

Abstract
A direct binding screen of 100 000 sp3-rich molecules identified a single diastereomer of a macrolactam core that binds specifically to myeloid cell leukemia 1 (MCL1). A comprehensive toolbox of biophysical methods was applied to validate the original hit and subsequent analogues and also established a binding mode competitive with NOXA BH3 peptide. X-ray crystallography of ligand bound to MCL1 reveals a remarkable ligand/protein shape complementarity that diverges from previously disclosed MCL1 inhibitor costructures.
Keywords: MCL1, myeloid cell leukemia 1, sp3-rich, biophysical validation
Evasion of programmed cell death, or apoptosis, is a hallmark of cancer that allows tumor cells to survive stresses that would kill a normal cell. Specifically, cell death-inducing mitochondrial permeabilization is prevented by tight sequestration of membrane-localized proteins by antiapoptotic members of the BCL-2 family, which include BCL-2, BCL-XL, BCL-W, A1, and MCL1.1−7 Human genetics points to a selective advantage of MCL1-amplified tumor cells. The analysis of over 3000 diverse human tumors indicates that MCL1 is among the top 10 most frequently amplified genes in human cancer.8,9
Consistent with its frequent amplification, MCL1 is highly expressed in many tumor types, and high expression levels of MCL1 contribute to tumor development and resistance to chemotherapy.10,11 As a result, there have been considerable efforts to develop small molecule therapeutics that target the function of MCL1 protein (Figure 1).12−16
Figure 1.

Representative MCL1 inhibitors.
To identify chemical starting points for targeting MCL1, we conducted a direct binding screen of 100 000 molecules synthesized at the Broad Institute.17−20 This chemical library contains a high degree of stereochemical and skeletal diversity that augments traditional screening collections. The enumeration of nonplanar, sp3-rich cores spanning four-membered ring azetidines to 14-membered macrocycles provides a rich source of three-dimensional space to sample for suitable protein/ligand shape complementarity (average core Fsp3 = 0.60).21 A particular strength of this library is the elaboration of individual diastereomers in optically enriched form, thereby allowing rapid stereochemical deconvolution of identified hits.
Differential scanning fluorimetry22,23 (DSF, or thermal shift assay) was selected as the biophysical method to detect ligand binding to MCL1. This direct binding technique is amenable to high-throughput screening formats and requires lower concentrations of protein relative to other methods (typically < 500 μg/mL). By screening for direct binders of MCL1, as compared to traditional peptide competition screens, we allow discovery of novel inhibitor binding modes (i.e., competitive and noncompetitive with BH3 peptide).
After screening over 13 000 stereochemically defined scaffolds, the macrolactam 5 emerged as a hit (ΔTm = +0.3 °C at 25 μM ligand and 100 μg/mL MCL1 protein). The synthetic pathway to assemble this macrolactam has been described previously.18 Importantly, only 1 of the 8 diastereomers that were screened caused a significant shift of MCL1 Tm, and it had the 5R,6S configuration in the macrolactam core (Figure 2). The strong preference for the 5R,6S core configuration suggests a highly specific binding event of 5 to MCL1. Meanwhile, identification of additional 5R,6S congeners revealed no consistent absolute configuration at C2 of the exocyclic side chain (see Supporting Information Figure SI-1), implying that this portion of the molecule does not directly engage the MCL1 protein surface. Molecular modeling of all eight diastereomers confirms that the C5 and C6 stereogenic centers largely dictate the spatial presentation of the sulfonamide and urea side chains, independent of the C2 stereochemistry (see Supporting Information Figure SI-2 for modeling data).
Figure 2.
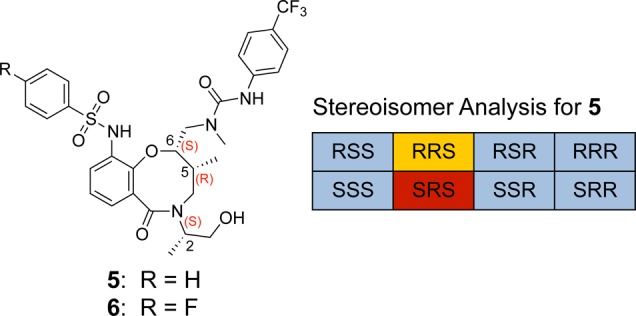
Identification of macrolactam hit 5 from DSF screening (stereochemistry labeling as C2C5C6; red, binding hit; blue, no binding; yellow, inconclusive).
