the idea that mismatch recognition and/or repair are targeted to the site of DNA replication in B. subtilis. It is not clear however, if the replication machinery is altered or influenced during the repair process or if other DNA replication proteins in addition to β clamp are involved in this process.
Here, we examined the subcellular localization of fluorescent fusions to six DNA replication proteins during MMR in vivo. We found that foci of the essential DNA polymerase DnaE-GFP decreased following the introduction of mismatches with 2-AP or with a proofreading deficient replicative DNA polymerase (polC mut-1) allele. We show that the loss of DnaE-GFP foci depends on the presence of MMR proteins, specifically MutS. Further investigation showed that a DNA damage-independent block to DNA synthesis caused DnaE-GFP foci to decrease in B. subtilis and that this effect was independent of the MMR pathway. These results suggest that loss of DnaE-GFP foci is an indicator of perturbations to DNA replication in B. subtilis. In addition, protein-blotting experiments show that MutS and MutL bind DnaE, which supports the hypothesis that the MMR proteins are physically coupled to the DNA replication machinery. It is hypothesized that in DNA methylation-independent MMR systems including human and S. cerevisiae, the MMR proteins utilize strand discontinuities to direct the repair process in vitro (Dzantiev et al., 2004; Genschel et al., 2002; Genschel and Modrich, 2003, 2006; Kadyrov et al., 2006; Kadyrov et al., 2007). It is tempting, therefore, to consider that a physical connection between MMR and the DNA replication machinery would provide a mechanism for the repair machinery to target strand termini, thereby aiding in the recognition of the mismatch-containing strand in organisms that lack a methylation directed signal, including B. subtilis.
RESULTS
DnaE-GFP foci decrease following mismatch formation
In B. subtilis, the DNA replication machinery forms foci at distinct subcellular positions characterized as midcell or future midcell positions (Berkmen and Grossman, 2006; Lemon and Grossman, 1998; Migocki et al., 2004). Previous analysis of the subcellular position of the replication protein DnaX-GFP (τ) showed no change in focus position or the percentage of cells with foci following challenge with the mismatch-inducing agent 2-AP (Smith et al., 2001). It is not clear if the subcellular position of DnaX-GFP is representative of the other replication proteins during MMR in B. subtilis.
To address whether addition of 2-AP alters the subcellular localization of other components of the DNA replication machinery, we individually analyzed the percentage of cells with foci bearing fusions to the replication sliding clamp, β clamp (DnaN-GFP), the polymerase dimerization and clamp loading component [DnaX-GFP (τ)], a component of the clamp loader complex [HolB-GFP (δ)], DNA single strand binding protein (SSB-GFP, also known as SsbA-GFP in B. subtilis), the leading and lagging strand DNA polymerase (PolC-GFP), and the essential lagging strand primer maturation DNA polymerase (DnaE-GFP), in addition to MutS-GFP and MutL-GFP (Figure 1). Cells expressing each GFP fusion protein were scored for focus formation untreated or with 2-AP added to the growth medium. For β clamp, DnaX, δ, PolC, MutS, and MutL, each fusion allele was integrated at its normal locus in the B. subtilis chromosome, placing expression of each fusion protein under control of its native promoters (Berkmen and Grossman, 2006; Lemon and Grossman, 1998; Simmons et al., 2008a; Smith et al., 2001) (see Table S1 for a list of strains). The ssb-gfp and dnaE-gfp fusion alleles were in merodiploid strains, where each was expressed from an ectopic locus with the native allele intact as described (Berkmen and Grossman, 2006; Dervyn et al., 2001). The ssb-gfp allele was under control of its native promoters at lacA and expression of the dnaE-gfp allele was controlled by a xylose inducible promoter from the amyE locus (Berkmen and Grossman, 2006; Dervyn et al., 2001). We also integrated dnaE-gfp or dnaE bearing a monomeric gfp variant (dnaE-mgfp) under control of its native promoter as the only copy of dnaE in the cell. The cells were viable and DnaE-GFP foci were observed with both fusion proteins, however the foci that formed were weak and difficult to characterize (data not shown). For this reason, we chose to study the xylose inducible dnaE-gfp allele with the native dnaE gene intact as described previously (Costes et al., 2010; Dervyn et al., 2001).
Figure 1. DnaE-GFP foci decrease following mismatch insertion.
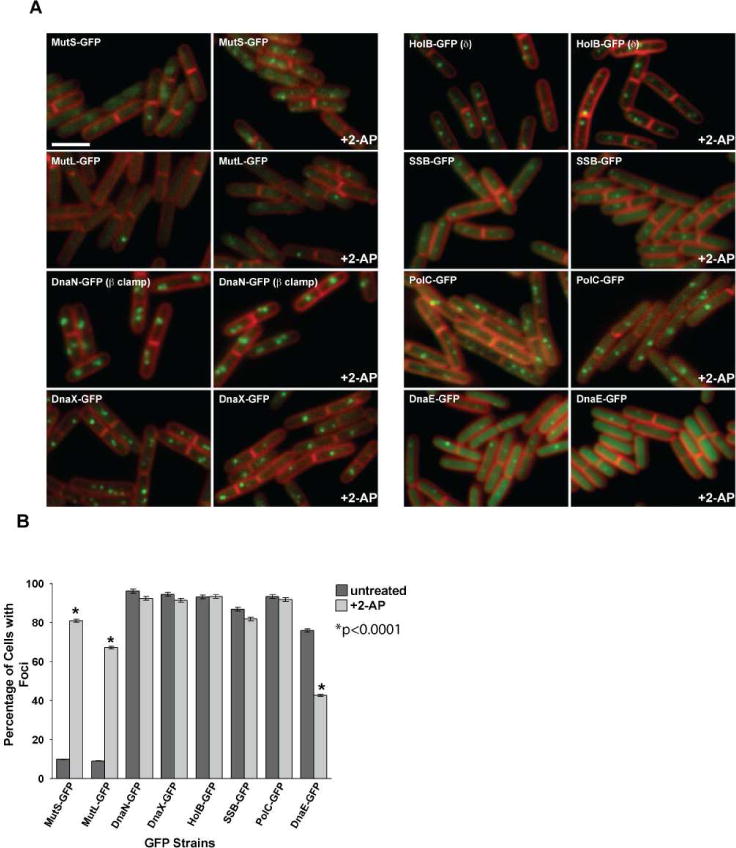
(A) Representative images of MMR proteins MutS-GFP and MutL-GFP and replication proteins DnaN-GFP (β clamp), DnaX-GFP, HolB-GFP (δ), SSB-GFP, PolC-GFP and DnaE-GFP are shown untreated and following 2-AP challenge. In each case, the panel on the left is untreated and the panel on the right shows cells challenged with 2-AP. The membrane is stained with the vital membrane stain FM4-64. The bar indicates 3 μm. (B) Scoring of the percent of cells with foci untreated (dark grey bars) and following 2-AP treatment (light grey bars). The error bars reflect the 95% confidence interval. The asterisk indicates the results are significant with p<0.0001. The bar graph represents a summary of the complete data set presented in Table 2.
The MMR proteins MutS-GFP and MutL-GFP formed foci at primarily midcell positions and focus formation was induced by the presence of 2-AP, confirming previous observations (Figure 1 and Table 1) (Dupes et al., 2010; Simmons et al., 2008a; Smith et al., 2001). Scoring of β clamp-GFP, DnaX-GFP, PolC-GFP, HolB-GFP and SSB-GFP foci showed virtually no change when we compared the percentage of untreated cells with foci to the percentage of cells with foci following challenge with 2-AP (Figure 1 and Table 1). We did, however, notice a striking loss in the percentage of cells with DnaE-GFP foci in the presence of 2-AP when compared to the untreated control (Figure 1 and Table 1). DnaE-GFP in the absence of 2-AP formed foci in ~76% of cells (n=2912). Following 2-AP challenge, DnaE-GFP foci were only observed in ~43% of cells (n=4131) (Figure 1 and Table 2; p<0.0001). Qualitatively, in cells that maintained foci, the fluorescence of DnaE-GFP foci in 2-AP treated cells were decreased and, in many cases, barely detectable (Figure 1). Because 2-AP is a base analog and it does not form a true mismatch, we assayed for DnaE-GFP foci in a strain that contained the mut-1 allele of polC (referred to here as polCexo−), which is defective in proofreading (Sanjanwala and Ganesan, 1991). This allele confers a substantial increase in mutation frequency due to an increase in DNA replication errors (Sanjanwala and Ganesan, 1991). Analysis of DnaE-GFP foci in the polCexo− showed a significant decrease in focus formation as compared with an isogenic polC+ strain (Figure S1). The loss of DnaE-GFP foci in the polCexo− strain was similar to what we observed following 2-AP addition, with p<0.0001 (compare Figure 1 and Figure S1). Thus, we observe both a quantitative and qualitative reduction in DnaE-GFP foci following the addition of 2-AP to the growth medium or in cells lacking a proofreading proficient polC allele. Therefore, we conclude that mismatches cause a decrease in the percentage of cells with DnaE-GFP foci.
Table 1.
Percentage of cells with MMR and replisome foci following 2-AP challenge.
| Percentage of cells with n foci
|
Avg. no. of foci | |||||||
|---|---|---|---|---|---|---|---|---|
| Fusion | 2AP | No. of cells | 0 | 1 | 2 | 3 | >4 | per cell |
| MutS-GFP | − | 1447 | 90.1 | 6.8 | 3.1 | 0 | 0 | 0.13 |
| + | 1589 | 19.0 | 18.8 | 33.1 | 19.2 | 9.8 | 1.82 | |
| MutL-GFP | − | 1592 | 90.9 | 9.1 | 0 | 0 | 0 | 0.09 |
| + | 2065 | 32.7 | 45.6 | 22.4 | 2.7 | 0.5 | 0.97 | |
| DnaN-GFP (β clamp) | − | 858 | 3.8 | 28.1 | 39.3 | 15.6 | 13.2 | 2.06 |
| + | 1072 | 7.6 | 23.8 | 45.0 | 16.4 | 7.2 | 1.92 | |
| DnaX-GFP (τ) | − | 1889 | 5.4 | 28.3 | 45.5 | 9.2 | 11.6 | 1.93 |
| + | 2045 | 8.4 | 41.1 | 39.8 | 5.3 | 5.3 | 1.58 | |
| HolB-GFP (δ) | − | 767 | 6.8 | 19.8 | 39.2 | 18.0 | 16.2 | 2.17 |
| + | 847 | 6.6 | 22.0 | 34.0 | 18.2 | 19.1 | 2.21 | |
| SSB-GFP | − | 1057 | 13.1 | 34.0 | 42.8 | 5.8 | 4.4 | 1.55 |
| + | 1569 | 18.1 | 35.4 | 37.7 | 6.2 | 2.5 | 1.40 | |
| PolC-GFP | − | 1283 | 6.6 | 37.2 | 37.1 | 9.4 | 9.7 | 1.78 |
| + | 1524 | 8.1 | 33.3 | 41.3 | 9.1 | 8.3 | 1.76 | |
| DnaE-GFP | − | 2912 | 23.9 | 51.9 | 19.3 | 4.6 | 0.4 | 1.06 |
| + | 4131 | 57.4 | 38.2 | 4.2 | 0.2 | 0 | 0.47 | |
Strains were grown in S750 minimal medium with 0.2% D-glucose except for strain AK74 harboring the amyE::Pxyl dnaE-gfp allele. AK74 was grown in 1% L-arabinose to allow for expression with the addition of 0.125% D-xylose. The numbers reported are from at least three independent experiments.
