

A full response to heat shock depends on the duration of HSF1 activation, which is controlled by the deacetylase HDAC6, known to bind ubiquitin residues, and by AAA ATPase p97/VCP. This new regulatory process relies on the extent of protein ubiquitination and directs the ability of cells to remain protected against heat-dependent apoptosis.
Abstract
After heat shock, HSF1 controls a major cellular transcriptional response involving the activation of early (HSP70) and late (HSP25) heat shock gene expression. Here we show that a full response to heat shock (activation of both HSP70 and HSP25) depends on the duration of HSF1 activation, which is itself controlled by HDAC6, a unique deacetylase known to bind monoubiquitin and polyubiquitin with high affinity. On the basis of a comparative analysis of the heat shock response in cells knocked out for HDAC6 or expressing HDAC6 mutants, we show that HDAC6 binding to ubiquitinated proteins controls the duration of HSF1 activation after heat shock. In cells expressing HDAC6 mutated in the ubiquitin-binding domain, the AAA ATPase factor p97/VCP mediates rapid inactivation of HSF1, precluding late activation of the HSP25 gene. In these cells, knockdown of p97/VCP rescues HSF1 from this rapid inactivation and restores HSP25 expression. We present here a new regulatory circuit that adjusts the duration of the heat shock response to the extent of protein ubiquitination after heat shock.
INTRODUCTION
Heat shock proteins (HSPs) prevent newly synthesized polypeptide chains and newly assembled subunits from aggregating into nonfunctional structures (Tyedmers et al., 2010; Ciocca et al., 2013). HSPs are therefore essential in the prevention of aggregate formation under stressing conditions such as proteasomal inhibition and heat shock (HS; Voellmy and Boellmann, 2007).
HSP expression is controlled by a specific transcription factor called heat-shock factor 1 (HSF1; Akerfelt et al., 2010). In unstressed cells, inactive HSF1 is present in both nuclear and cytoplasmic compartments and forms a hetero-oligomeric complex involving chaperones and other partners. On stress, HSF1 dissociates from its repressive interactors, undergoes several posttranslational modifications including hyperphosphorylation, trimerizes, and acquires its DNA-binding and transactivation competence (Cotto and Morimoto, 1999).
Of interest, we have identified histone deacetylase 6 (HDAC6) as a major player in the control of HSF1 activation upon proteasome inhibition (Boyault, Zhang, Fritah, et al., 2007). Compared to other HDACs, HDAC6 displays at least two specific features. In its N-terminal region, it contains two deacetylase domains with essential roles in deacetylating a variety of cytoplasmic substrates, including tubulin, and in its C-terminal region, it also contains a zinc finger ubiquitin domain (ZnF-UBP), capable of binding with high affinity to free ubiquitin and to monoubiquitinated and polyubiquitinated proteins (Boyault et al., 2006). HDAC6 is actively maintained in the cytoplasm (Verdel et al., 2000) and is partly associated with the microtubule network. It uses its ZnF-UBP domain to convey ubiquitinated proteins along microtubules to pericentriolar structures named aggresomes, helping cells to cope with abnormal accumulation of misfolded proteins (Kawaguchi et al., 2003; Iwata et al., 2005). HDAC6 is also essential in the formation of dynamic cytoplasmic structures named stress granules (SGs) that appear in response to various types of cellular stress. HDAC6 mediates the motor-protein-driven movement of these individual SGs components along microtubules (Kwon et al., 2007).
We found that in unstressed cells, HDAC6 is present within the inactive HSF1 complex involving the major cellular chaperone HSP90, valosin-containing protein p97/VCP, a multitask, chaperone-like AAA-ATPase, and other proteins (Seigneurin-Berny et al., 2001; Boyault, Zhang, Fritah, et al., 2007). Under proteasomal inhibition, HDAC6 binds to polyubiquitinated (poly-Ub) proteins via its ZnF-UBP domain. The binding of HDAC6 to polyubiquitinated proteins and to p97/VCP are mutually exclusive (Seigneurin-Berny et al., 2001). HDAC6 binding to polyubiquitinated proteins induces the dissociation of the HDAC6-p97/VCP complex. In turn, p97/VCP uses its segregase activity to dissociate the repressive HSP90-HSF1 complex. Conversely, after resorption of misfolded polyubiquitinated proteins after stress, VCP recovers free HDAC6, allowing the reformation of the inactive HSF1 complex (Boyault, Zhang, Fritah, et al., 2007). The central role played by HDAC6 in response to proteasome inhibition is therefore multiple (McConkey et al., 2012). Through binding to ubiquitinated protein aggregates, it coordinates the formation of aggresomes. In addition, through binding to ubiquitinated proteins, HDAC6 allows the activation of HSF1 with subsequent expression of HSP genes and chaperone accumulation. The essential role played by HDAC6 and p97/VCP in the context of proteasome inhibition is shown by the observation that HSF1 activation does not occur in the absence of HDAC6 or p97/VCP (Boyault, Zhang, Fritah, et al., 2007).
Surprisingly, in contrast to proteasome inhibition, heat shock leads to HSF1 activation even in the absence of HDAC6. This suggests that, unlike proteasome inhibition, heat-induced dissociation of HSF1 from HSP90 is independent of HDAC6 and of the segregase activity of p97/VCP (Boyault, Zhang, Fritah, et al., 2007). These data clearly illustrate that the mechanisms underlying HSF1 activation depend on the nature of the stress.
Here we show that HDAC6 plays a role in controlling the duration of heat shock response.
