

Abstract
In this article, we describe and show the application of some of the most advanced fluorescence superresolution techniques, STED AFM and STORM AFM microscopy towards imaging of cytoskeletal structures, such as microtubule filaments. Mechanical and structural properties can play a relevant role in the investigation of cytoskeletal structures of interest, such as microtubules, that provide support to the cell structure. In fact, the mechanical properties, such as the local stiffness and the elasticity, can be investigated by AFM force spectroscopy with tens of nanometers resolution. Force curves can be analyzed in order to obtain the local elasticity (and the Young's modulus calculation by fitting the force curves from every pixel of interest), and the combination with STED/STORM microscopy integrates the measurement with high specificity and yields superresolution structural information. This hybrid modality of superresolution-AFM working is a clear example of correlative multimodal microscopy.
Keywords: fluorescence, superresolution, STED AFM, correlative microscopy, STORM AFM
Introduction
Microscopy coexists with different biophysical techniques, which paved avenues for routing cellular and molecular biophysics into new directions and dimensions. The past two decades contributed to an outbreak of new optical methods, performing at the nanoscale, “to see” clearer, faster and to be more exact in the interpretations [Diaspro 2010,2011]. Fluorescence microscopy is one of the most relevant implementations in microscopy, allowing biophysical observations with a high degree of repeatability and reliability. Fluorescently tagged proteins and proteins that fluoresce themselves have opened up a new world full of images that depict the story of proteins with very good event localization [Lippincott-Schwartz et al., 2001; Hell et al., 2004]. Spatial resolution is physically limited by diffraction that can be circumvented as modern superresolution approaches demonstrate. The resolving power, as described by G.Toraldo di Francia [di Francia 1952], is not a well defined physical quantity and the fact that today, one is capable of imaging at subdiffraction resolution regime is related to the role of an important actor as the fluorescent probe. We will use the term superresolution, as commonly done, in a not formal way to point out that the spatial information can be accessed at a scale finer than 250 nm, this being the practical resolution limit in optical microscopy. Again, the Toraldo di Francia concept of superresolution [Sheppard 1988] is related to the fact that the information capacity [di Francia 1955; Cox and Sheppard 1986], which is the key-role for optical nanoscopy and superresolution methods, plays the control of at least two states—bright/dark, red/blue—of the probes being used. Fluorescence optical microscopy is still unique in offering three-dimensional imaging [Bianco and Diaspro 1989; Diaspro et al., 1996] of biological specimens [Diaspro et al., 1990]. Contemporary light microscopy methods [Schermelleh et al., 2010] include near-field methods like Near field Scanning Optical Microscopy [Betzig and Trautman 1992], illumination schemes like Structured Illumination Microscopy [Gustafsson 2000] and other far-field methods. Among them, the switching based technique RESOLFT (Reversible saturable optical fluorescence transitions) [Hofmann et al., 2005], STED (STimulated Emission Depletion Microscopy) [Hell and Wichmann 1994] and single molecule localization stochastic methods like STORM (STocastic Optical Reconstruction Microscopy) [Rust et al., 2006; Heilemann et al., 2008], PALM (Photo Activated Localization Microscopy) [Betzig et al., 2006; Hess et al., 2006], and other methods [Diaspro 2010; Hell 2007] are prominent. We aim to integrate fluorescence microscopy methods that allow gaining speed, imaging depth [Lavagnino et al., 2013] and more freedom and access in imaging a sample [Cella Zanacchi et al., 2011]. As well, we focused on integrating complementary techniques that could be used to get unequivocal datasets, thence removing artefacts [Harke et al., 2012] and providing biomanipulation at the nanoscale [Chacko et al., 2013]. The conception of a novel cellular level superresolution imaging technique is often demonstrated by imaging tubulin structures because of their structural splendour and sub resolution width (<250 nm) which poses a challenge for the technique.
Superresolution disciplines revealed details on cytoskeletal features with an impetus on the technique's ability of extracting new information, which is not possible by the conventional wide field imaging or confocal microscope. Recent advances within the superresolution field, such as multicolour and 3D imaging in a noninvasive manner at the nanometer scale makes these techniques suitable tools for imaging cytoskeletal structures in mammalian cells ranging from microtubules, actin filaments, intermediate filaments (keratin) and neurofilaments. For example, 3D imaging of actin has been performed using both STORM [Huang et al., 2008b; Xu et al., 2013] and PALM [Subach et al., 2010], stochasticity based superresolution techniques and structured illumination techniques [Zobel and Bogdan 2013]. STED microscopy has proven to be a golden solution to image keratin filaments [Vicidomini et al., 2011], neurofilaments [Urban et al., 2011] and primary cilia [Yang et al., 2013]. Furthermore in the line of STED, the key role played by cytoskeletal transient organization in signalling pathway has been recently investigated using STED microscopy [Tamarit et al., 2013]. Combined concepts in single molecule techniques, for example to track and image, enable us to watch live transport trajectories of lysosome on microtublin structures [Bálint et al., 2013]. Integrating alternative approaches like Fluorescence Correlation Spectroscopy into STED nanoscopic approaches proved advantageous in establishing cytoskeleton influenced membrane interactions [Mueller et al., 2011]. STED microscopy can also perform 3D resolution enhancement [Hein et al., 2008] which can be helpful to gain information from topmost structures and isolate them for a better correlation with surface imaging.