Several sulfonamide variants of the 5R,6S macrolactam core were identified as hits in the primary screen, with the 4-fluorophenyl sulfonamide 6 displaying the largest thermal shift (ΔTm = +0.6 °C at 25 μM ligand, 100 μg/mL MCL1 protein). To rule out HTS-derived assay artifacts, we investigated the binding of 6 using orthogonal biophysical methods. Differential scanning calorimetry (DSC) relies on a similar physical principle as DSF to determine the effect of ligand binding but directly measures the enthalpy of protein unfolding during a controlled temperature ramp without the use of a fluorescent reporter dye.24,25 When macrolactam 6 was tested for binding to MCL1 by DSC, a ΔTm = +1.9 °C was observed at 100 μM of ligand. Importantly, we again observed no binding of the other core stereoisomers by this second biophysical technique (see Supporting Information Figure SI-3).
A third approach to validate binding of putative hits is isothermal titration calorimetry (ITC).26−28 This method provides a direct measurement of binding enthalpy and also delineates entropic contributions to binding. In our initial experiments with 6, we were unable to detect binding at 800 μM ligand and 25 μM protein. We suspected that low aqueous solubility of 6 limited our ability to measure binding, even in the presence of 4% DMSO. To improve aqueous solubility, the exocyclic alcohol of 6 was oxidized to the corresponding acid 7 in two steps, with Dess–Martin oxidation conditions being optimal to minimize C2 epimerization (Scheme 1).29 With the more soluble macrolactam 7, we measured an MCL1 KD = 1.9 μM by ITC. Interestingly, the measured ΔH (enthalpy, −5.6 kcal/mol) and ΔS (entropy, −3.2 cal/mol/K) upon binding of macrolactam 7 were different from previously described MCL1 inhibitors. The indole carboxylate 1 reported by Fesik,13 for instance, derives much of its binding energy from enthalpic interactions with Arg263 of MCL1, as evidenced by the ΔH = −9.4 kcal/mol (ΔS for 1 = 1.7 cal/mol/K). The lower enthalpy of binding for macrolactam 7 suggests that the appended carboxylic acid does not engage the MCL1 protein and instead serves solely as a solubility handle.
Scheme 1. Oxidation of Exocyclic Alcohol to Afford Ligand 7.

Reaction conditions: 1. Dess–Martin periodinane, DCM, 88%; 2. NaClO2, NaH2PO4, 2-methyl-2-butene, tBuOH/CH3CN, 65%.
The 4-fluoro and 4-trifluoromethyl groups on the sulfonamide and urea side chains offer a ligand-based method to further confirm binding of macrolactam 7 to MCL1. Specifically, 19F NMR can be used to detect the binding of fluorinated ligands without requiring isotopically labeled protein.30,31 The 19F NMR resonance of the CF3 group, in particular, was significantly shifted in the presence of MCL1 (Δv = 5.4 Hz at 60 μM ligand, 10 μM protein), with significant line-broadening as expected for a slower-tumbling protein–ligand complex. Using 19F NMR detection, we measured an intrinsic KD = 6 μM for macrolactam 7 binding to MCL1, which is in good agreement with affinity measured by ITC.
To establish the binding mode for 7, we employed a fluorescence polarization (FP) assay that measures the ability of ligands to competitively bind in the presence of NOXA BH3 peptide. Macrolactam 7 inhibits BH3 peptide binding to MCL1 with IC50 = 4.5 μM. Importantly, compound 7 does not interfere with binding of BH3 peptides to antiapoptotic family members Bcl-2 and Bcl-xL (see Supporting Information Figure SI-4). Thus, this direct binding discovery effort has identified a novel macrolactam, which selectively binds in the hydrophobic BH3 binding groove of MCL1.
Having thus validated the binding of macrolactam 7 by several additional biophysical methods (DSC, ITC, 19F NMR, and FP), we were encouraged to explore the structure-binding relationships of this novel scaffold. Several analogues were synthesized in straightforward fashion to probe both the urea and sulfonamide portions of 7 (Tables 1 and 2). Phenyl ureas lacking an electron-withdrawing group (10) or bearing an electron-donating −OMe group (11) failed to bind MCL1. The para-CF3 phenyl substituent of the urea in 7 proved to be crucial for binding, as the ortho- or meta-CF3 analogues displayed no binding by DSC and FP (Table 1, compounds 7–9). The unique binding of the para-CF3 analogue suggests a specific lipophilic interaction, and not a general electronic effect, is required for binding to MCL1. Other lipophilic side chains including 1- and 2-naphthyl urea groups are apparently too large to effectively bind MCL1, as both showed a smaller thermal shift (ΔTm = −0.1 °C for 12; ΔTm = +0.8 °C for 13).