RESULTS
Mutation in the ubiquitin-binding domain of HDAC6 impairs HSP25 accumulation after heat shock
To assess the role of HDAC6 in the heat-induced accumulation of HSP proteins, we examined the profile of HSP25 and HSP70 accumulation in wild-type (WT) or in HDAC6-knockout (KO) mouse 3T3 cells reexpressing or not HDAC6 bearing inactivating mutations in the two catalytic deacetylase domains (HDm) or in the ubiquitin-binding domain (Ubm; Figure 1A). Similar amounts of HDAC6 were detected by Western blot analysis in these cells, whereas acetylated tubulin (a well-known substrate of HDAC6) was only detected in HDAC6 HDm and HDAC6 KO cells (Supplemental Figure S1).
FIGURE 1:

Absence of heat-induced HSP25 expression in cells expressing HDAC6 mutated in its ubiquitin-binding domain. (A) Schematic representation of HDAC6 and HDAC6 mutants. (B) Comparative analysis of HSF1 phosphorylation under HS and MG132 treatments. Proteins were extracted from WT cells and KO HDAC6 cells heat shocked or not for 1 h or incubated with MG132 inhibitor for 8 h. Phosphorylation of HSF1 was analyzed by Western blot using an anti-HSF1 antibody. Ponceau staining was used as a loading control. (C) Schematic representation of the heat shock kinetics. Cells were maintained at 37°C (NHS) or submitted to different conditions of recovery period (Rec) after 1 h of heat shock at 43°C. (D) HSP70 and HSP25 expression in HDAC6 mutants. Western blot analysis of HSP70 and HSP25 was performed in WT cells, HDAC6 KO cells, and cells expressing HDAC6 mutated in the ubiquitin-binding (Ubm) or HDAC domain (HDm). Tubulin (Tub) was used as a loading control. (E) HSP25 expression in HDAC6 mutants. Immunodetection of HSP25 was performed in the different cell lines submitted or not (NHS) to a recovery period of 8 h after heat shock. Bar, 10 μm.
We first confirmed that HSF1 activation was impaired in HDAC6 KO cells treated with a proteasome inhibitor and not in heat-shocked HDAC6 KO cells (Boyault, Zhang, Fritah, et al. 2007). To this end, HSF1 hyperphosphorylation, a characteristic to the active HSF1 fraction, was analyzed by Western blot. As expected, active HSF1 was detected in WT and HDAC6-KO heat-shocked cells (Figure 1B).
Western blots were also performed on extracts from cells exposed to a 1-h heat shock followed by different times of recovery (4.5–24 h; Figure 1C). As shown in Figure 1D, all four cell lines analyzed displayed a similar profile of HSP70 expression, with low expression of HSP70 in unstressed cells and a significant heat-induced accumulation of HSP70 already observed 4.5 h after heat shock. In comparison, heat-induced accumulation of HSP25 was only observed 8 h after heat shock in wild-type cells and in HDm HDAC6 cells. In cells knocked out for HDAC6, a higher level of constitutive expression of HSP25 was observed. Remarkably, in contrast to the other cell lines, almost no stress-induced accumulation of HSP25 was detected in cells expressing the HDAC6 Ubm mutant. Tubulin was used as a loading control.
Defect in stress-induced accumulation of HSP25 in Ubm HDAC6–expressing cells was confirmed by immunofluorescence (Figure 1E) in cells submitted to a 8-h recovery period after heat shock.
Taken together, the analyses of the profiles of HSP25 and HSP70 point to the important role of the Ub-binding domain of HDAC6 in the heat-induced accumulation of HSP25. Absence of a significant effect of Ubm HDAC6 expression on the profile of HSP70 accumulation rules out the existence of a global impairment of HSF1 activation in Ubm HDAC6–expressing heat- shocked cells.
Mutation in the ubiquitin-binding domain of HDAC6 impairs HSP25 transcription after heat shock
We then sought to better characterize the mechanisms underlying the loss of HSP25 accumulation in cells expressing Ubm HDAC6 after heat shock. The putative effect of HDAC6 mutations on the level of HSP70 and HSP25 transcripts was therefore examined by Northern blot analysis in cells exposed or not to a 1-h heat shock followed by a recovery period of 8 h at 37°C. Quantification of HSP25 and HSP70 transcripts detected by Northern blot was performed on the basis of two independent Northern blot experiments. As shown in Figure 2, A and B, accumulation of HSP70 transcripts after heat shock was observed in all four cell lines. Of interest, although HSP25 transcripts were also detected in wild-type and knockout HDAC6 heat-shocked cells, as well as in cells expressing HDm HDAC6, no HSP25 transcripts were detected in cells expressing Ubm HDAC6. No HSP25 transcripts were detected across the entire time scale of the kinetics of recovery (Supplemental Figure S2). Taken together, Western and Northern blot analyses clearly indicate that absence of heat-induced accumulation of HSP25 in Ubm HDAC6– expressing cells results from a lack of heat-induced transcriptional activation of the HSP25 gene.
FIGURE 2:

The ZnF-UBP domain of HDAC6 is essential for the heat-induced transcriptional activation of the HSP25 gene. (A) The different cell lines analyzed in Figure 1 were submitted (+) or not (–) to heat shock (HS). After 8 h of recovery, total RNA fractions were prepared and analyzed by Northern blot with probes specific for HSP25 and HSP70 transcripts. A probe specific for the 18S transcripts was used as control. (B) Quantification based on two different Northern blots. Normalization calculated with the signals obtained with probe 18S. (C) Analysis of HSF1 migration profiles in the different cell lines submitted (HS) or not (NHS) to different heat-shock durations display similar kinetics of HSF1 hyperphosphorylation. Whole-cell extracts were analyzed by Western blot with an HSF1 antibody. Tubulin was used as loading control.