Although optical techniques open ways to identify individual fibers and their interactions with surroundings, another appealing and popular method falls in the class of surface-probing microscopy, which reads the cytoskeletal features from the topology of the cell. A feasible, established approach in this regime to reach live cells is to use an atomic force (AFM) or scanning force microscope [Diaspro and Rolandi, 1997; Guo et al., 2012]. It had been shown that cytoskeletal features can be recognized and from their elastic properties stress fibers can be identified [Hofmann et al., 1997]. Even if AFM combines a nanometer-scale resolution with the ability to image biomolecular interactions in liquid environment it has not very often been used to study cytoskeleton structures due to tipi-filaments interaction problems [Hamon et al., 2010]. As well, total internal reflection fluorescence (TIRF) allows to study cellular events localized at the basal plasma membrane of adherent cells [Axelrod 2001] providing quantitative information on the organization of the cytoskeleton in cells [Grigoriev and Akhmanova 2010]. Coupling AFM with a superresolution method like STED could contribute to understand those mechanisms related to adhesion, mechanical forces sensing [Trache and Lim 2010] and how cells respond and adapt to a variable environment. In the past, an AFM was brilliantly integrated with TIRF microscopy and fast-spinning disk confocal microscopy [Grigoriev and Akhmanova 2010]. Significant rearrangement of the actin filaments and focal adhesions were demonstrated pushing on the need of merging innovative techniques to get a better understanding about cell restructuring and dynamics in response to mechanical forces. Our group successfully showed that the combined visualization of such a superresolution technique and a surface probing technique can open up different sorts of interpretations on force/elasticity maps [Harke et al., 2012].
In this article, we describe approaches, discuss advantages and limitations of the techniques, suitable to image cytoskeletal structures, introducing both STORM and STED as superresolution methods and integrated modalities including STORM AFM and STED AFM to retrieve multidimensional data. It is worth noting that coupling AFM with STED or other superresolution optical methods allows for design of a new class of experiments, towards answering unresolved biological questions.
Instrument
STORM Microscope
STORM imaging is performed using the NIKON N-STORM microscope. The system is equipped with four laser lines (Cube 405 nm Coherent Inc., Sapphire488 Coherent Inc., Sapphire561 Coherent Inc. and a readout laser 647 nm, MPB Communications Inc.) which were used for excitation of the reporter dye. The activation and the readout lasers can be maintained continuously running or either sequentially switched on in order to photo activate and image a sparse subset of molecules. To reduce the background signal due to the out of focus contributions, the system can work under a conventional wide field scheme, or under an inclined illumination or a TIRF regime. An EMCCD camera (Ixon 897BV, Andor Technology) at a frame rate of 50 Hz was used to collect fluorescence emission from single events. The position of each single molecule is determined through a Gaussian fitting procedure and the molecule positions are localized with a precision of 20–30 nm. Filtering based on the brightness and size of the imaged molecules allows us to reject aggregates' signal.
STED Microscope
The STED microscope is custom built as previously documented by our group [Harke et al., 2012]. Although STED is detailed and implemented in different architectures in our lab [Ronzitti et al., 2013; Bianchini et al., 2012; Galiani et al., 2012], we used a classic pulsed-STED setup because of the ready availability of pulsed laser within a multiphoton microscope setup (A1R-MP, Nikon Instruments) in use at our laboratory. Realizing a STED setup includes phase engineering a deexcitation beam (STED beam) in the shape of a doughnut and switching off fluorescence excitation to visualize the isolated fluorescence emission from subdiffraction scaled centre of the doughnut. This is made possible with help of a vortex phase plate (RPC photonics, Rochester, NY) which makes a helical phase on a Gaussian beam and suitably stretching the laser pulse from the femtosecond laser (Ultra II, Chameleon, Coherent Inc) to ensure complete shutting down of the fluorescence. We used a fiber coupled picosecond laser diode working at 637 nm to do the excitation (PDL800, Picoquant). Our dye of selection for these measurements was Abberior star 635P (Abberior GmbH, Germany) which is suitable for 635 nm excitation and 760 nm depletion wavelengths.
STORM AFM
A commercial AFM unit (JPK systems, Germany), was mounted on the N-STORM unit, which was used to study the cell topology and measure the force maps. We used a cantilever (NP-10, Bruker) for the measurements on fibroblast cells.
STED AFM
The above mentioned AFM unit was mounted on the optical setup [Harke et al., 2012], which was used to study the cell topology using a cantilever featured as sharp stylus (CSC38, Mikromasch) and to measure the force map measurements on Hela cells.
Methods
Sample Preparation
Cell Lines and Culture Conditions
Human Hela cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen) supplemented with 10% fetal bovine serum (FBS) (Invitrogen), 100 U/ml penicillin and 100 µg/ml streptomycin (pen/strep, Invitrogen) in a humidified 5% CO2 balanced-air atmosphere at 37°C. Hela cells were passaged at 80% confluency, split 1:7 in fresh media.
Immunofluorescence
For immunofluorescence assays, cells were plated on an 18-mm-diameter dish and cultured overnight in the maintenance medium. The next day, cells were rinsed three times with phosphate-buffered saline (PBS)(0.1 M, pH 7.4) and fixed with a combination of paraformaldehyde (3%) and glutaraldehyde (0.1%) in PBS for 15 min at room temperature. After fixation, the cells were washed three times in PBS for 5 min each and preincubated for 30 min at room temperature in PBS containing 3% normal bovine serum albumin and 0.1% Triton-X-100 to block nonspecific binding before the antibody treatment. The cells were then incubated with the monoclonal mouse anti-α-tubulin antiserum (Sigma Aldrich, St. Louis, MO) diluted in the blocking buffer (1:1000) for 1 h at room temperature. Cells were rinsed three times in PBS for 5 min before the secondary antibody incubation. α-tubulin antibody was revealed using Alexa Fluor 647 goat anti-mouse IgG (1:500, Molecular Probes, OR) for STORM imaging and using Abberior Star 635P goat anti-mouse IgG (1:1000, Abberior, Gottingen, Germany) for STED imaging. Finally, the cells were rinsed three times in PBS for 5 min and mounted in PBS medium. Cells were maintained in 20 mM sodium azide in PBS for long-term storage.