Table 1. Structure–Activity Relationship (SAR) on the Urea Domain of 7.
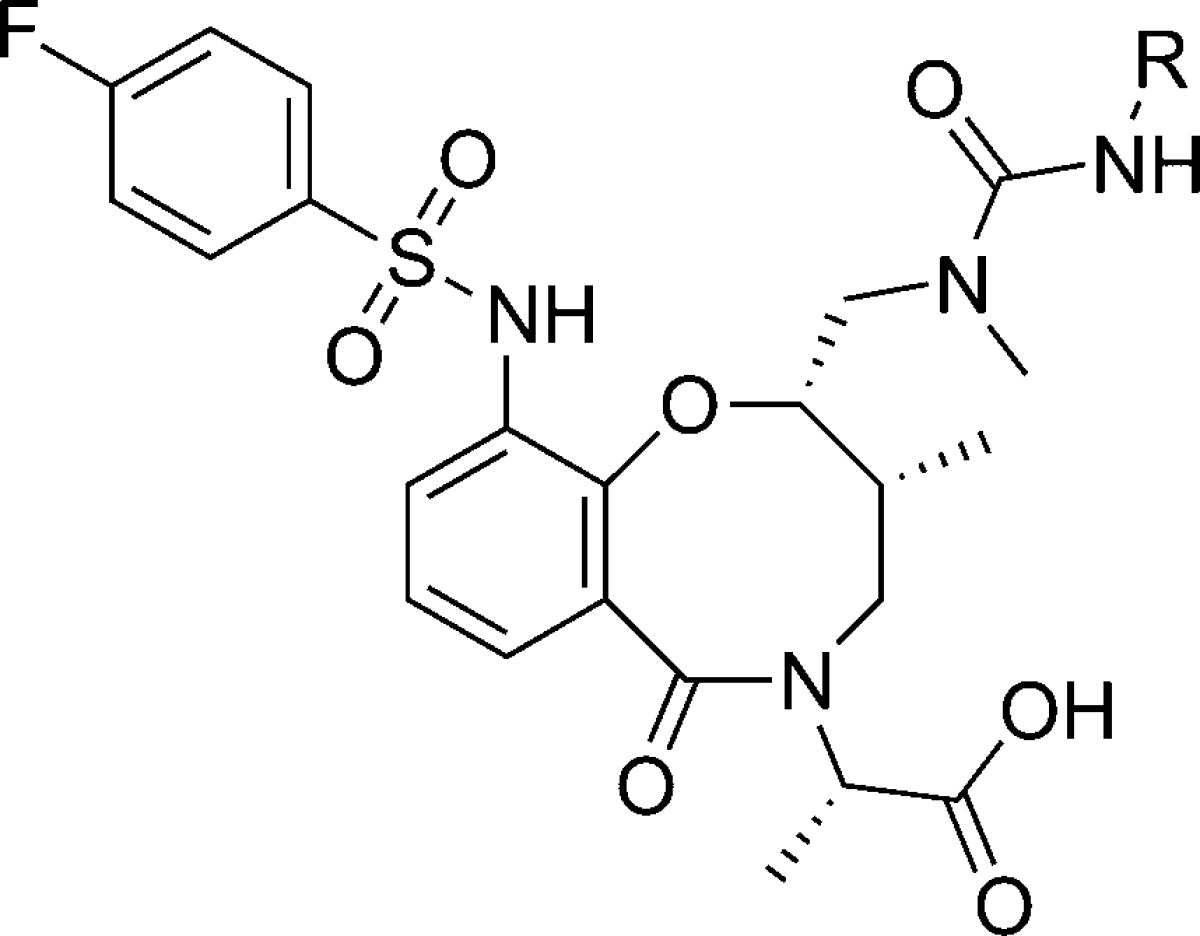
| compd | R | DSC ΔTm (°C)a | FP IC50 (μM)b |
|---|---|---|---|
| 7 | p-CF3C6H4 | 2.9 | 4.5 |
| 8 | m-CF3C6H4 | 0.2 | >25 |
| 9 | o-CF3C6H4 | 0.2 | >50 |
| 10 | Ph | 0.2 | >50 |
| 11 | p-MeOC6H4 | 0.2 | >50 |
| 12 | 1-Np | –0.1 | >50 |
| 13 | 2-Np | 0.8 | >25 |
Differential scanning calorimetry (DSC) assay: 250 μM compound and 25 μM MCL1 (173–329) in 25 mM Hepes, pH 7.4, 100 mM NaCl, 0.1 mM TCEP, and 4% DMSO.