Table 2.
Percentage of cells with DnaE-GFP foci.
| Percentage of cells with n foci
|
Avg. no. of foci | |||||||
|---|---|---|---|---|---|---|---|---|
| Strain (% xylose) | 2-AP | No. of cells | 0 | 1 | 2 | 3 | >4 | per cell |
| DnaE-GFP (0.125%) | − | 1082 | 15.9 | 52.9 | 23.2 | 7.0 | 1.0 | 1.24 |
| + | 1003 | 47.0 | 41.3 | 9.7 | 1.6 | 0.3 | 0.67 | |
| DnaE-GFP (0.025%) | − | 962 | 36.4 | 43.7 | 17.2 | 2.6 | 0.2 | 0.87 |
| + | 1414 | 56.3 | 37.0 | 6.4 | 0.4 | 0 | 0.51 | |
| DnaE-GFP (0.005%) | − | 1040 | 54.6 | 34.4 | 9.3 | 1.5 | 0 | 0.58 |
| + | 1671 | 80.3 | 18.4 | 1.3 | 0 | 0 | 0.21 | |
Strains were grown in defined S750 minimal medium with 1% arabinose with the amount of xylose indicated in parentheses. Cultures were split, followed by the addition of 2-AP (600 μg/mL) to one of the cultures for 1 hr prior to imaging (“Experimental Procedures”).
A decrease in DnaE-GFP foci could be directly caused by mismatches or could be a general response to DNA damage or incorporation of DNA replication errors. To address this concern, we challenged cells with mitomycin C (MMC) and monitored DnaE-GFP foci (Figure 2). MMC forms a mono adduct at the N2 position of guanine as well as inter- and intra-strand crosslinks [for review (Dronkert and Kanaar, 2001)]. We found that the percentage of cells with DnaE-GFP foci did not decrease following MMC challenge (Figure 2). To the contrary, we measured an increase in the percentage of cells with DnaE-GFP foci following MMC challenge (Figure 2, compare panels (B) and (C)). Thus, we found that loss of DnaE-GFP foci is not a widespread signal for DNA perturbations in B. subtilis.
Figure 2. The DNA damaging agent Mitomycin C increases the percentage of cells with DnaE-GFP foci.
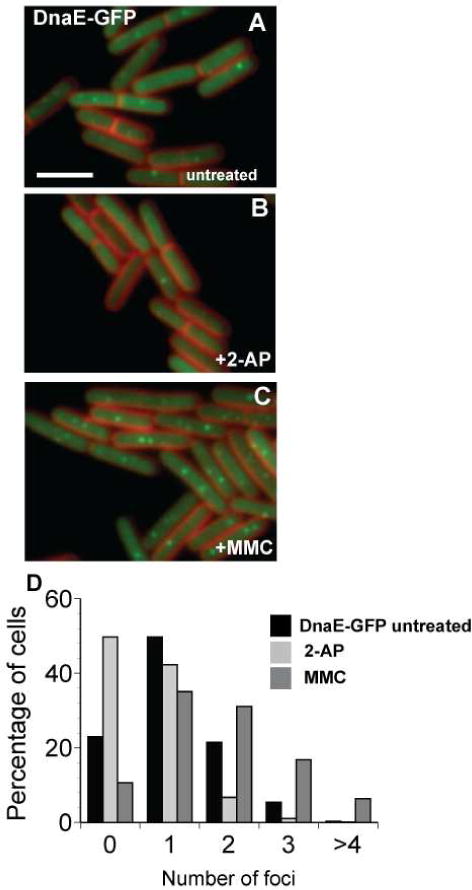
(A) Shown are cells untreated; (B) challenged with 2-AP (600 μg/ml); and (C) challenged with mitomycin C (MMC) (200 ng/ml). (D) The percentage of cells with 0, 1, 2, 3, and >4 DnaE-GFP foci per cell following the indicated growth condition are presented (the following number of cells were scored: untreated n= 1277, +2-AP n= 980, +MMC n= 881). The bar indicates 3 μm.
As discussed above, the dnaE-gfp fusion allele was ectopically expressed from a xylose inducible promoter (Pxyl) (Costes et al., 2010; Dervyn et al., 2001). We examined the level of DnaE-GFP expression relative to native DnaE and found that with 0.125% xylose the level of DnaE-GFP was comparable to the level of native DnaE in vivo (Figure S2). To determine if the level of DnaE-GFP protein in vivo contributed to the decrease in localization following 2-AP challenge, we scored the percentage of cells with DnaE-GFP foci grown in medium containing three different amounts of the inducer [xylose, at 0.125%, 0.025% and 0.005%] in the presence or absence of 2-AP. We found that the percentage of untreated cells with DnaE-GFP foci did decrease as the amount of xylose in the medium was reduced (Figure S3, Table 2). However, when cells at each xylose amount were examined following challenge with 2-AP, we observed a significant reduction in the percentage of cells with DnaE-GFP foci (Figure S3; p<0.0001 for each xylose concentration). We performed an immunoblot to detect DnaE-GFP levels in vivo with antibodies against the GFP moiety. We found that the levels of DnaE-GFP foci were indeed reduced as the amount of xylose was reduced (Figure S3C). We did not detect release of GFP from DnaE by proteolysis in vivo, as judged by immunoblotting (data not shown). Furthermore, at each percentage of xylose tested, the level of DnaE-GFP protein was unaffected by the addition of 2-AP. As a control, we analyzed β clamp (DnaN) levels in vivo and found that the level of β clamp was unchanged by the amount of xylose, or by the addition of 2-AP (Figure S3C). Thus, although the percentage of cells with DnaE-GFP foci is influenced by the amount of xylose added, 2-AP addition causes DnaE-GFP foci to decrease at each level of xylose we examined.
Complementary to these studies, we found that a strain expressing dnaE-mgfp from its native locus was also decreased for focus formation when 2-AP was added to the growth medium. In this experiment ~45% (n=201) of untreated cells showed DnaE-mGFP foci, while only ~15% (n=325) of cells showed DnaE-mGFP foci following 2-AP challenge, with p<0.0001 (data not shown). Taken together, under every experimental condition we examined, including the use of different fusions to DnaE and different levels of DnaE-GFP in the cell, 2-AP challenge caused a significant loss of DnaE-GFP foci in vivo.
Loss of DnaE-GFP foci requires the mismatch sensor protein MutS
Our results indicate that 2-AP challenge causes DnaE-GFP foci to decrease in vivo. To understand if loss of DnaE-GFP foci is dependent on the MMR pathway, we scored DnaE-GFP foci following 2-AP treatment in a strain disrupted for the mutS and mutL genes (Figure 3 and Table 3). We found that in cells disrupted for both mutS and mutL (mutSL::spc), DnaE-GFP foci were maintained following 2-AP addition (Figure 3 and Table 3) and the foci in these cells qualitatively resembled untreated cells (Figure 3). This experiment supports the hypothesis that a functional MMR pathway is required to observe a decrease of DnaE-GFP foci in vivo (Figure 3 and Table 3).
Figure 3. Loss of DnaE-GFP foci in response to DNA synthesis perturbations caused by mismatches requires MutS and MutL.
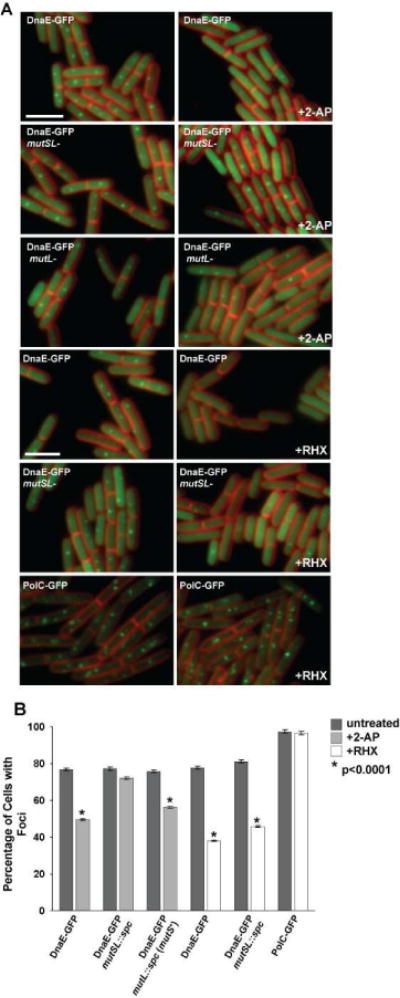
(A) Representative epi-fluorescence micrographs of DnaE-GFP in genetic backgrounds disrupted for mutSL::spc and mutL::spc, and either untreated (left column) or challenged with 2-AP or arginine hydroxamate (RHX) (right column). The membrane is stained with FM4-64 and the bar indicates 3 μm. (B) A bar graph quantifying the percentage of cells with DnaE-GFP foci under the conditions indicated in the panel is shown. The bar graph represents a summary of the complete data set shown in Table 4. The asterisk indicates p<0.0001 between treated and untreated samples. Error bars indicate the 95% confidence interval.
Table 3.
Percentage of cells with DnaE-GFP foci in strains containing or lacking mismatch repair genes, grown in the presence or absence of 2-aminopurine (2-AP) or arginine hydroxamate (RHX).
| Percentage of cells with n foci
|
Avg. no. of foci | |||||||
|---|---|---|---|---|---|---|---|---|
| Relevant characteristics | additive | No. of cells | 0 | 1 | 2 | 3 | >4 | per cell |
| DnaE-GFP | − | 1460 | 23.2 | 48.8 | 21.8 | 5.5 | 0.6 | 1.12 |
| 2-AP | 1710 | 50.4 | 37.0 | 11.2 | 1.1 | 0.1 | 0.64 | |
| DnaE-GFP, mutSL::spc | − | 1889 | 22.8 | 52.1 | 20.0 | 4.7 | 0.4 | 1.08 |
| 2-AP | 1356 | 28.0 | 52.5 | 16.5 | 2.8 | 0.2 | 0.95 | |
| DnaE-GFP, mutL::spc (mutS+) | − | 1082 | 24.3 | 48.9 | 20.0 | 6.0 | 0.7 | 1.10 |
| 2-AP | 2239 | 43.6 | 46.9 | 7.5 | 1.7 | 0.3 | 0.68 | |
|
| ||||||||
| DnaE-GFP | − | 1187 | 22.3 | 52.6 | 20.0 | 4.6 | 0.6 | 1.09 |
| RHX | 1273 | 62.0 | 31.4 | 5.9 | 0.7 | 0 | 0.45 | |
| DnaE-GFP, mutSL::spc | − | 928 | 18.9 | 56.6 | 21.6 | 2.5 | 0.6 | 1.09 |
| RHX | 872 | 54.2 | 36.1 | 8.3 | 1.3 | 0.1 | 0.57 | |
| PolC-GFP | − | 1051 | 2.7 | 20.2 | 45.2 | 13.6 | 18.4 | 2.25 |
| RHX | 1141 | 3.5 | 33.2 | 42.0 | 10.3 | 11.0 | 1.92 | |
Cells were grown in defined S750 minimal medium with 1% arabinose and 0.125% xylose where DnaE-GFP was imaged and in 0.2% glucose when PolC-GFP was imaged. 2-AP was used at a final concentration of 600 μg/mL and RHX was used at a final concentration of 0.5 mg/mL. Cells were imaged following treatment for 1 hr with RHX or 2-AP.