Imaging Buffer
Buffer used for STORM Measurement
Immediately before imaging cells, they were embedded in a suitable imaging buffer, freshly prepared before the use. An oxygen scavenger system is obtained by dissolving 14 mg of Glucose Oxidase in 200 µl 10 mM Tris (pH 8.0) supplemented with 50 mM NaCl and a gentle vortex. A 50 µl of catalase was added to Glucose Oxidase solution (GLOX) and centrifuged at 14000 rpm and only the supernatant of the GLOX solution is used. A MEA solution is prepared by dissolving 77 mg of MEA (Cysteamine) in 1 ml of 0.30N HCl. The final buffer solution is composed of 620 µl saline buffer, 7 µl GLOX solution and 70 µl MEA solution.
Microscope Mounting
STORM Imaging
The stained HeLa cells were plated on glass cover slips (18 mm diameter) and sealed in a Quick Change Chamber (Warner Instruments—18 mm Low Profile) in order to perform quick exchange of the imaging buffer solution. Stained fibroblast cells were plated on cover slips (18 mm) and used for STORM AFM measurements with above-mentioned STORM imaging buffer.
STED Imaging
The fixed and stained Hela cells were sealed with an embedding medium (Prolong Antifade reagent with DAPI, Molecular Probes, OR, USA). With this enclosure, we eliminate the option of topological maps on the sample, but it helps in keeping the cells for a longer time without fading and is suitable for only imaging. The STED alone images shown in this article are procured on these fixed, sealed samples. STED AFM measurements were carried out on unsealed cover slips with PBS buffer
AFM Imaging
AFM measurements were all done after mounting the samples on AFM cover slip holder (JPK Systems Germany) which allows us to work with high numerical aperture (NA) objectives for both STED and STORM. This chamber supports wet environment working of the AFM and hold the glass cover slip with an optional passage of PBS or CO2, aimed for long time recordings. Because of the fast targeting capability by the fluorescence microscope attached, all the measurements were done in a single buffer without any passaging.
Results
Individual Molecule Localization Microscopy
The resolution improvement provided by localization based techniques, such as STORM, dSTORM and PALM [Betzig et al., 2006; Hess et al., 2006; Rust et al., 2006; Heilemann et al., 2008; Fölling et al., 2008] does not rely on an effective reduction of the PSF size but it exploits the stochastic temporal separation between the emission of neighboring molecules. A spectral separation, induced by the photo activation process or by the transition to a dark metastable state, is used to collect photons emitted by a sparse subset of molecules. This allows precise localization of the molecule position. Despite different names and acronyms used for such class of superresolution techniques, we use the term individual molecule localization (IML) for generally referring to all of them. The highest localization precision theoretically achievable is related to the Cramér-Rao lower bound and is independent from the localization algorithm used. In case the emission of a single molecule could be described by the Gaussian approximation, the best localization precision achieved is given by:
where ‘ ' is the localization precision, ‘s' is the width of the point spread function and ‘N' the number of photons/molecule. As schematically shown in Fig. 1A, when a sufficient number of photons/molecule are collected the fluorophore's position can determined with a precision 10 times higher than the diffraction limit. A superresolution image can be obtained by repeating the localization process and mapping all the positions of sufficient number of molecules. Specifically, the localization precision strictly depends on the number of photons collected for each molecule and, for typical exposure times (20–30 ms), the value reached is around 20—30 nm. Such a value, which is approximately tenfold below the diffraction limit, guarantees a significant improvement of the radial resolution towards the investigation of cytoskeletal structures. Still, a high radial resolution itself does not permit the observation of sub resolved cellular structures if it is not coupled with a sub diffraction axial localization capability too. To this end several solution based on the point spread function(PSF) shaping, ranging from dual plane [Juette et al., 2008], astigmatism [Huang et al., 2008a], to double helix point spread function [Lee et al., 2012], have been successfully implemented to push the axial localization down to 60 nm. For 3D imaging in our STORM approach, a cylindrical lens is inserted in the detection path in order to discriminate the axial position of each molecule based on the ellipticity of the PSF. A 3D intensity distribution obtained by imaging single molecules or fiducial fluorescent markers, which is used to extract calibration curves in which the x and y coordinates are made explicit in terms of the z coordinate. The maximum localization precision reached is ∼20 nm along the radial direction and ∼60 nm along the axial one as demonstrated [Huang et al., 2008a,b]. 3D STORM imaging allows us to observe cytoskeletal structures in mammalian cells such as microtubules as shown in Fig. 1. Here, α-tubulin is immunostained with Alexa 647 anti-mouse secondary antibodies in order to perform direct STORM imaging on conventional fluorophores. The comparison between the conventional wide field image (Fig. 1B) and the superresolution image obtained using 3D storm (see Fig. 1C) shows a drastic resolution increase allowing a precise localization of microtubules in all three dimensions. The total number of frames acquired to collect the final storm image was 1000 frames and the exposure time for each frame was 20 ms. Thanks to the advent of photo-switchable dyes and photo-activatable proteins within a wide spectral range, this technique provides a comfortable solution for multicolor applications too [Bates et al., 2007]. Furthermore, several implementations of localization based techniques, based on array tomography, light sheet microscopy [Cella Zanacchi et al., 2011] and two photon excitation [Cella Zanacchi et al., 2013], have been recently developed to increase the imaging depth capabilities thus extending the application area of superresolution techniques to thicker samples.