We then reasoned that a lower level of expression of HSF1, or a less efficient activation of HSF1 in response to heat shock in cells expressing Ubm HDAC6, could have a more severe effect on the transcriptional activation of the HSP25 gene than on the transcriptional activation of the HSP70 gene. As previously described (Boyault, Zhang, Fritah, et al., 2007), no significant difference in the quantity of HSF1 was observed in the different cell lines examined. A comparative analysis of the kinetics of HSF1 activation in the cell lines submitted to a kinetic of continuous heat shock was performed to confirm these data. On stress, HSF1 is hyperphosphorylated, causing delayed electrophoretic mobility of active HSF1 in SDS–PAGE analysis. As shown in Figure 2C, no significant variation in the kinetics of HSF1 activation was observed in the different cell lines.
We next compared the DNA-binding capacity of HSF1 to HSP70 and HSP25 gene promoters in the four cell lines. To this end, chromatin immunoprecipitation (ChIP) analysis was carried out with an anti-HSF1 antibody in both unstressed and stressed cells exposed or not to a period of recovery of 4.5–12 h at 37°C. Binding of HSF1 to both HSP70 and HSP25 gene promoters was then assessed with sets of primers surrounding the heat shock elements (HSEs) present in the HSP70 and HSP25 gene promoters (Figure 3A). HSF1 binding to the U6 gene promoter was also analyzed as a negative control. The efficacy of HSF1 immunoprecipitation was confirmed by Western blot analysis with an anti-HSF1 antibody (Supplemental Figure S3), and the specificity of binding was controlled by ChIP using an immunoglobulin G (IgG) antibody (Figure 3B). In wild-type cells, delayed binding of HSF1 to the HSP25 gene promoter was observed compared with that of the HSP70 gene promoter, demonstrating delayed action of HSF1 on the HSP25 gene. In wild-type cells, HSF1 binding to the HSP70 promoter was already observed in cells submitted to a 1-h heat shock, whereas, in contrast, HSF1 binding to the HSP25 promoter was detected only in cells submitted to a recovery period of 4.5 h at 37°C. Of interest, a twofold decrease in the binding ability of HSF1 to the HSP70 gene promoter was observed in the KO HDAC6 cell line. This observation indicates that despite a decrease in HSF1 binding to HSP70 gene promoter in KO HDAC6 cells, HSF1 phosphorylation and HSP70 activation are not affected, which suggests that full activation of HSP70 can occur despite a lower level of HSF1 binding to HSP70 promoter. This observation also holds for cells expressing Ubm and HDm HDAC6 mutants, for which less HSF1 binding to HSP70 gene promoter was also observed.
FIGURE 3:

The ZnF-UBP domain of HDAC6 is essential for HSF1 binding to HSP25 promoter. (A) Schematic representation of the proximal regions of HSP70 and HSP25 promoters. The positions of transcription start sites (bent arrows, +1), heat shock elements (HSEs), and oligos (arrows) used in ChIP analysis are indicated. HSF1 ChIP fractions obtained from the different cell lines were analyzed by Western blot with an anti-HSF1 antibody (Supplemental Figure S3). (B) Relative enrichments of HSF1 at HSP70, HSP25, and U6 gene promoters were quantified by quantitative PCR analysis. HSF1 ChIP fractions were obtained from NHS and HS cells and from heat-shocked cells submitted to different conditions of recovery at 37°C (Rec). Error bars correspond to SDs from three independent ChIP experiments. A control experiment was also performed with IgG.
Strikingly, when HSP25 gene promoter was examined, in contrast to other cell lines, HSF1 was unable to bind to HSP25 gene promoter in Ubm HDAC6–expressing cells (Figure 3B), whereas in correlation with the higher level of expression of HSP25 in HDAC6 KO cells, HSF1 binding to the HSP25 gene promoter was found to be more efficient and to occur at an early stage of the recovery period. Our results reveal that lack of HSP25 accumulation in stressed cells expressing Ubm HDAC6 is caused by the absence of HSF1 binding to the HSP25 gene promoter. They also reveal that in cells knocked out for HDAC6, efficient and early binding of HSF1 to the promoter of the HSP25 gene occurs. Taken together, these data suggest that the timing of HSF1 binding to HSP70 and HSP25 gene promoters is tightly controlled by HDAC6.
Short-term activation of HSF1 occurs in the absence of HDAC6–ubiquitin interaction
Following our previous observations on the role of p97/VCP in the recovery of ubiquitin-bound HDAC6 (Boyault et al., 2006), we reasoned that after a heat shock stress, because of the absence of ubiquitin binding by HDAC6 Ubm, p97/VCP very rapidly recovers HDAC6, which in turn mediates the reformation of inactive HSF1. To test this hypothesis, we decided to knock down p97/VCP in HDAC6 Ubm cells. Our prediction was that we could rescue HSP25 expression by preventing premature recapture of HDAC6 by p97/VCP.
We first, tested the efficiency of sip97/VCP on the level of p97/VCP expression in unstressed and stressed cells by Western blot and found that, in agreement with our previous observations (Boyault, Zhang, Fritah, et al., 2007), sip97/VCP had no effect on heat-induced activation of HSF1. Delayed HSF1 migration in heat-shocked cells treated with sip97/VCP confirmed that HSF1 activation was not impaired in these cells, in agreement with the effective accumulation of HSP25 and HSP70 (Supplemental Figure S4).
In contrast, we observed complete rescue of HSP25 expression in heat-shocked HDAC6 Ubm–expressing cells treated with sip97/VCP (Figure 4).
