

Abstract
An extra-adrenal paraganglioma is a rare tumour derived from chromaffin cells of sympathetic ganglia. This report documents a rare case of a non-functional aortocaval paraganglioma in a 24-year-old woman with persistent abdominal pain. Computed tomography revealed a solid mass, measuring 2.5x3cms, localized between the celiac trunk and superior mesenteric artery in aortocaval location along with right ovarian cystic mass. A clinical diagnosis of malignant ovarian tumour with celiac nodal metastasis was made. Excision and pathological analysis of both revealed an aortocaval extra-adrenal paraganglioma and benign ovarian cyst. On serial follow-up the patient was in a good health, asymptomatic and without evidence of tumour recurrence. This case emphasizes the necessity of including extra-adrenal paraganglioma in the differential diagnosis and management of retroperitoneal tumours, despite its rarity.
Keywords: Aorta, venacava, lymph node, paraganglioma, surgery
Özet
Ekstra-adrenal paraganglioma sempatik ganglionların kromafin hücrelerinden gelişmiş nadir bir tümördür. Bu rapor; 24 yaşındaki sürekli karın ağrıları olan, nonfonksiyonel aortokaval bulunan nadir bir olgunun sunumudur. Tomografisinde sağ over kistik kitle ile birlikte aortokaval konumda çölyak gövde ve superior mezenterik arter arasında lokalize 2.5x3 cm boyutlarında ölçülen, solid bir kitle saptandı. Çölyak nodal metastazı olan malign over tümörün klinik tanısı konulmuştu. Eksizyon ve patolojik incelemenin her ikisi aortokaval ekstra-adrenal paraganglioma ve benign over kistinin olduğunu ortaya koydu. Seri takiple hasta asemptomatik ve tümör nüksü olmadan sağlığına kavuştu. Bu olgu, nadir olmasına rağmen, retroperitoneal tümörlerin ayırıcı tanısında ve tedavisinde ekstra-adrenal paragangliomanın dahil edilme gerekliliğini vurgulamaktadır.
Introduction
Paraganglioma is a rare chromaffin cell tumour that develops from the neural crest cells of the neuroendocrine system [1]. Retroperitoneal paragangliomas can represent a surgical challenge due to their close relation to large vessels. With the advances in imaging technology ultrasound, contrast enhanced computed tomography (CECT) and magnetic resonance imaging (MRI) may be useful in localizing the tumour, while I131-methyliodobenzylguanidine (I131-MIBG) and positron emission tomography (PET)-CT help to evaluate its function [2]. Clinical and histological distinction between benign and malignant cases is difficult. Complete surgical resection (either open or laparoscopic) is the treatment of choice for paragangliomas [3].
Case Report
A 24-year-old woman was evaluated for persistent abdominal pain of seven months duration. A detailed clinical examination was unremarkable. A CECT abdomen was performed, which revealed a solitary, necrotic, space occupying peripherally enhancing aortocaval cystic lesion 2.5x3 cms at L2 level between celiac trunk and superior mesenteric vessels in the preaortic region displacing fourth part of duodenum ventrally with no invasion of aorta or inferior vena cava (Figure 1). Multiple ovarian cysts were also present (Figure 2). A CT guided fine needle aspiration cytology (FNAC) of the aortocaval mass was done and was doubtful of a secondary deposit from an ovarian dysgerminoma. Complete excision of the aortocaval lesion (Figure 3) and right oophorectomy (Figures 3, 4) were executed. The final histopathology report revealed a well circumscribed encapsulated tumour composed of polygonal cells arranged in organoid pattern, nests and zellballen pattern with round to oval vesicular nuclei, moderate eosinophilic to clear cytoplasm with interspersed delicate branching vascular channels and fibrous septae. Areas of necrosis with ghost outlines of tumour cells with no significant mitosis were noted (Figure 5). Immunohistochemistry performed proved this to be an extra-adrenal paraganglioma. The ovarian specimen was reported to be a serous cystadenoma. The patient has been kept under close follow-up and has done well.
Figure 1.