' is the localization precision, ‘s' is the width of the point spread function and ‘N' the number of photons/molecule. As schematically shown in Fig. 1A, when a sufficient number of photons/molecule are collected the fluorophore's position can determined with a precision 10 times higher than the diffraction limit. A superresolution image can be obtained by repeating the localization process and mapping all the positions of sufficient number of molecules. Specifically, the localization precision strictly depends on the number of photons collected for each molecule and, for typical exposure times (20–30 ms), the value reached is around 20—30 nm. Such a value, which is approximately tenfold below the diffraction limit, guarantees a significant improvement of the radial resolution towards the investigation of cytoskeletal structures. Still, a high radial resolution itself does not permit the observation of sub resolved cellular structures if it is not coupled with a sub diffraction axial localization capability too. To this end several solution based on the point spread function(PSF) shaping, ranging from dual plane [Juette et al., 2008], astigmatism [Huang et al., 2008a], to double helix point spread function [Lee et al., 2012], have been successfully implemented to push the axial localization down to 60 nm. For 3D imaging in our STORM approach, a cylindrical lens is inserted in the detection path in order to discriminate the axial position of each molecule based on the ellipticity of the PSF. A 3D intensity distribution obtained by imaging single molecules or fiducial fluorescent markers, which is used to extract calibration curves in which the x and y coordinates are made explicit in terms of the z coordinate. The maximum localization precision reached is ∼20 nm along the radial direction and ∼60 nm along the axial one as demonstrated [Huang et al., 2008a,b]. 3D STORM imaging allows us to observe cytoskeletal structures in mammalian cells such as microtubules as shown in Fig. 1. Here, α-tubulin is immunostained with Alexa 647 anti-mouse secondary antibodies in order to perform direct STORM imaging on conventional fluorophores. The comparison between the conventional wide field image (Fig. 1B) and the superresolution image obtained using 3D storm (see Fig. 1C) shows a drastic resolution increase allowing a precise localization of microtubules in all three dimensions. The total number of frames acquired to collect the final storm image was 1000 frames and the exposure time for each frame was 20 ms. Thanks to the advent of photo-switchable dyes and photo-activatable proteins within a wide spectral range, this technique provides a comfortable solution for multicolor applications too [Bates et al., 2007]. Furthermore, several implementations of localization based techniques, based on array tomography, light sheet microscopy [Cella Zanacchi et al., 2011] and two photon excitation [Cella Zanacchi et al., 2013], have been recently developed to increase the imaging depth capabilities thus extending the application area of superresolution techniques to thicker samples.
Fig. 1.
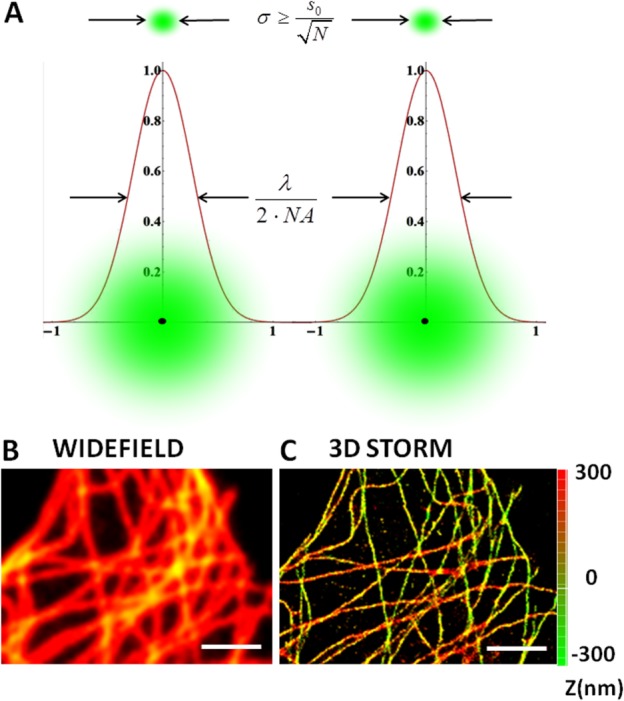
Direct 3D STORM images of microtubule network in Hela cells. Panel A illustrates the principle of localization precision (σ) which replaces diffraction limit by precisely points out the single molecule position by a Gaussian fit over a number of frames (N). In panels B and C, α-tubulin is immunostained with Alexa 647 anti-mouse secondary antibody in order to perform direct STORM imaging. The comparison between the conventional wide field image (1B) and the superresolution image obtained using 3D STORM (1C).The axial position is represented as a Z-color coded map and the scale bar is 2 µm. Image was reconstructed from 1000 frames of 20 ms exposure time to reach a localization precision ∼20 nm along the radial direction and ∼60 nm along the axial one.
STED Microscopy
STED microscopy provides immediate and direct tunable resolution enhancement [Harke et al., 2008] by carving a subdiffraction PSF from the diffraction limited confocal PSF. A selective removal of fluorescence by masking the diffraction limited fluorescence using a fluorescence depletion laser (STED laser) is the working principle of the technique [Klar et al., 2000; Hell et al., 2006]. It's adaptability to address imaging as well as spectroscopy has been well discussed in scientific literature [Eggeling et al., 2008; Vicidomini et al., 2011]. The final resolution gained in STED is tunable by controlling the depletion laser power [Westphal and Hell 2005]. The cartoons in Figs. 2A and 2B show the effective PSF observed as we increase the depletion power. We demonstrate STED imaging of microtubules structure to show that the resolution enhancement is simply power dependent and the observations can be better resolved by simply increasing the power of the depletion laser (Figs. 2D–2F). These fluorescence images are raw outputs without any image processing to prove that the resolution steering is a straightforward process. The inset zoom images and the adjoin line profiles show how individual microtubules can be separated. Same excitation laser power and same PMT gain settings were used for all the images. With this measurement, we exert the fact that STED imaging is a direct resolution enhancement option from confocal imaging. The brightness ranges for the individual images are adjusted to visualize which are shown alongside the color scale. The photons per pixel in the STED image looks lower because of the clustering effect of signal, previously described in literature [Kubitscheck 2013]. The temporal order of measurements is like 2E, 2C, 2F, 2D and hence the average brightness in 2D is lesser in Fig. 2E due to possible photo bleaching of the sample. Although the brightness seems fading by multiple scans, the increasing sharpness of the structures in the images is evident.