FIGURE 4:

p97/VCP is required to knock down HSP25 accumulation in Ubm heat-shocked cell line. WT and Ubm cells were submitted or not (NHS) to heat shock, followed by different conditions of recovery (Rec) at 37°C. Ubm HDAC6–expressing cells were transfected with a siRNA against p97/VCP. HSP25 expression was analyzed by Western blot with an anti-HSP25 antibody. The efficiency of the sip97/VCP knockdown was assessed by Western blot using an anti-p97/VCP antibody. Tubulin was used as loading control.
In conclusion, our data reveal that p97/VCP is not critical for HSF1 activation in heat-shocked cells but plays an important role in the reformation of the inactive HSF1 complex. We found that VCP accelerates the reformation of the repressive HSF1 complex when binding of HDAC6 to ubiquitinated residues no longer occurs (Figure 5).
FIGURE 5:

General model illustrating the role of the ubiquitin-binding domain of HDAC6 in the control of the duration of HSF1 activation. In unstressed cells, HSF1 is present in a dormant complex involving HDAC6, p97/VCP, and HSPs. On heat shock, HDAC6 dissociates from HSF1 and binds to polyubiquitinylated (Poly-Ub) proteins, allowing HSF1 binding to HSP70 gene (early response). The activation of the late inducible HSP25 gene (late response) depends on the duration of HSF1 activation and on the quantity of Poly-Ub proteins allowing (high amount) or not (low amount) the sustained activation of HSF1 during the recovery period from stress. Reformation of the inactive HSF1 dormant complex occurs either when poly-Ub proteins are no longer available or when binding of HDAC6 to poly-Ub proteins cannot occur due to a mutation in the Ub-binding domain of HDAC6 (bottom). Reformation of the HSF1 inactive complex requires p97/VCP, whose binding to HDAC6 only occurs when free, poly-Ub unbound HDAC6 is available. In KO cells, absence of VCP-HDAC6 complex impairs reformation of the inactive HSF1 complex, resulting in sustained activation of HSF1 and accumulation of HSP25.
The ubiquitin-binding domain of HDAC6 protects cells against stress-induced apoptosis. Because HSP25 is well known for its role in cell protection against stress-induced apoptosis (Huot et al., 1991; Lee et al., 2004), we next sought to determine whether down-regulation of HSP25 in Ubm HDAC6–expressing cells would have a negative effect on cell protection against heat-induced apoptosis. We thus measured and compared the percentage of apoptotic cells in wild-type cells and in HDAC6 mutant–expressing cells submitted or not to a heat shock followed by recovery of 12 and 24 h at 37°C (Figure 6A). As expected, the percentage of apoptotic cells 24 h after heat shock was significantly higher (35-fold increase) in HDAC6 Ubm–expressing cells than in unstressed wild-type cells and three times higher than in all other cell lines at the same time point. These results confirm the importance of the ubiquitin-binding domain of HDAC6 in the HSP25-mediated protection of cells against cell death.
FIGURE 6:
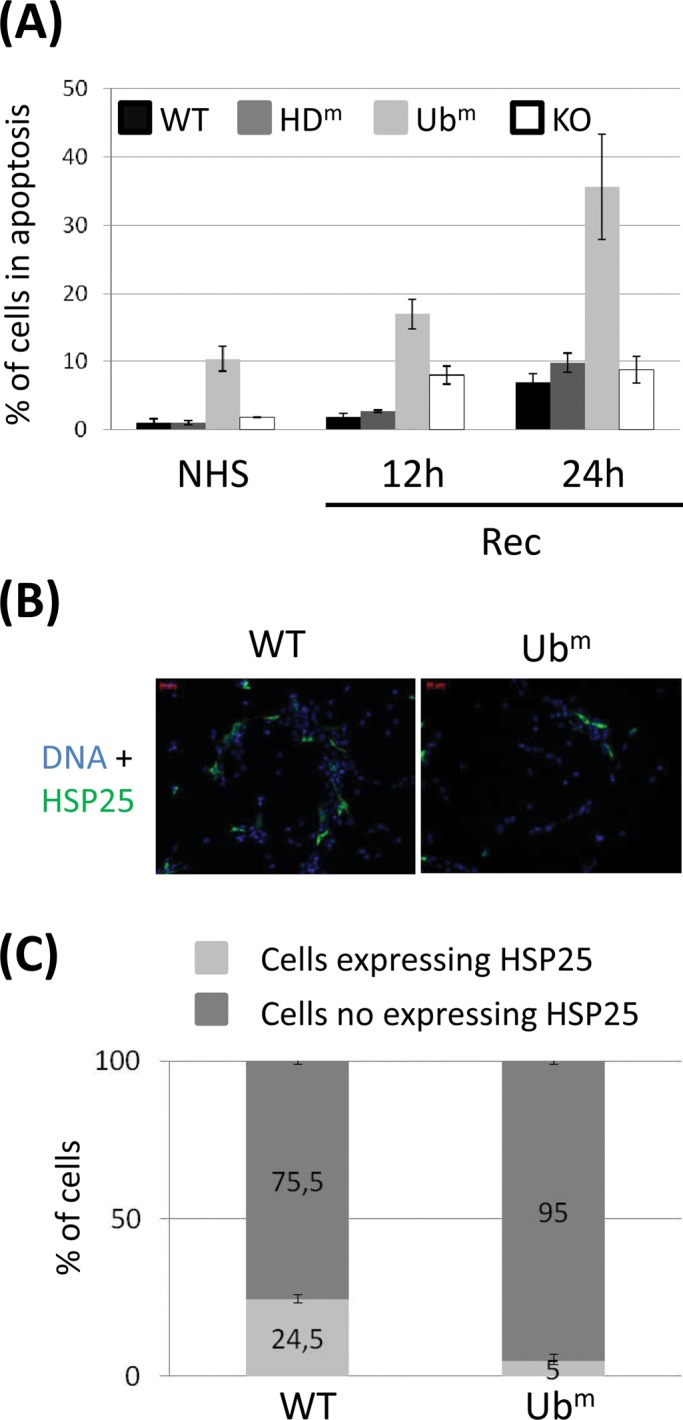
HDAC6 mutated in its ZnF-UBP domain increases the percentage of heat-induced apoptosis and prevents HSP25 accumulation in heat-shocked cells. (A) WT and HDAC6 mutant– expressing cells were submitted or not (NHS) to heat shock, followed by different times of recovery (Rec). The percentage of apoptotic cells was quantified by fluorescence-activated cell sorting through caspase 3 detection. Histograms represent average values determined from two independent experiments. The results were normalized with the wild-type point in unstressed conditions. (B) 3T3 cells were transiently transfected with the indicated HA-tagged HDAC6 expression vectors and submitted to a recovery period of 24 h after heat shock. Transfected cells were detected using an anti-HA antibody. (C) The proportion of cells expressing endogenous HSP25 was calculated using an anti-HSP25 antibody. The histograms represent values from three independent experiments.