FP assay: Serial dilutions of compound, with a top concentration of 50 μM, were preincubated for 30 min with 200 nM MCL1 and 25 nM TAMRA-Noxa in 25 mM Hepes, pH 7.4, 100 mM NaCl, 0.005% Tween 20, and 2% DMSO.
Table 2. SAR on the Sulfonamide Domain of 7.
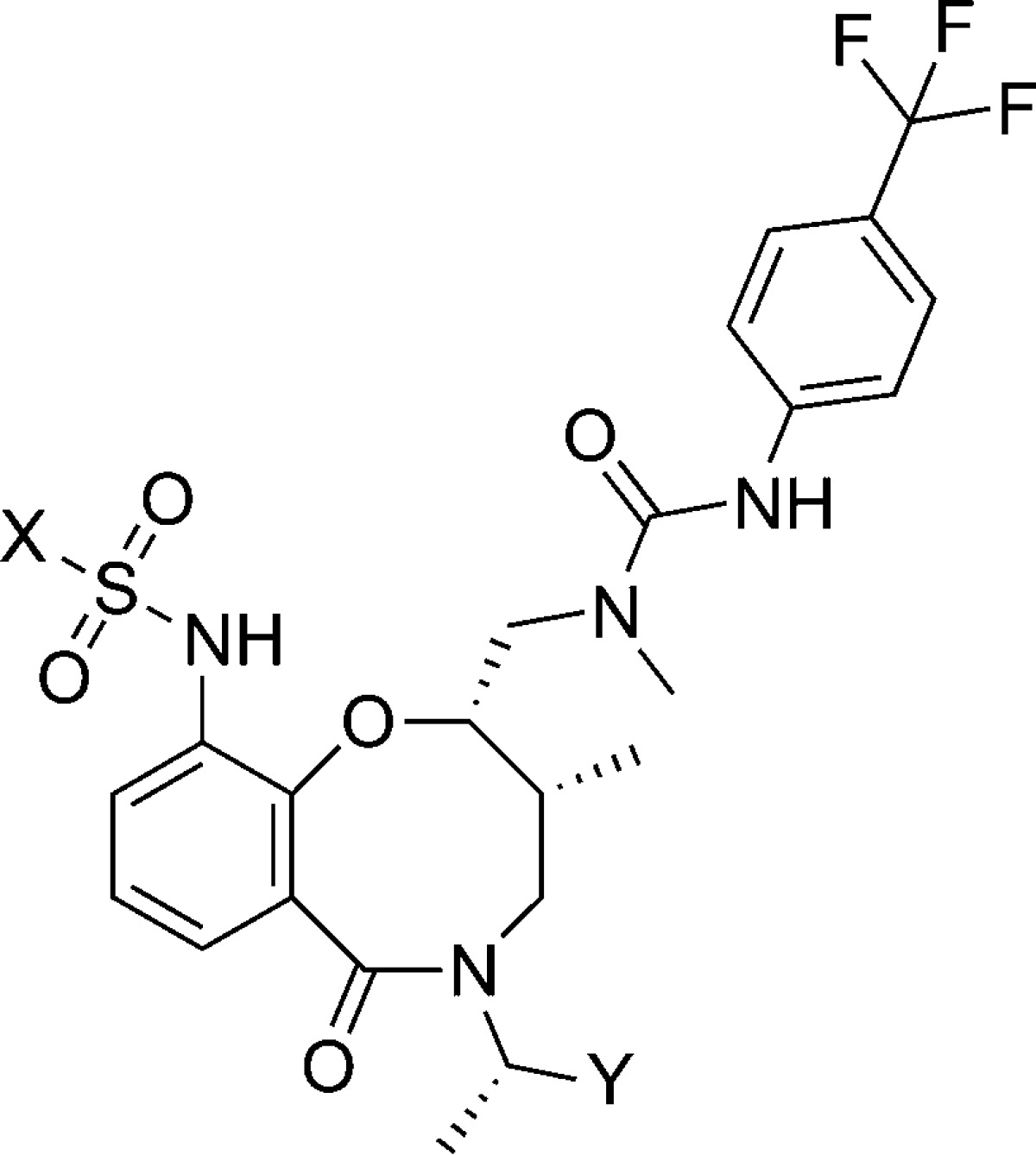
| compd | X | Y | DSC ΔTm (°C) | FP IC50 (μM) |
|---|---|---|---|---|
| 7 | p-F-C6H4 | CO2H | 2.9 | 4.5 |
| 14 | m-F-C6H4 | CO2H | 3.4 | 4.1 |
| 15 | o-F-C6H4 | CO2H | 1.9 | 21.3 |
| 16 | Ph | CO2H | 2.8 | 10 |
| 17 | PhCH2 | CH2OH | 1.4 | >50 |
| 18 | PhCH2CH2 | CH2OH | 0.4 | >50 |
A wider range of sulfonamide substitution patterns retained binding to MCL1 (Table 2). In contrast to the strong dependence on para substitution pattern observed in the urea portion, all three F-phenyl sulfonamide regioisomers (7, 14, and 15) and the unsubstituted phenyl analogue 16 bind MCL1 with similar degree of thermal shift and inhibition of BH3 peptide binding. Chain extension, meanwhile, appears to disfavor ligand binding, as both the benzyl (17) and phenethyl (18) sulfonamide show a smaller ΔTm and no inhibition of BH3 peptide binding.