To distinguish the component of the MMR pathway responsible for a decrease in DnaE-GFP foci, we scored DnaE-GFP in a strain lacking the mutL gene and expressing mutS in the presence and absence of 2-AP. Addition of 2-AP to this strain caused a decrease in DnaE-GFP foci, compared with untreated cells, with p<0.0001 (Figure 3 and Table 3). This result suggests that MutS is necessary for loss of DnaE-GFP foci following challenge with 2-AP. In an effort to further demonstrate that loss of DnaE-GFP foci in response to 2-AP treatment was mutS dependent, we tested the effect of 2-AP on DnaE-GFP foci in a strain disrupted for the mutSL operon, with mutL expression restored from an ectopic locus located at lacA (Figure 4). In this strain, mutS is not expressed (Figure 4A), and mutL expression is driven by IPTG at a concentration of 200 μM restoring MutL protein to wild type levels (Figure 4A). Also in this strain, DnaE-GFP expression is driven by xylose at a concentration of 0.125% as described above. Upon addition of 2-AP to cultures lacking mutS and expressing MutL and DnaE-GFP, we found virtually no change in DnaE-GFP foci in cells untreated or challenged with 2-AP (Figure 4B and C). With these data, we conclude that loss of DnaE-GFP foci upon treatment with 2-AP is dependent on MutS (see Discussion).
Figure 4. MutS and 2-AP are necessary and sufficient to cause loss of DnaE-GFP foci.
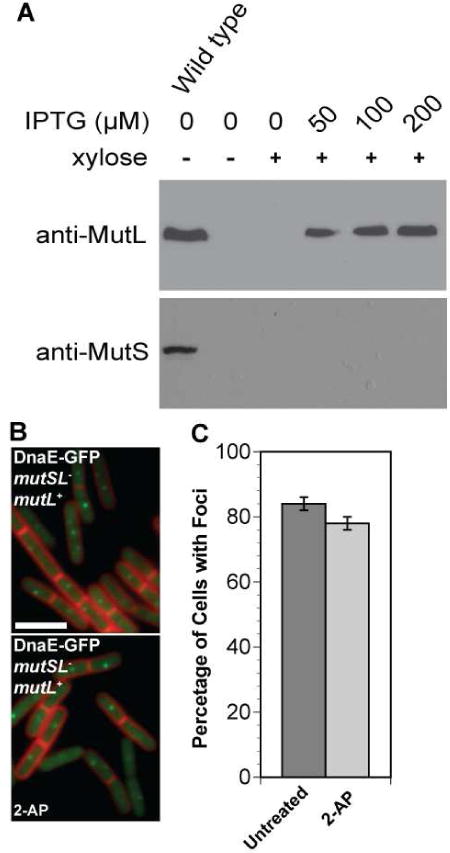
(A) Shown are immunoblots of whole cell extracts for strain JWS68 (relevant genotype: amyE::PxyldnaE-gfp; mutSL::kan; lacA::PspacmutL) bearing a mutSL deletion with mutL expression restored to wild type levels from the lacA locus. The first lane is an extract from PY79, while the remaining lanes are extracts probed for MutL from strain JWS68 with the indicated amount of IPTG. For xylose, the (+) symbol designates 0.125% xylose was added. (B) Representative images of DnaE-GFP are shown in the presence or absence of 2-AP with mutL expression restored from the lacA locus. (C) The bar graph quantifies the percentage of cells with DnaE-GFP foci untreated or following challenge with 2-AP. The number of cells scored for the untreated sample were as follows: untreated (n=1154) and 2-AP (n=1251). The error bars indicate the 95% confidence interval.
DNA replication arrest causes decrease of DnaE-GFP foci
To investigate if loss of DnaE-GFP foci is a consequence of inhibition of DNA replication, we used an established DNA damage-independent method to arrest DNA synthesis in B. subtilis (Wang et al., 2007). Challenge of B. subtilis cells with the amino acid analog arginine hydroxamate (RHX) causes amino acid starvation and induces the stringent response (Wang et al., 2007). The stringent response produces the small signaling molecules ppGpp and pppGpp [collectively referred to as (p)ppGpp] [for review (Paul et al., 2004; Srivatsan and Wang, 2008)] both of which bind and inhibit primase (DnaG), causing a rapid arrest in DNA synthesis. Using this method, nucleotide incorporation becomes undetectable within minutes of RHX addition [(Wang et al., 2007) and data not shown].
To determine if replication fork arrest results in a decrease in DnaE-GFP foci, we induced stringent response arrest of DNA replication following the addition of RHX. Indeed, we found that DnaE-GFP foci decreased in cells challenged with RHX (Figure 3A and Table 3), and that the stringent response-induced decrease of DnaE-GFP foci was more severe than the response we observed with 2-AP (compare +2-AP and +RHX panels in Figure 3A and the bar graph in Figure 3B).
A strain of B. subtilis lacking the MMR proteins MutS and MutL (mutSL::spc) still shows a decrease in DnaE-GFP foci in the presence of RHX, identical to the decrease of DnaE-GFP foci observed in a strain containing MutS and MutL (Figure 3). Therefore, the RHX effect on DnaE-GFP foci is independent of the MMR pathway, and is indicative of a general perturbation of the DNA replication fork that results in loss of DnaE-GFP foci in vivo.
The specificity of DnaE-GFP focus loss was evident when we compared the foci of a strain harboring PolC-GFP in the presence and absence of RHX. We scored PolC-GFP foci and found that the percentage of cells with PolC-GFP foci was nearly identical in cells that were growing normally and in cells that experienced a stringent response arrest of DNA replication (Figure 3). Therefore, only DnaE-GFP shows a dramatic loss of foci, indicating that DnaE undergoes a strong change following perturbations to DNA synthesis. This DNA replication disruption appears to cause a more striking effect than the perturbation exerted by MMR. However, it is clear that a disruption to DNA replication, such as that caused by mismatches, results in a MMR-dependent decrease to DnaE-GFP foci in vivo.
Protein levels of DnaE, MutL, MutS, and β clamp (DnaN) are unchanged following 2-AP challenge
We have shown that the percentage of cells with DnaE-GFP foci decreases in response to replication errors (Figures 1 – 4). One explanation for the loss of DnaE-GFP foci is that the MMR proteins, or DnaE, were degraded. Therefore, we performed immunoblots to determine if the levels of DnaE-GFP were altered following 2-AP challenge in both the presence and absence of the MMR gene products. Using monoclonal antibodies against GFP, we found that DnaE-GFP levels in cells growing normally or following 2-AP challenge were unchanged (Figure 5A). Furthermore, DnaE-GFP levels were unaffected by the presence or absence of the MMR proteins MutS and MutL (Figure 5A). Additionally, we also did not detect proteolytic release of GFP following 2-AP challenge (Figure 5B), nor did we detect an increase in the DnaE protein pool, which could result from degradation of the GFP moiety (Figure S2). Using antibodies generated against MutS, MutL, and β clamp (DnaN), we found that the levels of each of these proteins were unchanged in cells challenged with 2-AP. As controls, we show that MutS and MutL were not detected in whole cell extracts prepared from cells disrupted for their respective genes (Figure 5A). With these data, we conclude that the levels of MMR proteins, DnaE, and β clamp were unchanged following 2-AP challenge, and that the loss of DnaE-GFP foci does not result from degradation of DnaE, proteolytic release of the GFP moiety from DnaE or degradation of GFP from DnaE.
Figure 5. Cellular levels of replication and MMR proteins are unchanged following 2-AP challenge.
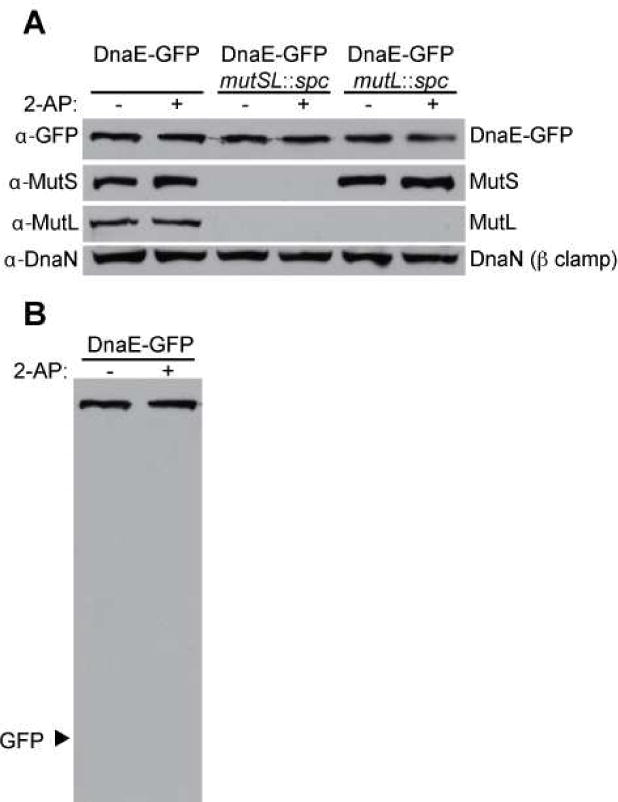
Shown is an immunoblot of DnaE-GFP, MutS, MutL and DnaN (β clamp). (A) The relevant genetic markers for the strains prepared for immunoblot analysis are indicated and cells treated with 2-AP are also indicated. Cultures with 2-AP were challenged for 1 hr with 600 μg/ml in LB medium. Each blot is representative of multiple independent experiments. (B) Shown is the full immunoblot of DnaE from the first two lanes of (A) with the expected position of proteolytically released GFP indicated.