Given the important effect of the ubiquitin-binding domain of HDAC6 on the level of stress-induced HSP25 accumulation, we further tested whether expressing the Ubm HDAC6 mutant in cells displaying constitutive expression of HSP25 could possibly affect the level of endogenous HSP25 through its competition with endogenous HDAC6. To this aim, an unrelated NIH/3T3 BALB/c cell line displaying a constitutive level of HSP25 expression was transiently transfected with wild-type or Ubm HDAC6–expressing plasmids. At 20 h after heat shock, the percentages of cells expressing HSP25 in both populations were assessed by immunofluorescence (Figure 6B). As shown in Figure 6C, a fivefold lower percentage of cells expressing HSP25 was obtained in cells transfected with the plasmid expressing Ubm HDAC6, supporting the idea that targeting the ubiquitin-binding domain of HDAC6 could represent an interesting strategy to down-regulate the level of HSP25.
DISCUSSION
The heat shock response includes a set of well-ordered and regulated reversible response to stress in the cell. Despite numerous data obtained in the past few decades concerning the molecular mechanisms involved in the control of HSF1 activation and inactivation (Voellmy, 2004; Westerheide and Morimoto, 2005), many questions remain. Heat shock proteins are known to play an important cytoprotective role through their transient binding to the misfolded proteins (Bukau et al., 2006). We identified a new mechanism involving the cytoplasmic histone deacetylase HDAC6 in response to proteasome inhibition (Boyault, Zhang, Fritah, et al., 2007). We found that binding of HDAC6 to ubiquitinated proteins triggers its dissociation from both the inactive HSP90-HSF1 complex and the chaperone-like p97/VCP AAA ATPase (Seigneurin-Berny et al., 2001; Boyault, Zhang, Fritah, et al., 2007). In this process, p97/VCP would facilitate in turn the dissociation of the HSP90-HSF1 complex, allowing an HSF1-dependent activation of HSP genes. Of interest, the occurrence of a dual mechanism involving HDAC6-dependent clearance of misfolded proteins aggregates (Kawaguchi et al., 2003) and HDAC6-dependent activation of HSF1 that we observed after proteasome inhibition was not found in heat-shocked cells.
The differences observed in the early steps of HSF1 activation after subjecting cells to the two types of stress, heat shock and proteasome inhibition, can be explained as follows. Heat-induced activation of HSF1 is a rapid process, occurring within minutes of heat-shock exposure, primarily mobilizing the nuclear fraction of HSF1 spatially unrelated to the HSP90-HDAC6 complex (Vujanac et al., 2005). In contrast, dissociation of the inactive HSF1 complex induced by MG132 is primarily triggered in the cytoplasm where HDAC6 is present and is a much slower process, relying on ubiquitinated protein binding to HDAC6. Second, heat is known to have a major effect on protein structure, likely sufficient to affect the stability of the HSF1-HSP90 complex in a p97/VCP- and HDAC6-independent manner. Therefore one can propose two distinct mechanisms underlying HSF1 activation: one involves direct disruption of the HSP90-HSF1 complex triggered by heat, and the other occurs in MG132-treated cells, elicited by HDAC6 binding to polyubiquitinated proteins and resulting in the p97/VCP-dependent driven segregation of the HSF1-HSP90 complex.
Delayed synthesis of HSP25 as compared with HSP70 in response to stress has already been observed (Klemenz et al., 1993), and the mechanisms underlying this different behavior are unclear (de Thonela et al., 2012). Structural differences between HSE binding sites present in the HSP25 and HPS70 gene promoters could explain the differences in their kinetics of activation. The HSEs (Gaestel et al., 1993) identified in the promoter of the HSP25 gene contains two inverted copies of the core nGAAn motif. In contrast, the two HSEs identified in the promoter of the HSP70 gene both contain three inverted tandem copies of this motif, with, in theory, a better affinity for HSF1. Specific regulatory elements could also account for the different behavior of HSP25 and HSP70 genes with regard to HSF1. Difference of affinity could set the basis for uncoupled expression of HSP25 and HSP70 genes in heat-shocked cells. Mild stressing conditions generating a low accumulation of ubiquitinated proteins could open a time window for HSF1 activation long enough for HSP70 gene activation and too short for HSP25 gene activation. In contrast, extensive accumulation of ubiquitinated proteins after a more severe stress extends HSF1 activation, also leading to a HSP25 gene activation.