Fig. 2.
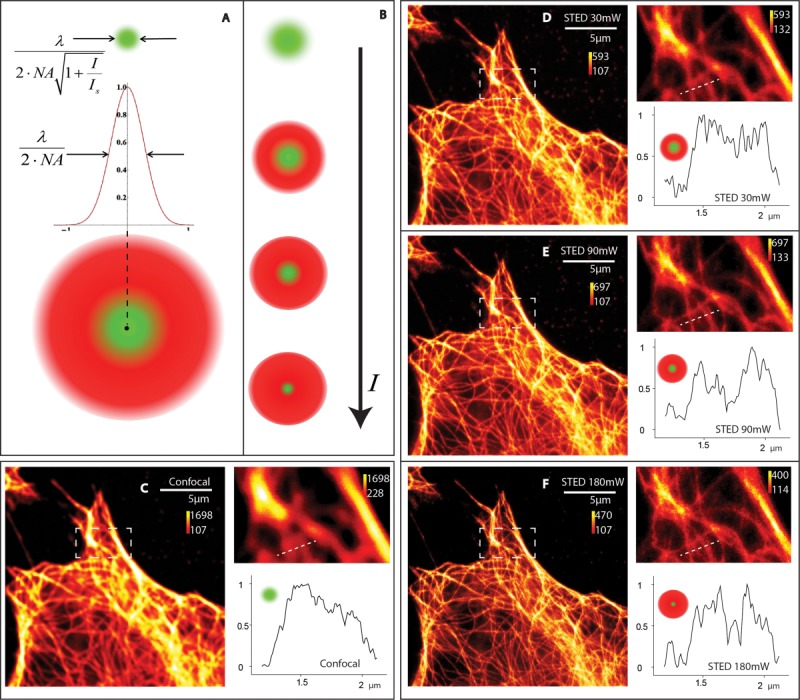
STED microscopy and its tunable resolution. Panel A illustrates how confocal and STED PSF overlay to give a smaller PSF and Panel B shows how this effective confocal PSF gets smaller as we increase the depletion power. Here λ is the fluorescence wavelength, NA the numerical aperture, I the light intensity and Isat is the saturation intensity. Panel C is the confocal image with a zoomed-in inset and a selected line profile of a dense tubulin structure. Panels D–F show the STED images made from the same area as we increase the power to 30 mW, 90 mW and 180 mW. The sample was a fixed Hela cell stained with Abberior Star 635P.
The conventional confocal imaging scheme is slowly being replaced by STED because of their very similarity in operation. The ability of STED imaging to work at the same speed and settings of confocal is made use in most of the labs to gain resolution by necessary modifications in their setup. We demonstrate this working versatility of STED by measuring a 3D stack shown in Supporting Information videos video1 (STED) and video2(confocal). An additional Z-stack video of DAPI stained nucleus of the same cell is also given as Supporting Information video video3(DAPI). A cell in a frame of 42 × 42 µm2 has been imaged for 46 steps of Z-axis spaced 120 nm apart, captured at the very same rate for confocal and STED. Can be noticed that the brightness of the STED images are a little lower than the confocal, though this can be compensated by a slower scanning and tuning depletion power for desired resolution. Figure 3 shows complementary information to the video content. Figure 3A shows the normalized cumulative histogram from all the z-stack. This histogram also shows that STED images are relatively dimmer than the confocal images, which is expected because of the isolation of total fluorescence into more number of pixels [Kubitscheck 2013]. Figure 3B shows the top and bottom slices of the cell where STED and confocal works similarly, but only with an higher resolution. Although the improvement in resolution can be appreciated throughout the sample slices (See Supporting Information video), color scales are adjusted to improve an increase in contrast in Fig. 3B. Figure 3C is a 3D visualization of the cell where α-tubulin is labeled (red) and nucleus are stained with DAPI (blue).
Fig. 3.
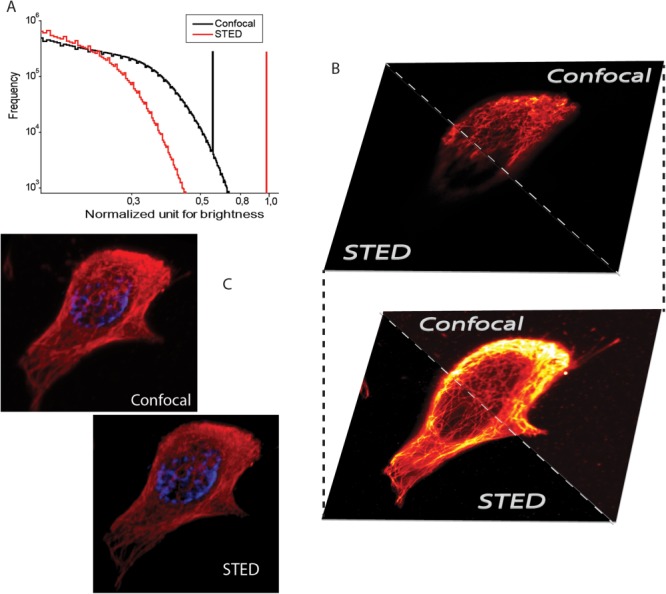
3D STED and Confocal imaging. STED, Confocal histogram from microtubule structure of a hell cell stained with Abberior Star 635P. Panel A shows a cumulative histogram from STED and Confocal showing the apparent decrease in brightness. Panel B shows the top and bottom slices of the cell in both STED and confocal modality. Panel C shows conventional 3D visualization of confocal and STED. These images are provided to show the STED imaging has the ability to redo the confocal without any changes in the scan settings.