Replication fork integrity is maintained during mismatch-induced perturbations to DNA replication
One explanation for the loss of DnaE-GFP foci upon 2-AP addition to growth medium is that the introduction of mismatches could destabilize the replication fork, causing fork collapse and subsequent changes to the subcellular localization of DnaE. To investigate the integrity of the replication fork during MMR, we analyzed the recruitment of RecA-GFP to the replication fork by fluorescence microscopy (Simmons et al., 2007; Simmons et al., 2009). In B. subtilis, RecA-GFP has been shown to form foci following replication fork stress (Bernard et al., 2010; Simmons et al., 2009). In addition, Wang and co-workers showed that stringent response arrest of DNA synthesis does not stimulate recruitment of RecA-GFP into foci in vivo even though DNA synthesis rapidly arrests (Wang et al., 2007). We examined the response of cells to 2-AP, and stringent arrest with RHX, and found no difference in the percentage of cells with RecA-GFP foci when compared to untreated cells (Figure 6). By contrast, the addition of the DNA-damaging agent mitomycin C (MMC) had a significant affect on the recruitment of RecA-GFP into foci; the percentage of cells with RecA-GFP foci was significantly higher when challenged with MMC, compared to unchallenged cells (Figure 6, p<0.0001). The percentage of cells with RecA-GFP foci compared between the untreated and 2-AP or RHX challenged groups was not significant (Figure 6). Therefore, perturbations in DNA replication caused by mismatches or amino acid starvation fails to recruit RecA-GFP, indicating a distinct correlation between conditions causing a decrease of DnaE-GFP but do not promote recruitment of RecA-GFP into foci. We suggest that loss of DnaE-GFP foci does not cause excess ssDNA to accumulate, which would have been indicative of replication fork stress exerted by MMR.
Figure 6. Replication fork integrity is maintained during MMR.
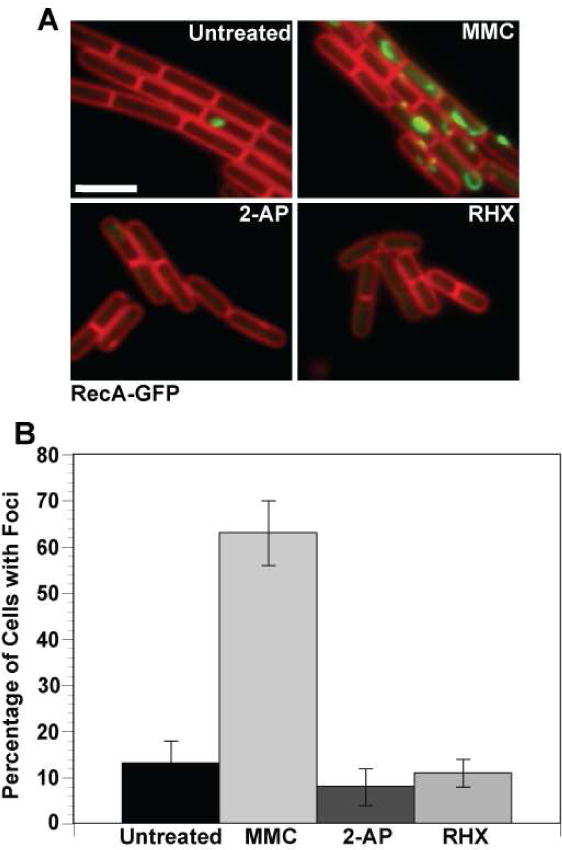
Shown are representative epi-fluorescence micrographs of RecA-GFP foci in cells (A) untreated; or challenged with MMC (100 ng/ml); 2-AP (600 μg/ml); or arginine hydroxamate (RHX). RecA-GFP foci are shown in green and the cell membrane is shown in red and was visualized using the vital membrane stain FM4-64. (B) A bar graph showing the percentage of cells with RecA-GFP foci following the indicated conditions. For each condition, the following numbers of cell were scored: untreated (n=201), MMC (n=168), 2-AP (n=210) and RHX (n=349). These data were analyzed using a two-tailed z test for significance between the untreated and treated groups. While the difference between untreated, +2-AP, and +RHX is insignificant, the +MMC bar has a significant difference from the other three (p<0.0001). Error bars reflect the 95% confidence interval.
MutS and MutL directly bind DnaE in vitro
We hypothesized that the mechanism resulting in loss of DnaE-GFP foci could be caused by a direct interaction between MMR proteins and the DNA replication machinery. Given that DnaE-GFP foci decreased in response to mismatches, we asked if MutS and MutL bound DnaE in vitro. To this end, we cloned, overexpressed, and purified MutS, MutL, β clamp and DnaE using an N-terminal polyhistidine tag that was cleaved following purification. We also purified His6-SSB for comparison (see “Experimental Procedures”). All five proteins were purified to near homogeneity and were judged as > 95% pure by SDS-PAGE (data not shown). Using these purified proteins, we affinity purified rabbit polyclonal antibodies that had been generated against MutS, MutL, and DnaE (see “Experimental Procedures”). To test the specificity of the affinity-purified antibodies, we spotted 40 pmol of each DNA replication protein, as well as BSA as a negative control, on a nitrocellulose membrane, followed by probing the membrane with one of three affinity-purified polyclonal antibodies against DnaE, MutL, or MutS. Anti-DnaE and anti-MutS antibodies were highly specific and detected only DnaE and MutS proteins, respectively (Figure 7A). For anti-MutL, we observed very slight cross-reactivity with high levels of purified MutS, but we did not observe any cross-reactivity with the remaining proteins examined (Figure 7A). Thus, the purified antibodies against MutS, MutL, and DnaE are specific and were used in the experiments that follow.
Figure 7. MutS and MutL directly bind DnaE.
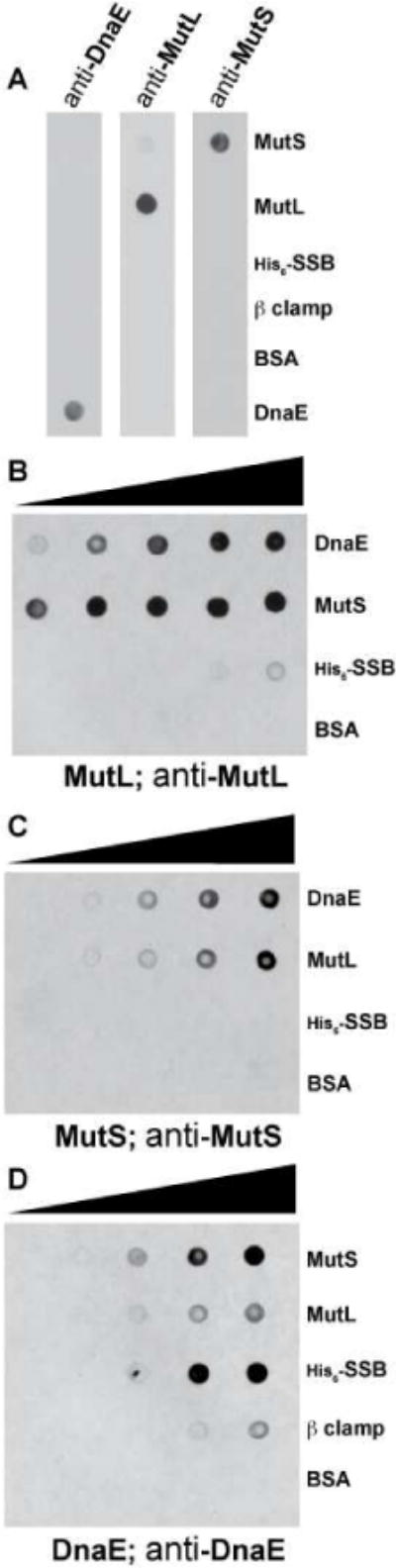
(A) Specificity test of the polyclonal antibodies against DnaE, MutL and MutS. 40 pmols of each protein was spotted onto a nitrocellulose membrane and probed with the indicated antibodies. (B–D) Immunodot blot binding analysis of the indicated proteins. Each protein was applied to a nitrocellulose membrane, following incubation with (B) 400 nM MutL; (C) 400 nM MutS; and (D) 400 nM DnaE, each membrane was then probed with the indicated polyclonal antibody against the indicated prey protein as described in “Experimental Procedures.”
To determine if MutS and MutL directly bind DnaE in vitro, we performed an immunodot blot (far-western) analysis. In this experiment, we spotted increasing amounts (2.5 to 40 pmol) of the indicated “bait” proteins on a nitrocellulose membrane. The membrane was then probed with the indicated “prey” protein, to allow for binding between the bait and prey proteins. Next, the membrane was probed with antibodies specific for the “prey” protein to determine the retention of the “prey” protein on the membrane and thus reveal interactions between the proteins examined.
We initially performed an immunodot blot experiment where DnaE and MutS were immobilized on the membrane, followed by probing with MutL. Strikingly, we found that MutL bound DnaE and MutS even down to the lowest amount of bait protein examined (2.5 pmol) (Figure 7B). As a control, we found that MutL did not bind BSA and interestingly showed some binding to His6-SSB (Figure 7B). We performed the reciprocal experiment, probing for retention of MutS on a membrane through interaction with DnaE and MutL. We found that MutS also bound DnaE and MutL with a comparable level of retention, while MutS did not bind BSA or His6-SSB at the protein concentrations tested (Figure 7C). We did however observe some interaction between MutS and His6-SSB when we probed the membrane with a higher concentration of MutS (data not shown).
We applied MutS and MutL to a nitrocellulose membrane to compare the retention of DnaE by each of these MMR proteins. We found that both MutS and MutL bound DnaE, and interestingly, MutS retained more DnaE than MutL (Figure 7D). We interpret these results to mean that MutS may have a stronger affinity for DnaE than does MutL. As a positive control, we detected a weak interaction between DnaE and β clamp, as described (Simmons et al., 2008a). We also found that DnaE bound His6-SSB, suggesting a strong interaction exists between these two DNA replication proteins (Figure 7D). Our finding that DnaE binds SSB has also been recently reported, and this work showed that DnaE binding to SSB is mediated through the C-terminal region of SSB (Costes et al., 2010). We conclude that the MMR proteins MutS and MutL directly interact with the essential DNA polymerase DnaE.
DISCUSSION
Here, we have fused GFP to components of the B. subtilis DNA replication machinery to investigate the effects of MMR on the subcellular organization of the DNA replication complex. We found that incorporation of mismatches induces loss of DnaE-GFP foci and that loss is dependent on the mismatch sensor MutS. Our analysis of six replication proteins indicates that the other five DNA replication proteins we examined were unaffected by 2-AP challenge, given that the percentage of cells with foci was unchanged between the conditions tested. It is striking that DnaE-GFP was the only replication protein altered following mismatch formation. For several years, it has been known in B. subtilis and in human cells that MutS homologs colocalize with the DNA replication machinery in vivo (Kleczkowska et al., 2001; Smith et al., 2001). Earlier studies in both organisms have centered on understanding the connection between MMR proteins and the replication sliding clamps: β clamp and PCNA in B. subtilis and human cells, respectively (Iyer et al., 2008; Kleczkowska et al., 2001; Pluciennik et al., 2010; Simmons et al., 2008a). Replication sliding clamps are known to bind MutS homologs in both systems and are known to have roles in regulating protein traffic at sites of DNA synthesis [for review (Lopez de Saro et al., 2004; Sutton, 2009)]. Until now, it has been unclear whether the DNA replication machinery itself is affected by MMR. Our results are perhaps the most striking example showing an intimate connection between the DNA replication machinery and the MMR pathway, as we found that MutS causes loss of DNA polymerase DnaE-GFP following mismatch formation in vivo.