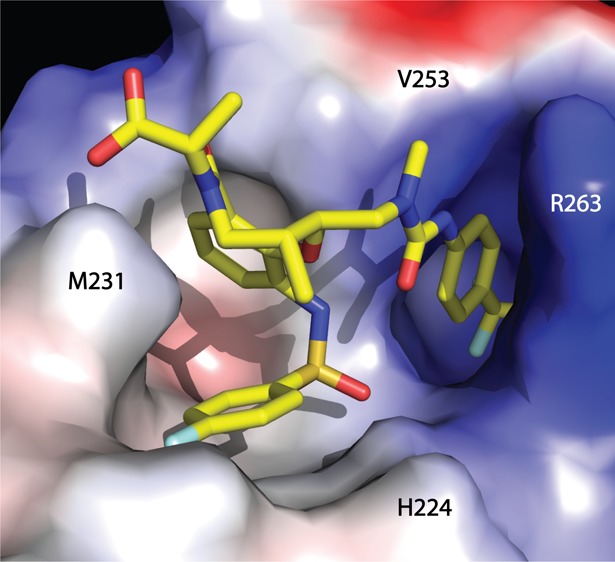
We have recently designed an MCL1 protein construct that can be readily crystallized in the presence and absence of well-validated MCL1 ligands.32 We were able to cocrystallize macrolactam 7 with this construct, and X-ray diffraction was used to generate a high-resolution (1.8 Å) structure of how this novel scaffold binds to MCL1 (Figure 3, PDB entry 4WGI; Supporting Information Table SI-1). We anticipated that the p-CF3-phenyl group of 7 would engage the large hydrophobic pocket near L246 seen in previous MCL1 structures.13−16 However, as 7 did not make a direct interaction with R263 of MCL1, both the arginine side chain, F254, and the surrounding residues 247–260 shift and reveal a new hydrophobic binding pocket (Figure 3). This marks the first cocrystallized ligand that binds MCL1 without engaging R263 via an ionic interaction and reveals a distinct binding mode relative to ligands such as 1. Any interaction with R263 with 7 is more subtle, with a Cδ–H interaction with the p-CF3-phenyl ring potentially contributing to binding. The movement of R263 and the 80 degree rotation of F254 (relative to 4HW2) also demonstrate the conformational flexibility of the MCL1 protein in response to rigid small molecule ligands.
Figure 3.
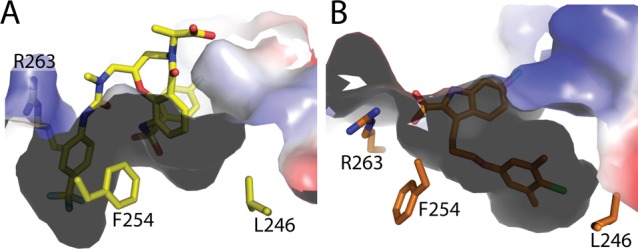
Structure of 7 and MCL1 at 1.8 Å (A) and its comparison to 4HW2 (B) reveals a new hydrophobic pocket. Superposition RMSD of MCL1 and PDB ID 4HW2 chain A, 0.58 Å overall, calculated on all common Cα.
The novel hydrophobic pocket easily accommodates a CF3 substitution at the para position of the phenyl ring; however, substitutions at the meta or ortho positions encounter steric clash with the main chain of MCL1, which explains the lack of binding for 8 and 9 as described in Table 1. The remaining portions of 7 interact with the binding pocket of MCL1 by shape complementarity, making no specific hydrogen bond or π-stacking interactions. This binding mode is consistent with the relative lack of binding enthalpy observed in ITC experiments. Interestingly, the conformation of macrolactam 7 when bound to MCL1 agrees well with the modeled conformation of free ligand, which suggests a low entropic penalty upon binding of 7 within the hydrophobic groove of MCL1.