HDAC6 catalytic activity is known to be essential for full steroid receptor activation (Kovacs et al., 2005). HDAC6 functions as a HSP90 deacetylase: inactivation of HDAC6 leads to HSP90 hyperacetylation and loss of chaperone activity. HSP90-dependent maturation of the glucocorticoid receptor is compromised in cells knocked down for HDAC6, resulting in glucocorticoid receptors defective in transcriptional activation. However, similar to what we observed in cells treated with MG132 (Boyault, Zhang, Fritah, et al., 2007), our observations provide additional evidence that the deacetylase domain of HDAC6 is dispensable for HSF1 activation in heat-shocked cells.
In stressing conditions such as heat shock, activation of the HSP25 gene plays an essential role in cell protection against apoptosis (Acunzo et al., 2012). The discovery reported here shows that it is the sensing of ubiquitinated proteins by the ZnF-UBP domain of HDAC6 that controls the duration of HSF1 activation. Our observation that late accumulation of HSP25 is impaired in Ubm HDAC6 reveals the role played by HDAC6 in the kinetics of reformation of the inactive HSF1 complex after heat shock. In Ubm HDAC6 cells, HDAC6 is not able to bind ubiquitinated proteins, leading to a premature reformation of the HSP90-HDAC6 platform and inactive HSF1 complex. Analysis of the kinetics of HSF1 dephosphorylation in the different cell lines indicates that HSF1 displays a similar kinetics of dephosphorylation in the recovery period from stress in WT and Ubm cell lines, suggesting a decoupling between the kinetics of HSF1 dephosphorylation and the reformation of the inactive HSF1 complex (Supplemental Figure S5). In cells knocked out for HDAC6, accumulation of HSP25 is observed, revealing the importance of HSP90-HDAC6 complex reformation in HSF1 inactivation. We have evidence for late inactivation of HSF1 after heat shock (Supplemental Figure S5), suggesting that alternative mechanisms of HSF1 inactivation exist in these cells.
Our data also show that p97/VCP, through its ability to recover the ubiquitin-bound HDAC6, plays an important role in controlling the duration of HSF1 activation after heat shock. Indeed, in the case of Ubm HDAC6 mutant, p97/VCP very rapidly reassociates with HDAC6 and reforms the inactive HSF1 complex. This activity of p97/VCP is clearly illustrated in the p97/VCP knockdown experiment in Ubm HDAC6 cells, which we show extends HSF1 activation and rescues HSP25 gene expression.
It is interesting to also note that in HDAC6 KO cells, an early-occurring heat-dependent accumulation of HSP25 is observed, correlating, in heat-shocked cells, with higher level of HSP25 transcripts. The reason why a higher constitutive level of HSP25 but not of HSP70 is present in HDAC6 KO cells is unclear. Based on ChIP experiments, it appears that a higher level of HSP25 does not result from increased binding of HSF1 to the HSP25 gene promoter. It is therefore possible that other transcription factors or posttranscriptional mechanisms induce a constitutive accumulation of HSP25 to compensate for the lack of HDAC6 and hence mediate cell adaptation to environmental stresses.
In conclusion, our work highlights how HDAC6, through its ubiquitin-binding domain, controls HSF1 inactivation in heat-shocked cells, restricting the activation of a major antiapoptotic pathway to cells with high levels of ubiquitinated residues. Finally, our work opens new perspectives in anticancer therapy (Santagata et al., 2011), clearly suggesting that the development of drugs targeting the ubiquitin-binding domain of HDAC6 could represent not only a good strategy to prevent stress-induced accumulation of HSP25, but also an efficient way to down-regulate HSP25 accumulation in HDAC6-expressing cancer cells, with seemingly no risk of activating compensatory mechanisms.
MATERIALS AND METHODS
Cell culture and treatments
Mouse 3T3 fibroblasts of WT, KO for HDAC6, or expressing mutated forms of HDAC6 (gift of P. Matthias, FMI, Basel, Switzerland) and BALB/c 3T3 cell line were grown in DMEM supplemented with 10% fetal bovine serum, 2% l-glutamine, and 1% penicillin/streptomycin in a humidified 5% CO2 atmosphere at 37°C. Heat shock treatments were performed at 43°C in a constant-temperature water bath. For proteasomal inhibition, cells were incubated with MG132 inhibitor (Calbiochem) at 2 μM for 8 h.
Western blot analysis
Western blot analysis was performed on 8 or 12% SDS–acrylamide gels with 10 μg of whole protein extracts prepared in 8 M urea with the following primary antibodies: anti-HSF1 (1:1000; ADI-SPA-901; Enzo Life Sciences), anti-tubulin (1:1000; T5168; Sigma-Aldrich), anti–acetylated tubulin (1:5000; T6793; Sigma-Aldrich), anti-HDAC6 (1:1000; 7612; Cell Signaling; or homemade), anti-HSP70 (1:1000; ab74084; Abcam), anti-VCP (1:5000; gift of N. K. Tonks, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY), or anti-HSP25 (1:500; ADI-SPA-801; Enzo Life Sciences), and horseradish peroxidase–conjugated anti-rabbit or anti-mouse antibodies (1:5000; GE Healthcare).
RNA interference
p97/VCP small interfering RNAs (siRNAs; forward, 5′-GAC GUU UGG AGA UUC UUC A11-3′; reverse, 5′-UGA AGA AUC UCC AAA CGU C11-3′) were purchased from Eurogentec. Transfections were performed using Lipofectamine RNAiMAX transfection reagent (Invitrogen). At 24 h after transfection, cells were heat shocked and then submitted or not to a recovery period.