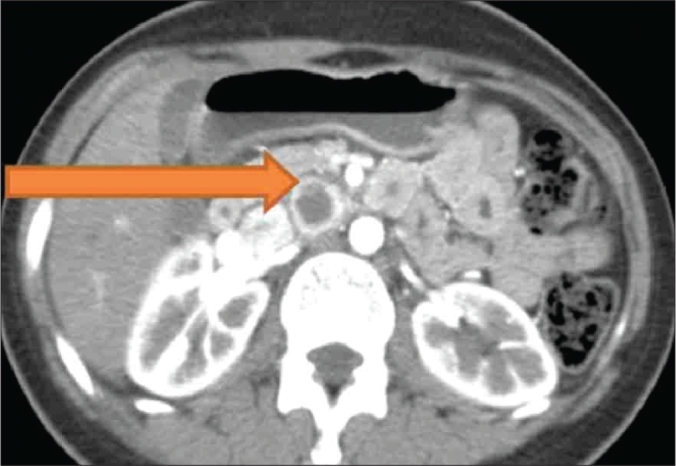
CECT showing a solitary, necrotic, space occupying peripherally enhancing aortocaval cystic lesion.
Figure 2.
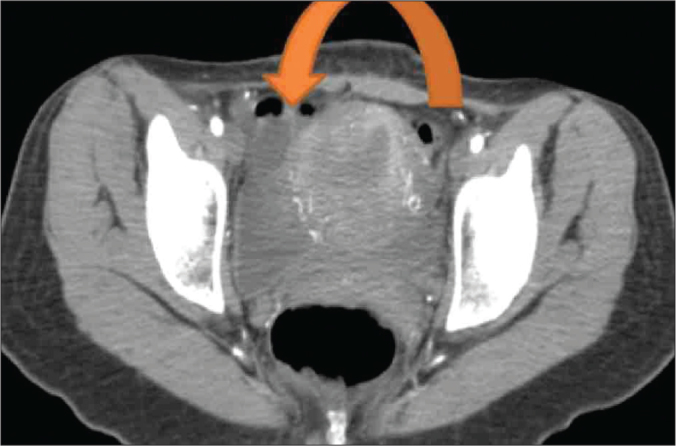
CECT showing a right ovarian cyst.
Figure 3.
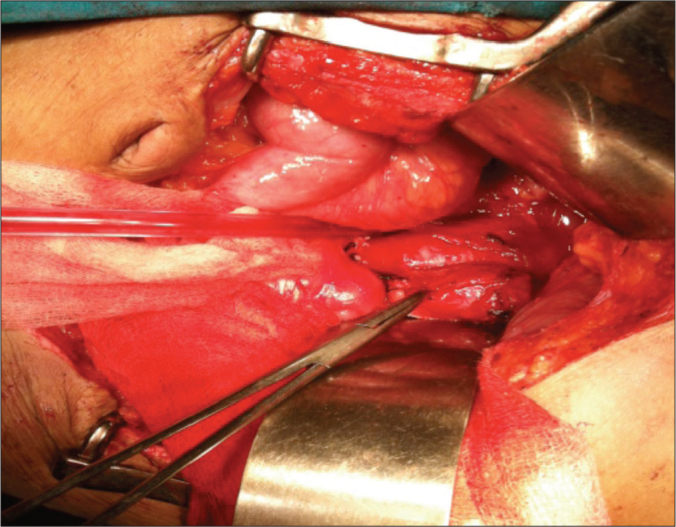
Intraoperative picture showing an aortocaval tumour.
Figure 4.
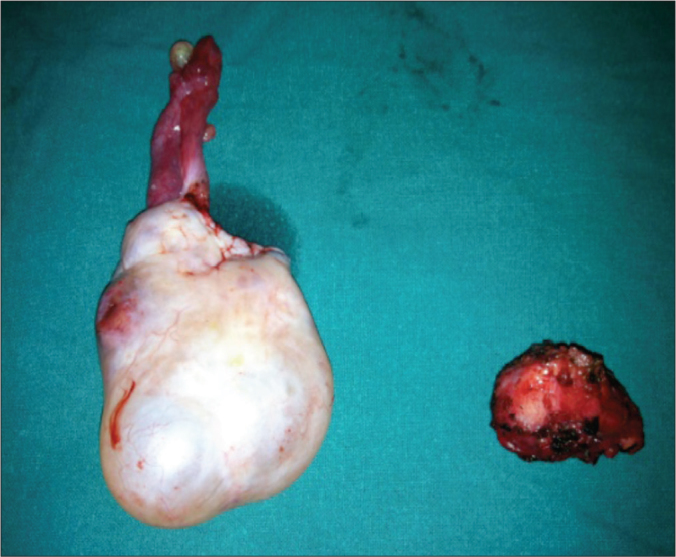
Ovarian mass with excised aortocaval tumour.
Figure 5.
