
Fig. 1. RAC-independent AKT-induced ROS caused oxidative DNA damage resulting in accumulation of imatinib-resistant clones.
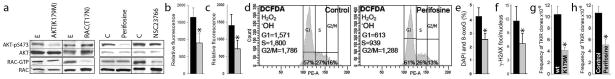
(a) BCR-ABL1-transformed 32Dcl3 cells transfected with AKT(K179M) and Rac(T17N) dominant-negative mutants or empty plasmids (E) 8,10, and Lin−CD34+ CML-CP cells treated with 10 μM AKT inhibitor perifosine, 25 μM NSC23766 or diluent (C) 8,11 were tested for activation of AKT and RAC. Western analyses detect AKT phosphorylated on serine 473 (AKT-pS473) and Rac bound to GTP as described before 8,15; total levels of AKT and RAC were also determined as loading controls. (b) ROS were measured with DCFDA in BCR-ABL1 -32Dcl3 cells transfected with empty plasmid (black bar) and AKT(K179M) mutant (grey bar). (c–f) Lin−CD34+ CML-CP cells were left untreated (black bars) or incubated with 10 μM perifosine (grey bars) in the presence of growth factors.8 (c) ROS were measured with DCFDA in annexin V-negative cells as described before 5,8. (d) ROS were detected by DCFDA in G1, S and G2/M phase determined by Vybrant DyeCycle Orange live cell staining (Invitrogen/Molecular Probes) as described before. 8 ROS measurements are at the left sides, and percentages of cells in cell cycle phases are indicated at the bottom. (e) 8-oxoG and (f) γ-H2AX detected by specific immunofluorescence as described before.8 (g, h) BCR-ABL1 –positive 32Dcl3 cells transfected with AKT(K179M) mutant or empty plasmid (g) and untreated (Control) or treated with 1 μM perifosine (h) were cultured for 10 weeks. The frequency of TKI resistant (TKIR) clones was determined as described before.8 *p<0.05.