Our results raise the question of why foci of DnaE-GFP are altered during repair. It is possible that the biological function of DnaE at a replication fork makes it susceptible to focus loss following perturbation to DNA replication. For example, the slower rate of DNA synthesis by DnaE (Sanders et al., 2010), or the hypothesis that the turnover of DnaE at replication forks is higher than other proteins, given that it switches with PolC during lagging strand synthesis (Sanders et al., 2010) could make DnaE more susceptible to a decrease in localization in response to DNA replication fork perturbations. Another reason that DnaE-GFP focus formation could be influenced is due to the fact that DnaE is involved in lagging strand maturation (Sanders et al., 2010) and MMR shows preference for repairing mismatches in the lagging strand of S. cerevisiae and E. coli (Fijalkowska et al., 1998; Pavlov et al., 2003). Although the overall mechanism causing a decrease in DnaE-GFP foci is not clear, loss of DnaE-GFP foci is signaled following mismatch detection by MutS. Since dnaE is essential and required for replication, we investigated the possibility that nascent strand synthesis was altered during MMR. We did measure a slight decrease in nascent strand synthesis using 3H-thymidine incorporation in vivo, but these results overall were not conclusive (data not shown). However, we cannot exclude the possibility that a brief MMR-dependent pause occurs in DNA synthesis that cannot be readily measured in 3H incorporation experiments.
It was recently shown that the C-terminus of SSB is involved in the recruitment of several proteins to the DNA replication machinery in B. subtilis, including DnaE (Costes et al., 2010). Strains bearing a truncated version of the SSB C-terminus (Δ35 or Δ6) grow slowly but are viable, yet DnaE-GFP foci are not observed in these cells (Costes et al., 2010). Since SSB interacts with a host of proteins involved in DNA metabolism, it was not determined if the slow growth phenotype results from the action of an individual protein or a protein complex. Moreover, ectopic expression of dnaE alone did not rescue the slow growth phenotype (Costes et al., 2010). Together these data suggest that the assembly of DnaE into a focus is not required for viability, although it could very well contribute to a normal growth rate in B. subtilis. One possibility is that MutS binding to DnaE prevents DnaE from interacting with SSB resulting in loss of DnaE-GFP foci.
Wang and co-workers have shown that amino acid starvation arrests DNA replication in B. subtilis by inhibiting primase (DnaG) (Wang et al., 2007). Amino acid starvation is a known method that causes DNA damage-independent perturbation to DNA replication (Wang et al., 2007). We found that stringent arrest of DNA replication indeed induced a loss of DnaE-GFP foci (Figure 3). In contrast, another replication protein, the predominant DNA polymerase PolC, was unaffected by treatment with RHX (Figure 3). These RHX results suggest that DnaE-GFP focus formation is a sensitive single cell assay for DNA damage-independent perturbations to DNA replication, based on our observations that mismatches and RHX arrest of DNA synthesis cause a loss in DnaE-GFP foci.
We speculate that following mismatch detection, MutS binds β clamp, thereby decreasing the amount of β clamp available for DnaE. The reduction of available β clamp could subsequently decrease the processivity of DnaE-dependent DNA synthesis (Sanders et al., 2010), resulting in a general perturbation to DNA replication and loss of DnaE-GFP foci. It has been shown that binding of MutS to β clamp is critical for repair and for the formation of MutS-GFP foci in response to mismatches in vivo (Dupes et al., 2010; Simmons et al., 2008a). We propose that following mismatch detection, MutS occupies the general protein binding site on β clamp, which would in turn prevent or reduce binding of DnaE to this site on β clamp and cause the loss of DnaE-GFP foci. This hypothesis is being tested through our attempts to isolate mutant forms of MutS that bind DnaE and are unable to bind β clamp. So far, we have yet to isolate such a MutS mutant with these characteristics. As mentioned above, another possibility is that binding of MutS to DnaE prevents interaction between DnaE and the C-terminal region of SSB, which would decrease the accumulation of DnaE at sites of DNA replication as described (Costes et al., 2010).
We also show that MutL binds DnaE in vitro, yet we show that the mutL gene in the absence of mutS is unable to cause a loss of DnaE-GFP foci in response to 2-AP. MutL is hypothesized to have a role in recruitment of a DNA polymerase for DNA synthesis following removal of the mismatch-containing strand. A possible role for the interaction we uncovered between MutL and DnaE could be for directing DnaE for resynthesis of the DNA during repair. We speculate that this interaction does not occur during initial mismatch detection and MMR activation as MutS and 2-AP are required for loss of DnaE-GFP foci in vivo.
B. subtilis uses a methylation-independent MMR mechanism like most bacteria and all eukaryotic systems (Culligan et al., 2000; Eisen, 1998; Eisen and Hanawalt, 1999). The mechanism that B. subtilis uses to identify the mismatch containing strand from the template strand is still unknown. It has been shown, however, that B. subtilis MutL is an endonuclease and the structure of the endonuclease domain has been solved (Pillon et al., 2010; Pillon et al., 2011). It has also been shown that integrity of the endonuclease active site is required for MMR in vivo and the endonuclease containing domain binds to β clamp (Pillon et al., 2010; Pillon et al., 2011). It is generally accepted that DNA strand discontinuities, including a nick, or a gap may be used to direct the repair machinery to the nascent strand in organisms that lack a methyl-directed signal [e.g. (Bruni et al., 1988; Dzantiev et al., 2004; Genschel et al., 2002; Genschel and Modrich, 2003; Holmes et al., 1990; Lacks et al., 1982) for review (Hsieh and Yamane, 2008; Kunkel and Erie, 2005; Larrea et al., 2010; Schofield and Hsieh, 2003)]. Furthermore, recent work suggests that endonuclease containing MutL proteins are capable of nicking a strand with a terminus (Kadyrov et al., 2007; Pluciennik et al., 2010). If strand discontinuities do indeed establish identification of the correct strand, it is not clear how the MMR machinery is targeted to strand termini at a replication fork.
It has been suggested that interactions between MutS homologs and their cognate processivity clamps could provide such a function (Dupes et al., 2010; Flores-Rozas et al., 2000; Kleczkowska et al., 2001; Lee and Alani, 2006; Lopez de Saro and O’Donnell, 2001; Lopez de Saro et al., 2006; Simmons et al., 2008a). This suggestion comes from the numerous published manuscripts showing that MutS and MutL homologs bind processivity clamps [e.g. (Dupes et al., 2010; Flores-Rozas et al., 2000; Kleczkowska et al., 2001; Lee and Alani, 2006; Simmons et al., 2008a)] and the observation that processivity clamps are loaded at strand termini [(Georgescu et al., 2008; Jeruzalmi et al., 2001) for review (Johnson and O’Donnell, 2005)]. Many studies on the physical connections between MMR and DNA replication proteins have centered on the processivity clamps in a variety of organisms.
The results we present here provide striking evidence for a perturbation to the replisome following mismatch detection by MutS in vivo and direct interaction between the mismatch repair proteins and the essential DNA polymerase DnaE. Collectively these data establish an intimate connection between MMR and the replication fork, and we suggest that MMR is integrated and actively engaged with the DNA replication complex to a greater extent than has previously been appreciated.
EXPERIMENTAL PROCEDURES
Bacteriological methods
The Bacillus subtilis strains used in this study are described in Table S1. Strains used here were constructed using standard procedures (Hardwood and Cutting, 1990); resulting progeny were grown on plates containing appropriate antibiotics correlating to the specific alleles to be transferred. Antibiotics were used at the following final concentrations: 100 μg/mL spectinomycin (spc); 5 μg/mL kanamycin (kan); 5 μg/mL chloramphenicol (cat); 5 μg/mL tetracycline (tet); 0.5 μg/ml erythromycin as described (Hardwood and Cutting, 1990; Klocko et al., 2010; Simmons et al., 2008a; Simmons et al., 2008b).
Live cell microscopy
Cells were prepared for live imaging essentially as described (Klocko et al., 2010). Briefly, strains were grown at 30°C in 1X S750 media supplemented with 0.2% glucose, except DnaE-GFP, which required 1% arabinose and xylose (typically 0.125%, except the concentrations indicated in Figure S3). Cells were grown to an optical density at 600 nm (OD600) of ~0.3 to 0.4. Cultures were split, and 2-aminopurine was added to one culture to a final concentration of 600 μg/mL. Cells were allowed to grow for an additional hour to form mismatches. The hour time point for visualization of DnaE-GFP foci was used because 2-AP incorporation into DNA is inefficient (Goodman et al., 1977; Grafstrom et al., 1988; Watanabe and Goodman, 1981) and because MutS-GFP and MutL-GFP have been shown to respond and form foci in approximately ~50% of cells following 1 hr of 2-AP treatment as described (Dupes et al., 2010; Klocko et al., 2010; Simmons et al., 2008a; Smith et al., 2001). Following incubation, 300 μL aliquots of cultures were incubated with the vital membrane stain FM4-64 and cells were allowed to settle on microscope slides containing 1% agarose pads prepared with 1X Spizizens salts (Simmons et al., 2008a; Simmons et al., 2009). Experiments performed with JWS68 were carried out as described, with 0.125% xylose and 200 μM IPTG in the culture medium to drive expression of mutL and dnaE-gfp. Cells were imaged using an Olympus BX61 microscope equipped with a Hamamatsu ORCAR2 CCD cooled camera and a Lumen 200 arc metal light source (Prior). An Olympus 100X oil immersion 1,45 NA TIRFM objective lens was used as described (Klocko et al., 2010). For detection of GFP and FM4-64 the following filter sets were used: FITC excitation 460–500, emission 510–560; TRITC excitation 510–560, emission 572–648. Images were captured and processed using Slidebook 4.2 (Advanced Imaging Software) and Photoshop (Adobe). Foci were scored using the software ImageJ (http://rsbweb.nih.gov/ij/), and quantification graphs were made using SigmaPlot (Systat) or Deltagraph 5 (Redrock Software). Images were assembled into figures using Illustrator (Adobe). Imaging for each strain was performed at least twice.