Coupling AFM and Superresolution Techniques
A correlative approach based on coupling far-field fluorescence superresolution techniques with AFM, allows us to perform experiments, which are able to combine topological information, local stiffness measurements and specific fluorescence imaging. Here we discuss the possibility to image cytoskeletal structures by combined AFM and superresolution approaches. Both STED and STORM approaches can be exploited to gain a direct and specific determination of the species with a lateral resolution in the range of the AFM. STED AFM working has been previously described in literature and was used for targeted nanomanipulation and targeted force maps of interested regions of a cell [Harke et al., 2012; Chacko et al., 2013]. Here we would like to demonstrate the limitations and advantages of the setup when working on a thicker cell sample. As seen in Fig. 4, confocal and STED images are compared for a single cell (4A, 4B) with an inset to demonstrate the resolution difference. The large scan area might hide the sharpness of the image. The area marked in 4B is imaged with AFM (Fig. 4C) and a detailed force mapping is done in the marked area of 4C as shown in (Figs. 4D and 4E). Young's moduli map shows the elasticity of the cell surface and for a fixed cell, arising from actin, tubulin, and other mechanically contributing elements of the cell seen close to the cell wall. When the cell is thick, AFM can see only the topology while STED can browse through the sample. We illustrate this fact through the slices of the Z stack image of α-tubulin in Fig. 4D. Figure 4H shows four representative slices from a Z stack (15 sections) within 3 µm range. We used a view space rendering (VolumeJ, ImageJ) to visualize the Z-stack into a 3D microtubule structure shown in Fig. 4G. Comparing the 3D image (Fig. 4G) to the topology of the cell shown in Fig. 4F, it is evident that the topology gets difficult to be interpreted in a thick sample, nonetheless the upper layers of tubulin mesh has a better correspondence to the topology.
Fig. 4.
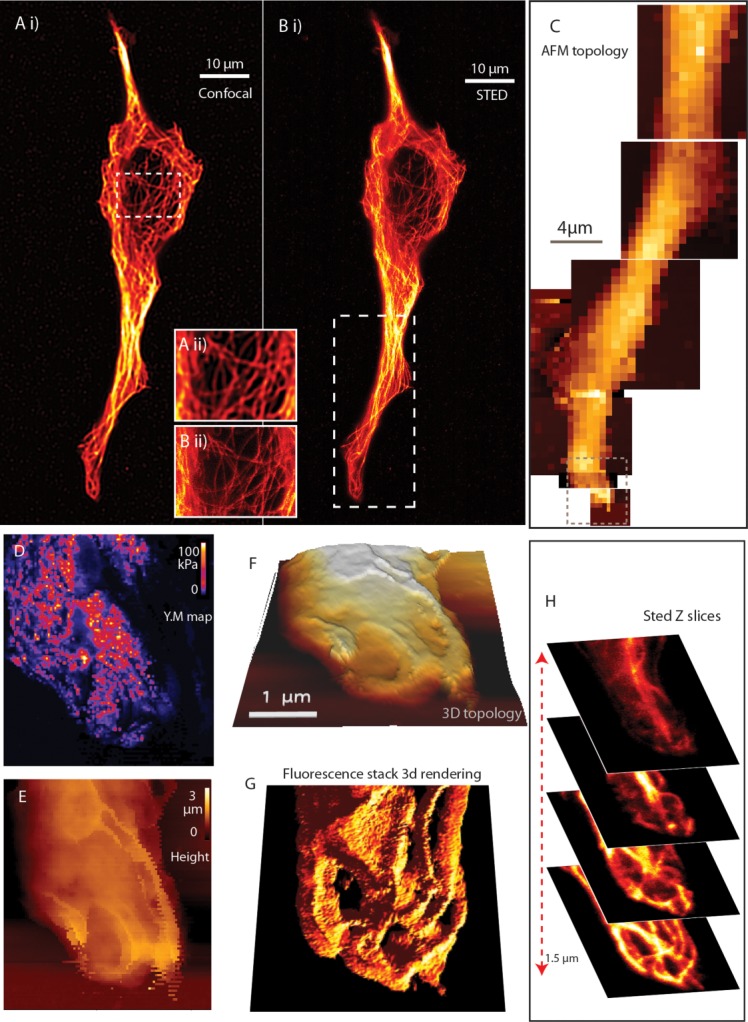
STED AFM. Panels A and B show STED and confocal aided with zoomed in frames to show that the image sharpness. Panel C is a mosaic of coarse AFM scan over a part of the cell to find suitable areas for probing. The demonstrated area in D and E by a force map, its topology can be visualized in 3 dimensions in F. Panel H shows representative four slices from the 3D stack made by STED, where the topology shows resemblance to the topmost layer and the underlying layers are hidden for surface probing, while STED reveals the mesh work. Panel G shows a 3D rendering of the Z-stack for easier comparison with the topology map panel F.