Transfection and immunofluorescence analysis
Unrelated BALB/c 3T3 cell line was transfected for 24 h with pcDNA3.1 vectors expressing hemagglutinin (HA)-tagged mWT HDAC6 and HA-mUbm HDAC6. For immunofluorescence, cells were fixed using 2% paraformaldehyde. Immunofluorescence was performed using an anti-HSP25 antibody (1:1000; SPA-801; Stressgen) and an Alexa 488–conjugated goat anti rabbit antibody (1:500; A11034; Invitrogen) or an anti-HA antibody (1:100; 3F10; Roche). Cells were stained with 250 ng/ml 4′,6-diamidino-2-phenylindole and visualized by epifluorescence microscopy. Four hundred cells from at least three independent series were quantified for each condition.
Measurement of caspase activity
Cells (106/point) were heat shocked and submitted to a recovery period of either 12 or 24 h before analysis. The percentage of apoptotic cells was then determined using active caspase-3 apoptosis kit (BD PharMingen) according to the manufacturer's instructions. Measures were performed by flow cytometry on an Accuri C6 machine. The results were normalized with the number of apoptotic cells in unstressed wild-type cells.
Northern blot analysis
RNA was isolated with TRI-Reagent (Sigma-Aldrich). We loaded 15 μg of RNA on 0.8% agarose and 2.2 M formaldehyde gel. RNA was transferred onto a nylon membrane and ultraviolet cross-linked with a Stratalinker. HSP70, HSP25, and 18S transcripts were detected with [α-32P]dCTP-labeled probes. HSP70 and HSP25 probes were amplified by PCR using the primers, for mHSP70, forward, 5′-CAAGATCACCATCACCAACG-3′, and reverse, 5′-ATGACCTCCTGGCACTTGTC-3′; and for mHSP25, forward, 5′-CCTCTTCGATCAAGCTTTCG-3′, and reverse, 5′-GCCTTCCTTGGTCTTCACTG-3′; on mouse cDNA. Hybridization signals were detected using a PhosphorImager PMI (Bio-Rad) and quantified (ImageJ). Total RNA samples were analyzed on the gel with ethidium bromide staining.
Chromatin immunoprecipitation
The 3T3 cells were cross-linked for 10 min in 1% formaldehyde. Chromatin samples were fragmented by sonication to an approximate size of 700 base pairs with BioRuptor Sonicator (Diagenode) at 4°C. Sonicated chromatin was centrifuged for 10 min at 4°C. Immunoprecipitations were performed with an antibody against HSF1 (sc-9144; Santa Cruz Biotechnology) at 4°C overnight. DNA was purified with an Invitek kit. The following primers were used for quantitative PCR: mHSP70-HSE, forward, 5′-CGCCCTGCGCCTTTAAG-3′, and reverse, 5′-GCACCAGCACTTCCCCA-3′; mHSP25-HSE, forward, 5′-ACGCCGTCATTTGTTTTCTT-3′, and reverse, 5′-CCAGCGTGCTTTTATGTGTG-3′; and mU6, forward, 5′-GAGGGATTTTCGTGGTCAGA-3′, and reverse, 5′-TCCAGTCTGGTCTGCTGTTG-3′. Quantifications were done by quantitative PCR (kit 4707516001; Roche). Values obtained from two independent ChIP experiments were normalized with the corresponding input values.
Supplementary Material
Acknowledgments
We thank C. Jolly, C. Boyault, C. Kretz, S. Fritah, and C. Caron for helpful discussions and comments and help in data analysis. We also thank P. Matthias for the gift of HDAC6 cell lines. This work was funded by Association pour la Recherche sur le Cancer Grant 3686 (to C.V.) and by the Institut National du Cancer EPISTRESS (C.V. and S.K.) and EpiSperm (S.K.). L.P. is supported by an Association Française contre les Myopathies fellowship.
Abbreviations used:
- HDm
HDAC6 mutated in the histone deacetylase domain
- HSE
heat shock element
- Ubm
HDAC6 mutated in the ubiquitin domain
- ZnF-UBP
C-terminal zinc finger ubiquitin domain.
Footnotes
*These authors contributed equally to this work.
The authors declare that they have no conflict of interest.
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-06-1032) on October 8, 2014.
REFERENCES
Boldface names denote co–first authors.