Immunoblotting
Immunodetection of MMR and DNA replication was performed as described (Rokop et al., 2004; Simmons et al., 2003; Simmons et al., 2007). Briefly, 10 mL cultures of DnaE-GFP cells were grown at 30°C in 1XS750 media supplemented with 1% arabinose and xylose (grown identically to imaging conditions) to an optical density of OD600~0.3–0.4, whereupon 2-aminopurine was added to 600 μg/mL final concentration. Cells were grown an additional hour at 30°C and 1.5 optical density 600 nm units of cells were removed, concentrated by centrifugation, frozen in liquid N2 and stored at −80°C. Whole cell lysates were prepared as described (Rokop et al., 2004). Lysates corresponding to an equivalent number of cells were separated in a 6% denaturing SDS-PAGE followed by transfer to a nitrocellulose membrane (Whatman) as described (Simmons et al., 2003). Membranes were probed with the indicated 1° Antibody (rabbit, typically 1:1,000 dilution), and goat anti-rabbit HRP conjugated secondary antibody (Pierce, typically 1:10,000 dilution) in 5% Milk + 1X TBST (1X Tris buffered saline + 0.02% Tween20 [Sigma]). Membranes were then exposed to film (BioExpress), images scanned into Photoshop (Adobe), and figures assembled in Illustrator (Adobe).
For the immunoblots shown in Figure 4, a single colony was inoculated into LB with spectinomycin, erythromycin and kanamycin, and grown to mid-log phase at 37°C, at which time they were diluted to OD600=0.05 in LB supplemented with the indicated amounts of IPTG and xylose. Cells were harvested at OD600=1, at which point 1.5 OD600 units of cells were centrifuged for use in immunoblots. Blots were then performed as above, with the following exceptions: secondary antibody was goat anti-rabbit-HRP at a 1:2000 dilution, and antibody binding and washing steps were performed in 5% milk in PBS-T (1x phosphate-buffered saline (150 mM NaCl) with 0.02% Tween-20).
Plasmid construction for protein expression
A derivative of pET28a, pET28aPB, encoding a PreScission protease cleavage site, was used for the overexpression of MutS, MutL, β clamp, DnaE, and SSB. Following protease cleavage, amino acids GPGS remain at the N-terminus of each protein as described (Schwartz et al., 1999a; Schwartz et al., 1999b; Schwartz et al., 1999c).
pBW18 was constructed by placing ssb (also known as ssbA) into plasmid pET28aPB by ligation following digestion with BamHI and XhoI. The primers used to amplify the 516 bp of ssb are as follows: forward, 5′- cgc gga tcc atg ctt aac cga gtt gta tta gtc gga aga; reverse 5′- ccg ctc gag cta gaa tgg aag atc atc atc cga gat gtc aat.
pBW1 was constructed by placing dnaN into plasmid pET28aPB by ligation following digestion with BamHI and XhoI. The primers used to amplify the 1134 bp of dnaN are as follows: forward, 5′- cgc gga tcc atg aaa ttc acg att caa aaa gat cgt ctt; reverse, 5′- ccg ctc gag cta ata ggt tct gac agg aag gat aag ctg tac.
pBW25 was constructed by placing dnaE into plasmid pET28aPB by ligation following digestion with BamHI and XhoI. The primers used to amplify the 3345 bp of dnaE are as follows: forward, 5′- cgc gga tcc atg tct ttt gtt cac ctg caa gtg cat agc; reverse, 5′- ccg ctc gag cta cca ctg ttt taa aac gac gtt ttt ttg acc.
pLS126 was generated for overexpression of mutL. The mutL gene was PCR amplified followed by digestion with BamHI and XhoI and ligation into pET28aPB. The following primers were used for amplification of the mutL gene: forward, 5′- cgc gga tcc gtg gca aaa gtc atc caa ctg tca gat gag; reverse, 5′- ccg ctc gag tta cat cac gcg ttt gaa cat ctt ttc cat ctc ata.
pLS128 was constructed for overexpression of mutS. The mutS gene was PCR amplified followed by digestion with BamHI and XhoI and ligation into plasmid pET28aPB following digestion of pET28aPB with the same restriction enzymes. The following primers were used for amplification of the mutS gene: forward 5′ cgc gga tcc atg gcc ggt tat acg cct atg ata cag caa; reverse 5′ ccg ctc gag tta atg taa ttt ctt ttg cag ctt gta cat ttc.
pJS33 was constructed for the purpose of inserting mutL under control of the Pspac at the lacA locus via double crossover integration. The mutL coding region was PCR amplified and digested with SphI and SalI and inserted between the same sites of pA-spac (Hartl et al., 2001). The resulting plasmid, pJS33, was then used to transform B. subtilis and followed by verification of double crossover integration by diagnostic PCR amplification of the lacA locus (data not shown). Primers used to amplify mutL were as follows: forward, 5′ - gtg gtc gac taa gga ggt ata cat gtg gca aaa gtc atc caa c; reverse, 5′ - gtg gca tgc tta tta cat cac gcg ttt gaa cat c.
All constructs were sequenced prior to use (University of Michigan core sequencing facility).
Protein purification
Following cloning of MutS, MutL, DnaE, and β clamp into pET28aPB (fusing an N-terminal His6 tag), these proteins were overexpressed in E. coli BL21DE3 cells using standard procedures. Cell pellets containing overexpressed proteins were resuspended in cell lysis buffer (50 mM Tris HCl [pH 7.6], 10% sucrose, 200 mM NaCl, 20 mM SpCl3) and lysed by french press at 4°C. The lysates were then clarified by centrifugation. All subsequent steps were conducted at 4°C. Equilibrated Ni2+-agarose beads (Qiagen, Hilden, Germany) were incubated with the prepared supernatant for one hour and mixed by rotation. The supernatant was drained in a Poly-Prep chromatography column (Bio-Rad, Hercules, CA) washed with Ni2+-wash buffer (Tris-HCl [pH 7.6], 150 mM imidazole, 0.5 M NaCl, and 15% glycerol), and eluted with elution buffer (Tris-HCl [pH 6.0], 400 mM imidazole, 0.5 M NaCl, and 15% glycerol). MutS and β clamp were dialyzed for 16 hours in 20 mM Tris HCl (pH 7.6), 15% glycerol, 60 mM NaCl, and 1 mM DTT. DnaE was dialyzed for 16 hours in 20 mM Tris HCl (pH 7.6), 20% glycerol, 200 mM NaCl, and 1 mM DTT. MutL was dialyzed for 16 hours in 20 mM Tris HCl (pH 7.6), 15% glycerol, 300 mM NaCl, and 1 mM DTT. The N-terminal His-tag was removed with PreScission protease (2 units per 100 μg fusion protein) (GE Healthcare, United Kingdom) during dialysis. Protein solutions were then applied to a Ni2+-agarose column (prepared as stated earlier), and the flow through of proteins lacking the polyhistadine tag was collected. All proteins were quantified using extinction coefficients derived at ExPASy proteomics server (http://expasy.org/) using a 50-bio UV visible spectrophotometer (Varian, Palo Alto, CA) and purity was verified as >95% using SDS-PAGE (data not shown). Proteins were subsequently aliquoted into usable amounts, flash frozen with liquid N2 and stored at −80°C prior to use.
Affinity purification of antibodies
All primary serum was prepared through Covance (Denver, PA), and was raised against the indicated antigen that was purified as described above. To affinity purify the DnaE, MutS, and MutL antiserum further, 100 μg of purified protein (untagged) was electrophoresed per lane on a 6% SDS-PAGE. Proteins were then transferred to nitrocellulose (Whatman, Dassel, Germany) using standard procedures (Simmons et al., 2003). The membrane was stained with PonceauS and the portion representing the targeted protein was excised and blocked in PBS +5% milk for 20 min, and then rinsed with PBS. The membrane was then incubated in 500 μL of antisera for one hour at room temperature and then rinsed with PBS. The targeted antibodies were removed from the membrane with a strip solution (5 mM glycine, 150 mM NaCl [pH 2.4]) and then neutralized with 1M NaPO4 at pH 8.0.
Immunodot blotting
For immunodot blotting, 16 μM protein stocks were diluted 1:2 into immunoblotting binding buffer (50 mM Tris, pH 8.0; 300 mM NaCl; 10% glycerol) five times, giving protein concentration solutions of 8.0 μM, 4.0 μM, 2.0 μM, and 1.0 μM. Protein dilutions (2.5 μL) were then blotted onto nitrocellulose membranes (Whatman), giving the indicated protein amounts (40 pmol to 2.5 pmol). Following blocking of the membrane in 5% Milk + 1X TBST (20 mM Tris-HCl pH 7.6, NaCl 300 mM, 0.1% Tween 20), the indicated prey protein (400 nM final concentration) was incubated with the membrane in 5% milk with binding buffer (50 mM Tris, pH 8.0; 300 mM NaCl; 10% glycerol) overnight at 4°C. Membranes were washed 3X for at least 5 minutes with wash buffer [4 mM KH2PO4, 16 mM Na2HPO4 (pH 7.6), 300 mM NaCl], and subsequently probed with affinity purified 1° antibody (rabbit) (1:2,000) and goat anti-rabbit HRP conjugated secondary antibody (1:2,000) (Pierce) identically to immunoblotting. Control blots in Figure 7A were performed in an identical manner, except that only bait protein was used and the prey protein step was omitted to determine the cross-reactivity of the antiserum with the other proteins used in the assay.
Statistical analysis
Bar charts are presented with error bars representing the 95% confidence interval, and statistical significance was determined using a two-tailed z-test, as described (Utts and Heckard, 2006).
Supplementary Material
Acknowledgments
We thank Drs. Etienne Dervyn, S. Dusko Ehrlich (Génétique Microbienne, Institut National de la Recherche Agronomique, Jouy en Josas, France), and Alan Grossman for strains (Massachusetts Institute of Technology). We thank Drs. Amy Klocko and Eric Nielsen (University of Michigan) for anti-GFP antibodies (Abcam). We thank Drs. Jim Bardwell, Janine Maddock, Robert Bender, Stefan Walter, and Matthew Chapman (University of Michigan) for use of equipment and reagents. We are indebted to Heather Schroeder for help with statistical analysis and we thank Kaleena Crafton for initiating this work. We also thank members of the Simmons lab for comments on the manuscript. This study was supported by start-up funds from the College of Literature, Science & Arts, from the Department of Molecular, Cellular, and Developmental Biology at the University of Michigan. This work was also supported by a grant from the Wendy Will Case Cancer Fund and grant MCB1050948 from the National Science Foundation to L.A.S.