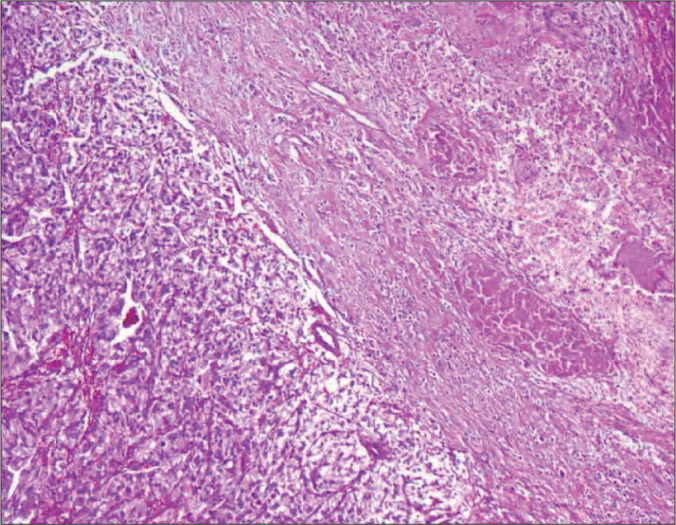
Photomicrograph showing a tumour composed of polygonal cells arranged in an organoid pattern, nests and zellballen pattern with round to oval vesicular nuclei, moderate eosinophilic to clear cytoplasm with interspersed delicate branching vascular channels and fibrous septae.
Discussion
Paragangliomas are tumours of the neural crest-derived paraganglia that exist throughout the body along the distribution of the sympathetic and parasympathetic nervous system [1]. Approximately 20% are malignant and surgical resection is considered the primary treatment when possible. The optimal systemic treatment for advanced disease is undefined, due in part to the lack of effective chemotherapeutic agents [3].
The current World Health Organization (WHO) classification reserves the term pheochromocytoma for intra-adrenal tumours, whereas tumours arising in extra-adrenal paraganglia are classified as paragangliomas [4]. These tumours can either be sporadic or occur in the context of hereditary cancer syndromes such as VHL disease, multiple endocrine neoplasia type 2 (MEN2), neurofibromatosis type 1 or germline succinate dehydrogenase complex subunit B (SDHB), succinate dehydrogenase complex subunit C (SDHC), or succinate dehydrogenase complex subunit D (SDHD) [2–5].
Extra-adrenal paraganglia are divided into two categories: those related to the parasympathetic system and those connected with the orthosympathetic system. The former, usually nonchromaffin, lie in the head and neck including the carotid body, intravagal, jugulotympanic, mediastinal and aorticopulmonary paraganglia [6]. The latter are chromaffin, associated with the peripheral sympathetic nervous system, and secrete catecholamines in response to sympathetic neural stimulation. They lie in the para-axial region of the trunk close to the para and prevertebral ganglia or in the connective tissue adjacent to pelvic organs, predominating along the thoracolumbar para-aortic region in the retroperitoneum [7, 8]. Extra-adrenal paragangliomas of the sympathoadrenal neuroendocrine system consist 5–15% of sporadic pheochromocytomas and are located anywhere extending from the upper neck to the pelvic floor, parallel to the autonomic nervous system [6, 7]. Extra-adrenal, abdominal paragangliomas are divided into three groups: superior para-aortic, inferior para-aortic, and urinary bladder tumours [7]. Rarely, extra-adrenal paragangliomas can occur aberrantly outside this distribution.
Histologically, paraganglia contain nests of neuroendocrine cells surrounded by sustenacular cells. This pattern is most clearly seen in parasympathetic paraganglia as the characteristic ‘zellballen’. In sympathetic paraganglia, neuroendocrine cells are referred to as chromaffin cells and in parasympathetic as chief cells. These cells are immunopositive for neuroendocrine markers including synaptophysin and chromogranin A and for several neuropeptides. Features that may predict malignancy include extra-adrenal location, confluent tumour necrosis, vascular invasion, local invasion, coarse nodularity, and absence of hyaline globules [8]. Moreover, decreased expression of neuropeptides and, particularly, negative staining for enkephalins, somatostatin, pancreatic polypeptide, and Vasoactive intestinal poly-peptide (VIP) have been associated with malignancy [9]. Although the presented tumour was extra-adrenal, the presence of hyaline globules and the absence of confluent tumour necrosis, vascular invasion, local invasion, and coarse nodularity along with the positive staining for the above-mentioned polypeptides show the benign nature of this lesion.