The competitive binding behavior of compound 7 with regards to BH3 peptide can be examined in more detail by alignment with the NOXA-bound structure PDB ID 2NLA (Figure 4A). The hydrophobic binding pocket induced by the p-CF3 phenyl urea aligns with G82 of NOXA, while the p-F phenyl substituent occupies the space filled by L78 (H2 residue) of NOXA. The displacement of BH3 peptides by compound 7 can thus be explained by at least two perturbations: (1) insertion of a hydrophobic moiety into a region where only glycine is presented by BH3 binding partners; and (2) the mimicry of a BH3 hydrophobic residue by the p-F phenyl substituent of 7.
Figure 4.
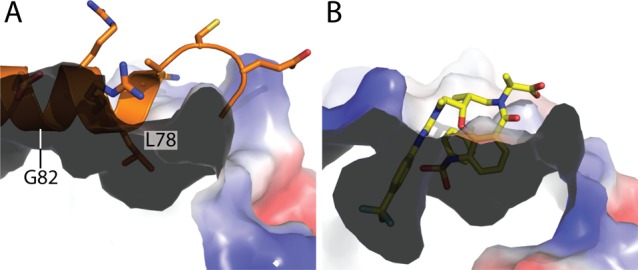
Structure of MCL1 bound to 7 at 1.8 Å aligned to MCL1 bound to the NOXA peptide (PDB ID 2NLA).
Meanwhile, the lipophilicity of macrolactam 7 (clogP = 4.25)33 prompted us to identify the minimal structural elements required for binding to MCL1. The urea and sulfonamide side chains were tested for their ability to bind MCL1 (19 and 20, Figure 5). Surprisingly, even at ligand concentrations up to 1000 μM, we were unable to detect measurable binding of either fragment to MCL1 (see Supporting Information Figure SI-5). Our inability to measure binding of these side chains to MCL1 may be attributed to at least two factors: (1) insufficient sensitivity of our biophysical techniques with respect to the intrinsic solubility of these fragments and/or (2) the requisite display of these side chains from a stereochemically unique, rigid core that enabled their discovery. Thus, scaffold-based display of potential binding elements offers a complementary approach to fragment-based discovery methods.
Figure 5.

Side chain fragments derived from 7 that fail to bind MCL1.
In summary, we have discovered a novel macrolactam that binds to MCL1 in a manner distinct from previously described chemotypes. A suite of biophysical methods reveal the largely entropic mode of binding, which was ultimately confirmed by X-ray crystallography. The selective binding of a single core diastereomer highlights the importance of screening with stereochemically diverse scaffolds to adequately sample three-dimensional space while also suggesting a necessary complement to fragment-based screening methods for tackling difficult biological targets.
Acknowledgments
The authors would like to acknowledge Acme Biopharma for preparing a key intermediate for macrolactam assembly.
Glossary
Abbreviations
- MCL1
myeloid cell leukemia 1
- DSF
differential scanning fluorimetry
- DSC
differential scanning calorimetry
- ITC
isothermal titration calorimetry
- FP
fluorescence polarization
Supporting Information Available
Synthesis and characterization data for all new compounds; biological and computational methods to establish ligand binding. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was conducted as part of the Slim Initiative for Genomic Medicine, a project funded by the Carlos Slim Health Institute in Mexico. Additional support was provided by the Robertson Foundation.
The authors declare no competing financial interest.