The nature of STORM technique, able to provide noninvasive subdiffraction imaging like STED, demands for a STORM-AFM coupling [Anne et al., 2013] which should give advantages as in the STED AFM case. There is a wide interest in high resolution pinpointing of structures and fluorescence signatures in order to understand an AFM image made on cell wall elongation and identifying cytoskeletal structures [Turner et al., 2013]. This can be carried out with STORM AFM, because of its adored adaptive ability to perform multicolor and 3D imaging without big changes in the microscope setup. These approach can be extensively applied for inorganic structures and new localization methods like surface enhanced fluorescence photo localization microscopy SEF-PALM [Lin et al., 2012]. Here we demonstrate how STORM AFM unveils microtubule structure in a fibroblast cell. Although actin structure is well studied and represented by morphological studies, we chose tubulin in order to give a comparison to STED AFM images. Because of the higher sensitivity to local mechanical properties, mammalian cells which are soft can give delusive results [Dufrêne and Pelling 2013]. This is made difficult due to a softer cell wall, complete cytoskeletal architecture consisting of tubulin, actin, vimentin, other intermediate filaments and the adhesion to extracellular matrix [Dufrêne and Pelling 2013; Takai et al., 2005].
In Fig. 5A, we show AFM overlaid on a cell of interest, followed by highly magnified image Fig. 5B where microtubules close to cell periphery is chosen for detailed examination. Figure 5C shows the AFM image of the respective cell and inset images 5C.i and 5C.ii shows two selected areas of the cell in which the details of cytoskeleton can be seen eg: separation of a fiber bundle in Fig. 5C.ii. The nonlabeled filament can be seen in the AFM while only a background illumination is visible in the fluorescence image(shown by arrows in Figs. 5B and 5C). In Figs. 5D and 5E, wide field and STORM images of the same area for showing tubulin structure localized by individual α-tubulin proteins. For an ideal comparison of size and shape an AFM morphological image is shown in Fig. 5F overlaid on the STORM image of the same area. Although parts of the microtubules structure can be seen, the evident ambiguity of the measurement is due to the presence of other filaments and structures. We selected an area of the cell for the AFM force map from the coarse topology measured on the cell. The fluorescence STORM image of individual tubulin molecules, topological map and the force map are compared in Fig. 5G–5I, respectively.
Fig. 5.
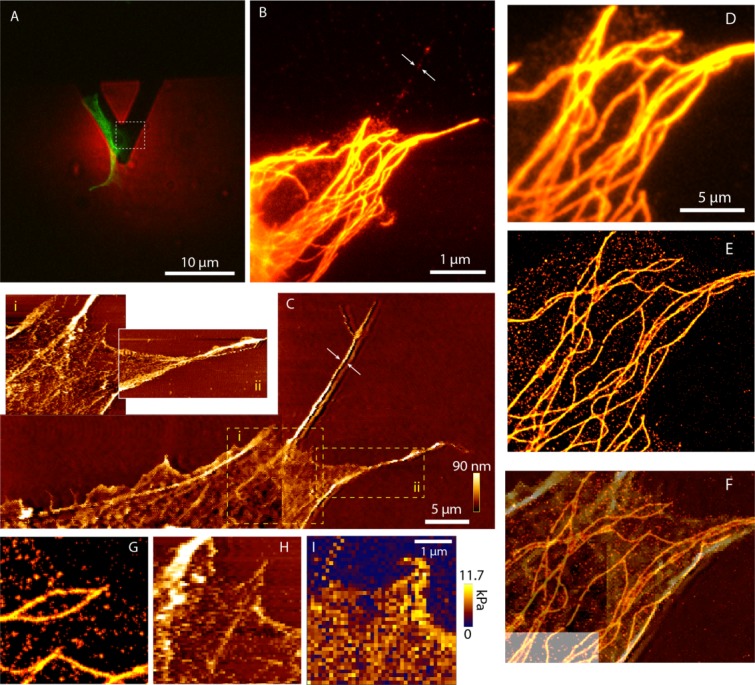
STORM AFM. Panel A shows the cell overlaid with the image of the tip. This is imaged with a 10× objective. A selected fibroblast cell is shown in the green channel and AFM tip in the red. Panel B is the wide field image of a part of the same cell with a 100× objective and C is the respective AFM image. The arrows in B and C show the same area. It is a nonlabeled part of the cell which is prominent in the AFM image. The insets 5C.i, 5C.ii shows higher resolution image where the topology and single fiber bundle of the cell is seen. Panels D and E show the wide field and STORM image comparison of the same area. Panel F is the AFM topological image corresponding to the same region overlaid on the STORM image. Panels G–I show STORM, topological and Young's modulii map of the same area.
Discussion and Conclusions
In recent years, superresolution microscopy saw an exponential growth because of its unique capability to splice subdiffraction resolving power and noninvasive imaging of biological samples under physiological conditions. Following the dream of an unlimited resolution, several techniques were proposed (RESOLFT/STED, STORM/PALM and structured illumination); but all of them feature different advantages and/or drawbacks[Schermelleh et al., 2010]. Between them STED/RESOLFT provides subdiffraction resolution coupled with a good imaging speed, a feature strongly required for imaging sub cellular and cytoskeletal structures in live cells. Single molecule localization based techniques, on the other side, provide insights within cells at the molecular scale and thus represent a suitable tool for imaging the cytoskeletal structures such as actin and tubulin filaments. The high resolution reached (around tens of nanometers) makes STORM an elected method for multicolor/3D imaging at the molecular level but the limited temporal resolution in the conventional implementation, often restricts its application to fixed samples. New fast implementations [Jones et al., 2011] recently smoothed the way to the access of multilevel information. Meanwhile, AFM is a widely used technique for the reconstruction of biomaterials topography with a nanometric resolution. Despite the high resolution provided, the nonspecific nature of the imaging provided by this technique does not allow to answer the biological queries focused on specific molecular targets within cellular and/or cytoskeletal compartments. On the other side, fluorescence based superresolution techniques provides precise localization of selected molecular species with a resolution far below the diffraction limit. Within this scenario, one of the most advanced implementation of AFM is shown by the advantages of correlative approaches STED/AFM, demonstrating a better comprehension of the biological problem and a strong integration of fluorescence imaging with the label free nature of the morphological information provided by AFM.