References
- Ahrends R, Kosinski J, Kirsch D, Manelyte L, Giron-Monzon L, Hummerich L, Schulz O, Spengler B, Friedhoff P. Identifying an interaction site between MutH and the C-terminal domain of MutL by crosslinking, affinity purification, chemical coding and mass spectrometry. Nucleic Acids Res. 2006;34:3169–3180. doi: 10.1093/nar/gkl407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkmen MB, Grossman AD. Spatial and temporal organization of the Bacillus subtilis replication cycle. Mol Microbiol. 2006;62:57–71. doi: 10.1111/j.1365-2958.2006.05356.x. [DOI] [PubMed] [Google Scholar]
- Bernard R, Marquis KA, Rudner DZ. Nucleoid occlusion prevents cell division during replication fork arrest in Bacillus subtilis. Mol Microbiol. 2010;78:866–882. doi: 10.1111/j.1365-2958.2010.07369.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruni R, Martin D, Jiricny J. d(GATC) sequences influence Escherichia coli mismatch repair in a distance-dependent manner from positions both upstream and downstream of the mismatch. Nucleic Acids Res. 1988;16:4875–4890. doi: 10.1093/nar/16.11.4875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chopra I, O’Neill AJ, Miller K. The role of mutators in the emergence of antibiotic-resistant bacteria. Drug Resist Updat. 2003;6:137–145. doi: 10.1016/s1368-7646(03)00041-4. [DOI] [PubMed] [Google Scholar]
- Costes A, Lecointe F, McGovern S, Quevillon-Cheruel S, Polard P. The C-terminal domain of the bacterial SSB protein acts as a DNA maintenance hub at active chromosome replication forks. PLoS Genet. 2010;6:e1001238. doi: 10.1371/journal.pgen.1001238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox EC, Degnen GE, Scheppe ML. Mutator gene studies in Escherichia coli: the mutS gene. Genetics. 1972;72:551–567. doi: 10.1093/genetics/72.4.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culligan KM, Meyer-Gauen G, Lyons-Weiler J, Hays JB. Evolutionary origin, diversification and specialization of eukaryotic MutS homolog mismatch repair proteins. Nucleic Acids Res. 2000;28:463–471. doi: 10.1093/nar/28.2.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denamur E, Bonacorsi S, Giraud A, Duriez P, Hilali F, Amorin C, Bingen E, Andremont A, Picard B, Taddei F, Matic I. High frequency of mutator strains among human uropathogenic Escherichia coli isolates. J Bacteriol. 2002;184:605–609. doi: 10.1128/JB.184.2.605-609.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dervyn E, Suski C, Daniel R, Bruand C, Chapuis J, Errington J, Janniere L, Ehrlich SD. Two essential DNA polymerases at the bacterial replication fork. Science. 2001;294:1716–1719. doi: 10.1126/science.1066351. [DOI] [PubMed] [Google Scholar]
- Dronkert ML, Kanaar R. Repair of DNA interstrand cross-links. Mutat Res. 2001;486:217–247. doi: 10.1016/s0921-8777(01)00092-1. [DOI] [PubMed] [Google Scholar]
- Dupes NM, Walsh BW, Klocko AD, Lenhart JS, Peterson HL, Gessert DA, Pavlick CE, Simmons LA. Mutations in the Bacillus subtilis beta clamp that separate its roles in DNA replication from mismatch repair. J Bacteriol. 2010;192:3452–3463. doi: 10.1128/JB.01435-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzantiev L, Constantin N, Genschel J, Iyer RR, Burgers PM, Modrich P. A defined human system that supports bidirectional mismatch-provoked excision. Mol Cell. 2004;15:31–41. doi: 10.1016/j.molcel.2004.06.016. [DOI] [PubMed] [Google Scholar]
- Eisen JA. A phylogenomic study of the MutS family of proteins. Nucleic Acids Res. 1998;26:4291–4300. doi: 10.1093/nar/26.18.4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen JA, Hanawalt PC. A phylogenomic study of DNA repair genes, proteins, and processes. Mutat Res. 1999;435:171–213. doi: 10.1016/s0921-8777(99)00050-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fijalkowska IJ, Jonczyk P, Tkaczyk MM, Bialoskorska M, Schaaper RM. Unequal fidelity of leading strand and lagging strand DNA replication on the Escherichia coli chromosome. Proc Natl Acad Sci U S A. 1998;95:10020–10025. doi: 10.1073/pnas.95.17.10020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishel R, Lescoe MK, Rao MRS, Copeland NG, Jenkins NA, Garber J, Kane M, Kolodner R. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis cancer. Cell. 1993;75:1027–1038. doi: 10.1016/0092-8674(93)90546-3. [DOI] [PubMed] [Google Scholar]
- Flores-Rozas H, Clark D, Kolodner RD. Proliferating cell nuclear antigen and Msh2p-Msh6p interact to form an active mispair recognition complex. Nat Genet. 2000;26:375–378. doi: 10.1038/81708. [DOI] [PubMed] [Google Scholar]
- Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis: Second Edition. Washington, DC: American Society for Microbiology; 2006. pp. 389–433. [Google Scholar]
- Genschel J, Bazemore LR, Modrich P. Human exonuclease I is required for 5′ and 3′ mismatch repair. J Biol Chem. 2002;277:13302–13311. doi: 10.1074/jbc.M111854200. [DOI] [PubMed] [Google Scholar]
- Genschel J, Modrich P. Mechanism of 5′-directed excision in human mismatch repair. Mol Cell. 2003;12:1077–1086. doi: 10.1016/s1097-2765(03)00428-3. [DOI] [PubMed] [Google Scholar]
- Genschel J, Modrich P. Analysis of the excision step in human DNA mismatch repair. Methods Enzymol. 2006;408:273–284. doi: 10.1016/S0076-6879(06)08017-7. [DOI] [PubMed] [Google Scholar]
- Georgescu RE, Kim SS, Yurieva O, Kuriyan J, Kong XP, O’Donnell M. Structure of a sliding clamp on DNA. Cell. 2008;132:43–54. doi: 10.1016/j.cell.2007.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginetti F, Perego M, Albertini AM, Galizzi A. Bacillus subtilis mutS mutL operon: identification, nucleotide sequence and mutagenesis. Microbiology. 1996;142(Pt 8):2021–2029. doi: 10.1099/13500872-142-8-2021. [DOI] [PubMed] [Google Scholar]
- Goodman MF, Hopkins R, Gore WC. 2-Aminopurine-induced mutagenesis in T4 bacteriophage: a model relating mutation frequency to 2-aminopurine incorporation in DNA. Proc Natl Acad Sci U S A. 1977;74:4806–4810. doi: 10.1073/pnas.74.11.4806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grafstrom RH, Amsterdam A, Zachariasewycz K. In vivo studies of repair of 2-aminopurine in Escherichia coli. J Bacteriol. 1988;170:3485–3492. doi: 10.1128/jb.170.8.3485-3492.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall MC, Jordan JR, Matson SW. Evidence for a physical interaction between the Escherichia coli methyl-directed mismatch repair proteins MutL and UvrD. Embo J. 1998;17:1535–1541. doi: 10.1093/emboj/17.5.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall MC, Matson SW. The Escherichia coli MutL protein physically interacts with MutH and stimulates the MutH-associated endonuclease activity. J Biol Chem. 1999;274:1306–1312. doi: 10.1074/jbc.274.3.1306. [DOI] [PubMed] [Google Scholar]
- Hamilton SR, Liu B, Parsons RE, Papadopoulos N, Jen J, Powell SM, Krush AJ, Berk T, Cohen Z, Tetu B, et al. The molecular basis of Turcot’s syndrome. N Engl J Med. 1995;332:839–847. doi: 10.1056/NEJM199503303321302. [DOI] [PubMed] [Google Scholar]
- Hardwood CR, Cutting SM. Molecular Biological Methods for Bacillus. Chichester: John Wiley & Sons; 1990. pp. 27–446. [Google Scholar]
- Hartl B, Wehrl W, Wiegert T, Homuth G, Schumann W. Development of a new integration site within the Bacillus subtilis chromosome and construction of compatible expression cassettes. J Bacteriol. 2001;183:2696–2699. doi: 10.1128/JB.183.8.2696-2699.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman MJ, Samson LD. Role of DNA mismatch repair and p53 in signaling induction of apoptosis by alkylating agents. Proc Natl Acad Sci U S A. 1999;96:10764–10769. doi: 10.1073/pnas.96.19.10764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman MJ, Samson LD. Apoptotic signaling in response to a single type of DNA lesion, O(6)-methylguanine. Mol Cell. 2004;14:105–116. doi: 10.1016/s1097-2765(04)00162-5. [DOI] [PubMed] [Google Scholar]
- Holmes J, Jr, Clark S, Modrich P. Strand-specific mismatch correction in nuclear extracts of human and Drosophila melanogaster cell lines. Proc Natl Acad Sci U S A. 1990;87:5837–5841. doi: 10.1073/pnas.87.15.5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh P, Yamane K. DNA mismatch repair: molecular mechanism, cancer, and ageing. Mech Ageing Dev. 2008;129:391–407. doi: 10.1016/j.mad.2008.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer RR, Pohlhaus TJ, Chen S, Hura GL, Dzantiev L, Beese LS, Modrich P. The MutSalpha-proliferating cell nuclear antigen interaction in human DNA mismatch repair. J Biol Chem. 2008;283:13310–13319. doi: 10.1074/jbc.M800606200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeruzalmi D, O’Donnell M, Kuriyan J. Crystal structure of the processivity clamp loader gamma (gamma) complex of E. coli DNA polymerase III. Cell. 2001;106:429–441. doi: 10.1016/s0092-8674(01)00463-9. [DOI] [PubMed] [Google Scholar]
- Johnson A, O’Donnell M. Cellular DNA replicases: components and dynamics at the replication fork. Annu Rev Biochem. 2005;74:283–315. doi: 10.1146/annurev.biochem.73.011303.073859. [DOI] [PubMed] [Google Scholar]
- Kadyrov FA, Dzantiev L, Constantin N, Modrich P. Endonucleolytic function of MutLalpha in human mismatch repair. Cell. 2006;126:297–308. doi: 10.1016/j.cell.2006.05.039. [DOI] [PubMed] [Google Scholar]
- Kadyrov FA, Holmes SF, Arana ME, Lukianova OA, O’Donnell M, Kunkel TA, Modrich P. Saccharomyces cerevisiae MutLalpha is a mismatch repair endonuclease. J Biol Chem. 2007;282:37181–37190. doi: 10.1074/jbc.M707617200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleczkowska HE, Marra G, Lettieri T, Jiricny J. hMSH3 and hMSH6 interact with PCNA and colocalize with it to replication foci. Genes Dev. 2001;15:724–736. doi: 10.1101/gad.191201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klocko AD, Crafton KM, Walsh BW, Lenhart JS, Simmons LA. Imaging mismatch repair and cellular responses to DNA damage in. Bacillus subtilis J Vis Exp. 2010:1–4. doi: 10.3791/1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel TA, Erie DA. DNA mismatch repair. Annu Rev Biochem. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- Lacks SA, Dunn JJ, Greenberg B. Identification of base mismatches recognized by the heteroduplex-DNA-repair system of Streptococcus pneumoniae. Cell. 1982;31:327–336. doi: 10.1016/0092-8674(82)90126-x. [DOI] [PubMed] [Google Scholar]