Supplementary Material
References
- Adams J. M.; Cory S. The Bcl-2 protein family: arbiters of cell survival. Science 1998, 281, 1322. [DOI] [PubMed] [Google Scholar]
- Cory S.; Adams J. M. The Bcl2 family: regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2002, 2, 647. [DOI] [PubMed] [Google Scholar]
- Youle R. J.; Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat. Rev. Mol. Cell. Biol. 2008, 9, 47. [DOI] [PubMed] [Google Scholar]
- Reed J. C. Double identity for proteins of the Bcl-2 family. Nature 1997, 387, 773. [DOI] [PubMed] [Google Scholar]
- Kelly P. N.; Strasser A. The role of Bcl-2 and its pro-survival relatives in tumourigenesis and cancer therapy. Cell Death Differ. 2011, 18, 1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fesik S. W. Promoting apoptosis as a strategy for cancer drug discovery. Nat. Rev. Cancer 2005, 5, 876. [DOI] [PubMed] [Google Scholar]
- Liu W.; Bulgaru A.; Haigentz M.; Stein C. A.; Perez-Soler R.; Mani S. The BCL2-family of protein ligands as cancer drugs: the next generation of therapeutics. Curr. Med. Chem. Anticancer Agents 2003, 3, 217. [DOI] [PubMed] [Google Scholar]
- Beroukhim R.; Mermel C. H.; Porter D.; Wei G.; Raychaudhuri S.; Donovan J.; Barretina J.; Boehm J. S.; Dobson J.; Urashima M.; McHenry K. T.; Pinchback R. M.; Ligon A. H.; Cho Y. J.; Haery L.; Greulich H.; Reich M.; Winckler W.; Lawrence M. S.; Weir B. A.; Tanaka K. E.; Chiang D. Y.; Bass A. J.; Loo A.; Hoffman C.; Prensner J.; Liefeld T.; Gao Q.; Yecies D.; Signoretti S.; Maher E.; Kaye F. J.; Sasaki H.; Tepper J. E.; Fletcher J. A.; Tabernero J.; Baselga J.; Tsao M. S.; Demichelis F.; Rubin M. A.; Janne P. A.; Daly M. J.; Nucera C.; Levine R. L.; Ebert B. L.; Gabriel S.; Rustgi A. K.; Antonescu C. R.; Ladanyi M.; Letai A.; Garraway L. A.; Loda M.; Beer D. G.; True L. D.; Okamoto A.; Pomeroy S. L.; Singer S.; Golub T. R.; Lander E. S.; Getz G.; Sellers W. R.; Meyerson M. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei G.; Margolin A. A.; Haery L.; Brown E.; Cucolo L.; Julian B.; Shehata S.; Kung A. L.; Beroukhim R.; Golub T. R. Chemical genomics identifies small-molecule MCL1 repressors and BCL-xL as a predictor of MCL1 dependency. Cancer Cell. 2012, 21, 547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B.; Gojo I.; Fenton R. G. Myeloid cell factor-1 is a critical survival factor for multiple myeloma. Blood 2002, 99, 1885. [DOI] [PubMed] [Google Scholar]
- Wertz I. E.; Kusam S.; Lam C.; Okamoto T.; Sandoval W.; Anderson D. J.; Helgason E.; Ernst J. A.; Eby M.; Liu J.; Belmont L. D.; Kaminker J. S.; O’Rourke K. M.; Pujara K.; Kohli P. B.; Johnson A. R.; Chiu M. L.; Lill J. R.; Jackson P. K.; Fairbrother W. J.; Seshagiri S.; Ludlam M. J. C.; Leong K. G.; Dueber E. C.; Maecker H.; Huang D. C. S.; Dixit V. M. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 2011, 471, 110. [DOI] [PubMed] [Google Scholar]
- Belmar J.; Fesik S. W. Small molecule Mcl-1 inhibitors for the treatment of cancer. Pharmacol. Ther. 2014, 10.1016/j.pharmthera.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friberg A.; Vigil D.; Zhao B.; Daniels R. N.; Burke J. P.; Garcia-Barrantes P. M.; Camper D.; Chauder B. A.; Lee T.; Olejniczak E. T.; Fesik S. W. Discovery of potent myeloid cell leukemia 1 (Mcl-1) inhibitors using fragment-based methods and structure-based design. J. Med. Chem. 2013, 56, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petros A. M.; Swann S. L.; Song D.; Swinger K.; Park C.; Zhang H.; Wendt M. D.; Kunzer A. R.; Souers A. J.; Sun C. Fragment-based discovery of potent inhibitors of the anti-apoptotic MCL-1 protein. Bioorg. Med. Chem. Lett. 2014, 24, 1484. [DOI] [PubMed] [Google Scholar]