- Acunzo J, Katsogiannou M, Rocchi P. Small heat shock proteins HSP27 (HspB1), β-crystallin (HspB5) and HSP22 (HspB8) as regulators of cell death. Int J Biochem Cell Biol. 2012;44:1622–1631. doi: 10.1016/j.biocel.2012.04.002. [DOI] [PubMed] [Google Scholar]
- Akerfelt M, Morimoto RI, Sistonen L. Heat shock factors: integrators of cell stress, development and lifespan. Nat Rev Mol Cell Biol. 2010;11:545–555. doi: 10.1038/nrm2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyault C, Gilquin B, Zhang Y, Rybin V, Garman E, Meyer-Klaucke W, Matthias P, Müller CW, Khochbin S. HDAC6-p97/VCP controlled polyubiquitin chain turnover. EMBO J. 2006;25:3357–3366. doi: 10.1038/sj.emboj.7601210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyault C, Zhang Y, Fritah S, Caron C, Gilquin B, Kwon SH, Garrido C, Yao TP, Vourc’h C, Matthias P, Khochbin S. HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genes Dev. 2007;21:2172–2181. doi: 10.1101/gad.436407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukau B, Weissman J, Horwich A. Molecular chaperones and protein quality control. Cell. 2006;125:3443–3451. doi: 10.1016/j.cell.2006.04.014. [DOI] [PubMed] [Google Scholar]
- Ciocca DR, Arrigo AP, Calderwood SK. Heat shock proteins and heat shock factor 1 in carcinogenesis and tumor development: an update. Arch Toxicol. 2013;87:19–48. doi: 10.1007/s00204-012-0918-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotto JJ, Morimoto RI. Stress-induced activation of the heat-shock response: cell and molecular biology of heat-shock factors. Biochem Soc Symp. 1999;64:105–118. [PubMed] [Google Scholar]
- de Thonela A, Le Mouël A, Mezger V. Transcriptional regulation of small HSP-HSF1 and beyond. Int J Biochem Cell Biol. 2012;44:1593–1612. doi: 10.1016/j.biocel.2012.06.012. [DOI] [PubMed] [Google Scholar]
- Gaestel M, Gotthardt R, Müller T. Structure and organisation of a murine gene encoding small heat-shock protein Hsp25. Gene. 1993;128:279–283. doi: 10.1016/0378-1119(93)90575-n. [DOI] [PubMed] [Google Scholar]
- Huot J, Roy G, Lambert H, Chrétien P, Landry J. Increased survival after treatments with anticancer agents of Chinese hamster cells expressing the human Mr 27,000 heat shock protein. Cancer Res. 1991;51:5245–5252. [PubMed] [Google Scholar]
- Iwata A, Riley BE, Johnston JA, Kopito RR. HDAC6 and microtubules are required for autophagic degradation of aggregated Huntingtin. J Biol Chem. 2005;280:40282–40292. doi: 10.1074/jbc.M508786200. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115:727–738. doi: 10.1016/s0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- Klemenz R, Andres AC, Fröhli E, Schäfer R, Aoyama A. Expression of the murine small heat shock proteins Hsp 25 and alpha B crystallin in the absence of stress. J Cell Biol. 1993;120:639–645. doi: 10.1083/jcb.120.3.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs JJ, Murphy PJ, Gaillard S, Zhao X, Wu JT, Nicchitta CV, Yoshida M, Toft DO, Pratt WB, Yao TP. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell. 2005;18:601–607. doi: 10.1016/j.molcel.2005.04.021. [DOI] [PubMed] [Google Scholar]
- Kwon S, Zhang Y, Matthias P. The deacetylase HDAC6 is a novel critical component of stress granules Involved in the stress response. Genes Dev. 2007;21:3381–3394. doi: 10.1101/gad.461107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YJ, Cho HN, Jeoung DI, Soh JW, Cho CK, Bae S, Chung HY, Lee SJ, Lee YS. HSP25 overexpression attenuates oxidative stress-induced apoptosis: roles of ERK1/2 signaling and manganese superoxide dismutase. Free Radic Biol Med. 2004;36:429–444. doi: 10.1016/j.freeradbiomed.2003.11.009. [DOI] [PubMed] [Google Scholar]
- McConkey DJ, White M, Yan W. HDAC inhibitor modulation of proteotoxicity as a therapeutic approach in cancer. Adv Cancer Res. 2012;116:131–163. doi: 10.1016/B978-0-12-394387-3.00004-5. [DOI] [PubMed] [Google Scholar]
- Santagata S, Xu YM, Wijeratne EM, Kontnik R, Rooney C, Perley CC, Kwon H, Clardy J, Kesari S, Whitesell L, et al. Using the heat-shock response to discover anticancer compounds that target protein homeostasis. ACS Chem Biol. 2011;7:340–349. doi: 10.1021/cb200353m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seigneurin-Berny D, Verdel A, Curtet S, Lemercier C, Garin J, Rousseaux S, Khochbin S. Identification of components of the murine histone deacetylase 6 complex: link between acetylation and ubiquitination signaling pathways. Mol Cell Biol. 2001;21:8035–8044. doi: 10.1128/MCB.21.23.8035-8044.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyedmers J, Mogk A, Bukau B. Cellular strategies for controlling protein aggregation. Nat Rev Mol Cell Biol. 2010;11:777–788. doi: 10.1038/nrm2993. [DOI] [PubMed] [Google Scholar]
- Verdel A, Curtet S, Brocard MP, Rousseaux S, Lemercier C, Yoshida M, Khochbin S. Active maintenance of mHDA2/mHDAC6 histone-deacetylase in the cytoplasm. Curr Biol. 2000;10:747–749. doi: 10.1016/s0960-9822(00)00542-x. [DOI] [PubMed] [Google Scholar]
- Voellmy R. On mechanisms that control heat shock transcription factor activity in metazoan cells. Cell Stress Chaperones. 2004;9:122–133. doi: 10.1379/CSC-14R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voellmy R, Boellmann F. Chaperone regulation of the heat shock protein response. Adv Exp Med Biol. 2007;594:89–99. doi: 10.1007/978-0-387-39975-1_9. [DOI] [PubMed] [Google Scholar]
- Vujanac M, Fenaroli A, Zimarino V. Constitutive nuclear import and stress-regulated nucleocytoplasmic shuttling of mammalian heat-shock factor 1. Traffic. 2005;6:214–215. doi: 10.1111/j.1600-0854.2005.00266.x. [DOI] [PubMed] [Google Scholar]
- Westerheide SD, Morimoto RI. Heat shock response modulators as therapeutic tools for diseases of protein conformation. J Biol Chem. 2005;280:33097–33100. doi: 10.1074/jbc.R500010200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.