Clinical and histological distinction between benign and malignant cases is difficult. The only proof of malignancy is the presence of metastases or local invasion, while in localized tumours there are no absolute criteria for predicting malignant potential. This principle has the drawback that some cases will initially be classified as benign and then reclassified as malignant if metastases are identified at follow-up [8, 9]. There is, therefore, a clear need for additional diagnostic tools to enable detection of malignant cases at initial surgery. Additional diagnostic markers would be valuable, but till to-date, no reliable prognostic markers have been identified [9].
The treatment of choice for paragangliomas is complete surgical resection. Traditional treatment consists of open exploration and resection. However, laparoscopic management is nowadays increasingly being used for their management due to the advances in laparoscopic technique and refined preoperative imaging even though few studies regarding the laparoscopic treatment of extra-adrenal intra-abdominal paragangliomas exist in the literature because of the rarity of this clinical entity [3]. Intraoperative ultrasound is considered an invaluable tool for laparoscopic surgeons in these cases. In these studies of laparoscopic management, encouraging results are presented; it should be mentioned, however, that all tumours were less than 4 cm [9].
Footnotes
Conflict of Interest: No conflict of interest was declared by the authors.
Peer-review: Externally peer-reviewed.
Informed Consent: Written informed consent was obtained from patients who participated in this case.
Author Contributions: Concept - G.R., N.G.; Design - G.R., N.G.; Supervision - G.R., G.K.; Materials - G.R., N.G.; Data Collection and/or Processing - G.R., R.P.; Analysis and/or Interpretation - G.R., L.R.; Literature Review - G.R., N.G.; WriterN.G., G.R.; Critical Review - L.R., G.K.
Financial Disclosure: The authors declared that this study has received no financial support.
References
- 1.Noguchi K, Okada K, Kawamura H, Ishizu H, Honma S. Nonfunctional retroperitoneal paraganglioma simulating pancreatic tumor: report of a case. Am Surg. 2012;78:E244–5. [PubMed] [Google Scholar]
- 2.Manabe O, Oyama-Manabe N, Alisa K, et al. Multimodality evaluation of cardiac paraganglioma. Clin Nucl Med. 2012;37:599–601. doi: 10.1097/RLU.0b013e3182485204. [DOI] [PubMed] [Google Scholar]
- 3.Disick GI, Palese MA. Extra-adrenal pheochromocytoma: diagnosis and management. Curr Urol Rep. 2007;8:83–8. doi: 10.1007/s11934-007-0025-5. [DOI] [PubMed] [Google Scholar]
- 4.Tischler AS, Kimura N, Mcnicol AM. Pathology of pheochromocytoma and extra-adrenal paraganglioma. Ann N Y Acad Sci. 2006;1073:557–70. doi: 10.1196/annals.1353.059. [DOI] [PubMed] [Google Scholar]
- 5.Astuti D, Douglas F, Lennard TW, et al. Germline SDHD mutation in familial phaeochromocytoma. Lancet. 2001;357:1181–2. doi: 10.1016/S0140-6736(00)04378-6. [DOI] [PubMed] [Google Scholar]
- 6.Lack EE, Cubilla AL, Woodruff JM. Paragangliomas of the head and neck region. A pathologic study of tumors from 71 patients. Hum Pathol. 1979;10:191–218. doi: 10.1016/s0046-8177(79)80008-8. [DOI] [PubMed] [Google Scholar]
- 7.Lack EE, Cubilla AL, Woodruff JM, Lieberman PH. Extra-adrenal paragangliomas of the retroperitoneum: a clinicopathologic study of 12 tumors. Am J Surg Pathol. 1980;4:109–20. doi: 10.1097/00000478-198004000-00002. [DOI] [PubMed] [Google Scholar]
- 8.Davidovic LB, Djukic VB, Vasic DM, Sindjelic RP, Duvnjak SN. Diagnosis and treatment of carotid body paraganglioma: 21 years of experience at a clinical center of Serbia. World J Surg Oncol. 2005;3:10. doi: 10.1186/1477-7819-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parenti G, Zampetti B, Rapizzi E, Ercolino T, Giachè V, Mannelli M. Updated and new perspectives on diagnosis, prognosis, and therapy of malignant pheochromocytoma/paraganglioma. J Oncol. 2012;2012:872713. doi: 10.1155/2012/872713. [DOI] [PMC free article] [PubMed] [Google Scholar]