- Abulwerdi F.; Liao C.; Liu M.; Azmi A. S.; Aboukameel A.; Mady A. S.; Gulappa T.; Cierpicki T.; Owens S.; Zhang T.; Sun D.; Stuckey J. A.; Mohammad R. M.; Nikolovska-Coleska Z. A novel small-molecule inhibitor of Mcl-1 blocks pancreatic cancer growth in vitro and in vivo. Mol. Cancer. Ther. 2014, 13, 565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard D. J.; Lena R.; Bannister T.; Blake N.; Pierceall W. E.; Carlson N. E.; Keller C. E.; Koenig M.; He Y.; Minond D.; Mishra J.; Cameron M.; Spicer T.; Hodder P.; Cardone M. H. Hydroxyquinoline-derived compounds and analoguing of selective Mcl-1 inhibitors using a functional biomarker. Bioorg. Med. Chem. 2013, 21, 6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber S. L. Organic synthesis toward small-molecule probes and drugs. Proc. Natl. Acad. Sci. U.S.A. 2011, 26, 6699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcaurelle L. A.; Comer E.; Dandapani S.; Duvall J. R.; Gerard B.; Kesavan S.; Lee M. D.; Liu H.; Lowe J. T.; Marie J.-C.; Mulrooney C. A.; Pandya B. A.; Rowley A.; Ryba T. D.; Suh B.-C.; Wei J.; Young D. W.; Akella L. B.; Ross N. T.; Zhang Y.-L.; Fass D. M.; Reis S. A.; Zhao W.-N.; Haggarty S. J.; Palmer M.; Foley M. A. An aldol-based build/couple/pair strategy for the synthesis of medium- and large-sized rings: discovery of macrocyclic histone deacetylase inhibitors. J. Am. Chem. Soc. 2010, 132, 16962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe J. T.; Lee M. D.; Akella L. B.; Davoine E.; Donckele E. J.; Durak L.; Duvall J. R.; Gerard B.; Holson E. B.; Joliton A.; Kesavan S.; Lemercier B. C.; Liu H.; Marie J. C.; Mulrooney C. A.; Muncipinto G.; Welzel-O’Shea M.; Panko L. M.; Rowley A.; Suh B.-C.; Thomas M.; Wagner F. F.; Wei J.; Foley M. A.; Marcaurelle L. A. Synthesis and profiling of a diverse collection of acetidine-based scaffolds for the development of CNS-focused lead-like libraries. J. Org. Chem. 2012, 77, 7187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerald B.; Duvall J. R.; Lowe J. T.; Murillo T.; Wei J.; Akella L. B.; Marcaurelle L. A. Synthesis of a stereochemically diverse library of medium-sized lactams and sultams via SNAr cycloetherification. ACS Comb. Sci. 2011, 13, 365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovering F.; Bikker J.; Humblet C. Escape from flatland: increasing saturation as an approach to improving clinical success. J. Med. Chem. 2009, 12, 6752. [DOI] [PubMed] [Google Scholar]
- Niesen F. H.; Berglund H.; Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2007, 2, 2212. [DOI] [PubMed] [Google Scholar]
- Senisterra G. A.; Finerty P. J. High throughput methods of assessing protein stability and aggregation. Mol. Biosyst. 2009, 5, 217. [DOI] [PubMed] [Google Scholar]
- Sturtevant J. M. Biochemical applications of differential scanning calorimetry. Annu. Rev. Phys. Chem. 1987, 38, 463. [Google Scholar]
- Weber P. C.; Salemme F. R. Applications of calorimetric methods to drug discovery and the study of protein interactions. Curr. Opin. Struct. Biol. 2003, 13, 115. [DOI] [PubMed] [Google Scholar]
- Jelesarov I.; Bosshard H. R. Isothermal titration calorimetry and differential scanning calorimetry as complementary tools to investigate the energetics of biomolecular recognition. J. Mol. Recognit. 1999, 12, 3. [DOI] [PubMed] [Google Scholar]
- Leavitt S.; Freire E. Direct measurement of protein binding energetics by isothermal titration calorimetry. Curr. Opin. Struct. Biol. 2001, 11, 560. [DOI] [PubMed] [Google Scholar]
- Ladbury J. E.; Chowdhry B. Z. Sensing the heat: the application of isothermal titration calorimetry to thermodynamic studies of biomolecular interactions. Chem. Biol. 1996, 3, 791. [DOI] [PubMed] [Google Scholar]
- Oxidation of 6 with Parikh–Doering conditions gave a 1:1 mixture of epimers for the corresponding aldehyde.
- Vulpetti A.; Hommel U.; Landrum G.; Lewis R.; Dalvit C. Design and NMR-based screening of LEF: a library of chemical fragments with different local environment of fluorine. J. Am. Chem. Soc. 2009, 131, 12949. [DOI] [PubMed] [Google Scholar]
- Jordan J. B.; Poppe L.; Xia X.; Cheng A. C.; Sun Y.; Michelsen K.; Eastwood H.; Schnier P. D.; Nixey T.; Zhong W. Fragment based drug discovery: practical implementation based on 19F NMR spectroscopy. J. Med. Chem. 2012, 55, 678. [DOI] [PubMed] [Google Scholar]
- Clifton M. C.; Dranow D. M.; Leed A.; Fulroth B.; Fairman J. W.; Abendroth J.; Atkins K. A.; Wallace E.; Fan D.; Xu G.; Ni Z. J.; Daniels D.; Van Drie J.; Wei G.; Burgin A. B.; Golub T. R.; Hubbard B.; Serrano-Wu M. H. A maltose-binding protein fusion construct yields a robust crystallography platform for MCL1. Submitted. [DOI] [PMC free article] [PubMed]
- cLogP was calculated using ChemAxon.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.