- Lahue RS, Au KG, Modrich P. DNA mismatch correction in a defined system. Science. 1989;245:160–164. doi: 10.1126/science.2665076. [DOI] [PubMed] [Google Scholar]
- Larrea AA, Lujan SA, Kunkel TA. SnapShot: DNA mismatch repair. Cell. 2010;141:730 e731. doi: 10.1016/j.cell.2010.05.002. [DOI] [PubMed] [Google Scholar]
- Lee SD, Alani E. Analysis of interactions between mismatch repair initiation factors and the replication processivity factor PCNA. J Mol Biol. 2006;355:175–184. doi: 10.1016/j.jmb.2005.10.059. [DOI] [PubMed] [Google Scholar]
- Lemon KP, Grossman AD. Localization of bacterial DNA polymerase: evidence for a factory model of replication. Science. 1998;282:1516–1519. doi: 10.1126/science.282.5393.1516. [DOI] [PubMed] [Google Scholar]
- Li F, Liu Q, Chen YY, Yu ZN, Zhang ZP, Zhou YF, Deng JY, Bi LJ, Zhang XE. Escherichia coli mismatch repair protein MutL interacts with the clamp loader subunits of DNA polymerase III. Mutat Res. 2008;637:101–110. doi: 10.1016/j.mrfmmm.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Li GM. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008;18:85–98. doi: 10.1038/cr.2007.115. [DOI] [PubMed] [Google Scholar]
- Lopez de Saro F, Georgescu RE, Leu F, O’Donnell M. Protein trafficking on sliding clamps. Philos Trans R Soc Lond B Biol Sci. 2004;359:25–30. doi: 10.1098/rstb.2003.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez de Saro FJ, O’Donnell M. Interaction of the beta sliding clamp with MutS, ligase, and DNA polymerase I. Proc Natl Acad Sci USA. 2001;98:8376–8380. doi: 10.1073/pnas.121009498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez de Saro FJ, Marinus MG, Modrich P, O’Donnell M. The beta sliding clamp binds to multiple sites within MutL and MutS. J Biol Chem. 2006;281:14340–14349. doi: 10.1074/jbc.M601264200. [DOI] [PubMed] [Google Scholar]
- Mechanic LE, Frankel BA, Matson SW. Escherichia coli MutL loads DNA helicase II onto DNA. J Biol Chem. 2000;275:38337–38346. doi: 10.1074/jbc.M006268200. [DOI] [PubMed] [Google Scholar]
- Migocki MD, Lewis PJ, Wake RG, Harry EJ. The midcell replication factory in Bacillus subtilis is highly mobile: implications for coordinating chromosome replication with other cell cycle events. Mol Microbiol. 2004;54:452–463. doi: 10.1111/j.1365-2958.2004.04267.x. [DOI] [PubMed] [Google Scholar]
- Modrich P. Mechanisms in eukaryotic mismatch repair. J Biol Chem. 2006;281:30305–30309. doi: 10.1074/jbc.R600022200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nystrom-Lahti M, Perrera C, Raschle M, Panyushkina-Seiler E, Marra G, Curci A, Quaresima B, Costanzo F, D’Urso M, Venuta S, Jiricny J. Functional analysis of MLH1 mutations linked to hereditary nonpolyposis colon cancer. Genes Chromosomes Cancer. 2002;33:160–167. [PubMed] [Google Scholar]
- Paul BJ, Ross W, Gaal T, Gourse RL. rRNA transcription in Escherichia coli. Annu Rev Genet. 2004;38:749–770. doi: 10.1146/annurev.genet.38.072902.091347. [DOI] [PubMed] [Google Scholar]
- Pavlov YI, Mian IM, Kunkel TA. Evidence for preferential mismatch repair of lagging strand DNA replication errors in yeast. Curr Biol. 2003;13:744–748. doi: 10.1016/s0960-9822(03)00284-7. [DOI] [PubMed] [Google Scholar]
- Peltomaki P. Lynch syndrome genes. Fam Cancer. 2005;4:227–232. doi: 10.1007/s10689-004-7993-0. [DOI] [PubMed] [Google Scholar]
- Pillon MC, Lorenowicz JJ, Uckelmann M, Klocko AD, Mitchell RR, Chung YS, Modrich P, Walker GC, Simmons LA, Friedhoff P, Guarne A. Structure of the endonuclease domain of MutL: unlicensed to cut. Mol Cell. 2010;39:145–151. doi: 10.1016/j.molcel.2010.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillon MC, Miller JH, Guarne A. The endonuclease domain of MutL interacts with the beta sliding clamp. DNA Repair (Amst) 2011;10:87–93. doi: 10.1016/j.dnarep.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluciennik A, Burdett V, Lukianova O, O’Donnell M, Modrich P. Involvement of the beta clamp in methyl-directed mismatch repair in vitro. J Biol Chem. 2009;284:32782–32791. doi: 10.1074/jbc.M109.054528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluciennik A, Dzantiev L, Iyer RR, Constantin N, Kadyrov FA, Modrich P. PCNA function in the activation and strand direction of MutLalpha endonuclease in mismatch repair. Proc Natl Acad Sci U S A. 2010;107:16066–16071. doi: 10.1073/pnas.1010662107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayssiguier C, Thaler DS, Radman M. The barrier to recombination between Escherichia coli and Salmonella typhimurium is disrupted in mismatch-repair mutants. Nature. 1989;342:396–401. doi: 10.1038/342396a0. [DOI] [PubMed] [Google Scholar]
- Rokop ME, Auchtung JM, Grossman AD. Control of DNA replication initiation by recruitment of an essential initiation protein to the membrane of Bacillus subtilis. Mol Microbiol. 2004;52:1757–1767. doi: 10.1111/j.1365-2958.2004.04091.x. [DOI] [PubMed] [Google Scholar]
- Sanders GM, Dallmann HG, McHenry CS. Reconstitution of the B. subtilis replisome with 13 proteins including two distinct replicases. Mol Cell. 2010;37:273–281. doi: 10.1016/j.molcel.2009.12.025. [DOI] [PubMed] [Google Scholar]
- Sanjanwala B, Ganesan AT. Genetic structure and domains of DNA polymerase III of Bacillus subtilis. Mol Gen Genet. 1991;226:467–472. doi: 10.1007/BF00260660. [DOI] [PubMed] [Google Scholar]
- Schofield MJ, Nayak S, Scott TH, Du C, Hsieh P. Interaction of Escherichia coli MutS and MutL at a DNA mismatch. J Biol Chem. 2001;276:28291–28299. doi: 10.1074/jbc.M103148200. [DOI] [PubMed] [Google Scholar]
- Schofield MJ, Hsieh P. DNA mismatch repair: molecular mechanisms and biological function. Annu Rev Microbiol. 2003;57:579–608. doi: 10.1146/annurev.micro.57.030502.090847. [DOI] [PubMed] [Google Scholar]
- Schwartz T, Lowenhaupt K, Kim YG, Li L, Brown BA, 2nd, Herbert A, Rich A. Proteolytic dissection of Zab, the Z-DNA-binding domain of human ADAR1. J Biol Chem. 1999a;274:2899–2906. doi: 10.1074/jbc.274.5.2899. [DOI] [PubMed] [Google Scholar]
- Schwartz T, Rould MA, Lowenhaupt K, Herbert A, Rich A. Crystal structure of the Zalpha domain of the human editing enzyme ADAR1 bound to left-handed Z-DNA. Science. 1999b;284:1841–1845. doi: 10.1126/science.284.5421.1841. [DOI] [PubMed] [Google Scholar]
- Schwartz T, Shafer K, Lowenhaupt K, Hanlon E, Herbert A, Rich A. Crystallization and preliminary studies of the DNA-binding domain Za from ADAR1 complexed to left-handed DNA. Acta Crystallogr D Biol Crystallogr. 1999c;55:1362–1364. doi: 10.1107/s090744499900582x. [DOI] [PubMed] [Google Scholar]
- Simmons LA, Felczak M, Kaguni JM. DnaA Protein of Escherichia coli: oligomerization at the E. coli chromosomal origin is required for initiation and involves specific N-terminal amino acids. Mol Microbiol. 2003;49:849–858. doi: 10.1046/j.1365-2958.2003.03603.x. [DOI] [PubMed] [Google Scholar]
- Simmons LA, Grossman AD, Walker GC. Replication is required for the RecA localization response to DNA damage in Bacillus subtilis. Proc Natl Acad Sci U S A. 2007;104:1360–1365. doi: 10.1073/pnas.0607123104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons LA, Davies BW, Grossman AD, Walker GC. Beta clamp directs localization of mismatch repair in Bacillus subtilis. Mol Cell. 2008a;29:291–301. doi: 10.1016/j.molcel.2007.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons LA, Grossman AD, Walker GC. Clp and Lon proteases occupy distinct subcellular positions in Bacillus subtilis. J Bacteriol. 2008b;190:6758–6768. doi: 10.1128/JB.00590-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons LA, Goranov AI, Kobayashi H, Davies BW, Yuan DS, Grossman AD, Walker GC. Comparison of responses to double-strand breaks between Escherichia coli and Bacillus subtilis reveals different requirements for SOS induction. J Bacteriol. 2009;191:1152–1161. doi: 10.1128/JB.01292-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith BT, Grossman AD, Walker GC. Visualization of mismatch repair in bacterial cells. Mol Cell. 2001;8:1197–1206. doi: 10.1016/s1097-2765(01)00402-6. [DOI] [PubMed] [Google Scholar]
- Srivatsan A, Wang JD. Control of bacterial transcription, translation and replication by (p)ppGpp. Curr Opin Microbiol. 2008;11:100–105. doi: 10.1016/j.mib.2008.02.001. [DOI] [PubMed] [Google Scholar]
- Sutton MD. Coordinating DNA polymerase traffic during high and low fidelity synthesis. Biochim Biophys Acta. 2009;1804:1167–1179. doi: 10.1016/j.bbapap.2009.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utts JM, Heckard RF. Mind on Statistics. Duxbury Press; 2006. [Google Scholar]
- Viswanathan M, Burdett V, Baitinger C, Modrich P, Lovett ST. Redundant exonuclease involvement in Escherichia coli methyl-directed mismatch repair. J Biol Chem. 2001;276:31053–31058. doi: 10.1074/jbc.M105481200. [DOI] [PubMed] [Google Scholar]
- Wang JD, Sanders GM, Grossman AD. Nutritional control of elongation of DNA replication by (p)ppGpp. Cell. 2007;128:865–875. doi: 10.1016/j.cell.2006.12.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe SM, Goodman MF. On the molecular basis of transition mutations: frequencies of forming 2-aminopurine.cytosine and adenine.cytosine base mispairs in vitro. Proc Natl Acad Sci U S A. 1981;78:2864–2868. doi: 10.1073/pnas.78.5.2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshioka K, Yoshioka Y, Hsieh P. ATR kinase activation mediated by MutSalpha and MutLalpha in response to cytotoxic O6-methylguanine adducts. Mol Cell. 2006;22:501–510. doi: 10.1016/j.molcel.2006.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.