In particular, we also demonstrate that localization based approaches can also be coupled with the AFM topological information. Although the resolution provided in a crowded environment with local stiff structures can make the matching of the individual contributions very difficult, especially considering that fluorescence is identifying only one protein/cellular structure in the sample while the local AFM fluctuations are modulated by multiple components. In this case, the 3D imaging capabilities and multicolor STORM could represent a suitable tool for a better overlay of the AFM and fluorescence information. 3D STORM allows us to select the structures from the external layer of the cell and the multicolor imaging can separate different type of proteins from the unspecific AFM topology. Similarly, 3D and multicolor STED can also can be applied for getting isolated information content that can suit the correlated techniques perform better. The new developments in STED techniques allows one to work at lower depletion laser powers [Vicidomini et al., 2011] and build cost efficient systems which also suggests that correlative methods based on STED microscopy will have an important role for membrane and cytoskeleton morphological studies.
We believe that these 3D and multicolor modalities will play a key role in improving performances of correlative approaches, helping to overcome the eventual limitations that can occur when the structures in crowded environments are imaged. The tip probing based approach opens the way to the possibility of mechanical nanomanipulation of cellular structures: the combined approach of AFM with the most advanced superresolution methods provides a golden opportunity for simultaneous nanomanipulation and imaging (with tens of nanometer resolution) of effects produced on specific molecule of interest in the cytoskeletal compartments of the cell. These techniques, based on correlative approaches, will smooth the way to a new generation of experiments able to couple topological information, local stiffness measurements and specific fluorescence imaging close to the molecular level.
Acknowledgments
We thank Dr. Benjamin Harke and Dr.Claudio Canale for the help and critical discussions. We also thank Dr. Marta D'Amora for the necessary help in cell sample preparations. This work is partially funded by Italian Programmi di ricerca di rilevante interesse nazionale 2010JFYFY2_002 grant.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supplementary
References
- Madl H, Winfried JR. 2013. Anne JPK Instruments AG - Application Reports / Technical Reports - AFM_STORM, Available at: http://www.jpk.com/afm.230.en.html. Accessed on 25 August, 2013.
- Axelrod D. Total internal reflection fluorescence microscopy in cell biology. Traffic. 2001;2:764–774. doi: 10.1034/j.1600-0854.2001.21104.x. [DOI] [PubMed] [Google Scholar]
- Bálint Š, Vilanova IV, Álvarez ÁS, Lakadamyali M. Correlative live-cell and superresolution microscopy reveals cargo transport dynamics at microtubule intersections. PNAS. 2013;110:3375–3380. doi: 10.1073/pnas.1219206110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates M, Huang B, Dempsey GT, Zhuang X. Multicolor super-resolution imaging with photo-switchable fluorescent probes. Science. 2007;317:1749–1753. doi: 10.1126/science.1146598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betzig E, Trautman JK. Near-field optics: Microscopy, spectroscopy, and surface modification beyond the diffraction limit. Science. 1992;257:189–195. doi: 10.1126/science.257.5067.189. [DOI] [PubMed] [Google Scholar]
- Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- Bianchini P, Harke B, Galiani S, Vicidomini G, Diaspro A. Single-wavelength two-photon excitation–stimulated emission depletion (SW2PE-STED) superresolution imaging. PNAS. 2012;109:6390–6393. doi: 10.1073/pnas.1119129109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianco B, Diaspro A. Analysis of three-dimensional cell imaging obtained with optical microscopy techniques based on defocusing. Cell Biophys. 1989;15:189–199. doi: 10.1007/BF02989683. [DOI] [PubMed] [Google Scholar]
- Cella Zanacchi F, Lavagnino Z, Donnorso MP, Del Bue A, Furia L, Faretta M, Diaspro A. Live-cell 3D super-resolution imaging in thick biological samples. Nat Methods. 2011;8:1047–1049. doi: 10.1038/nmeth.1744. [DOI] [PubMed] [Google Scholar]
- Cella Zanacchi F, Lavagnino Z, Faretta M, Furia L, Diaspro A. Light-sheet confined super-resolution using two-photon photoactivation. PloS one. 2013;8:e67667. doi: 10.1371/journal.pone.0067667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacko JV, Canale C, Harke B, Diaspro A. Sub-diffraction nano manipulation using STED AFM. PLoS ONE. 2013;8:e66608. doi: 10.1371/journal.pone.0066608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox IJ, Sheppard CJR. Information capacity and resolution in an optical system. J Opt Soc Am A. 1986;3:1152–1158. [Google Scholar]
- Diaspro A, editor. Nanoscopy and Multidimensional Optical Fluorescence Microscopy. 1st ed. London: Chapman and Hall/CRC; 2010. [Google Scholar]
- Diaspro A. Optical Fluorescence Microscopy: from the Spectral to the Nano Dimension. Heidelberg: Springer; 2011. [Google Scholar]
- Diaspro A, Sartore M, Nicolini C. 3D representation of biostructures imaged with an optical microscope: Part A: Digital optical sectioning. Image Vis Comput. 1990;8:130–141. [Google Scholar]
- Diaspro A, Beltrame F, Fato M, Ramoino P. Characterizing biostructures and cellular events in 2D/3D [using wide-field and confocal optical sectioning microscopy] IEEE Eng Med Biol Mag. 1996;15:92–100. [Google Scholar]
- Diaspro A, Rolandi R. Atomic force microscopy. [Guest Editorial] Engineering in Medicine and Biology Magazine. IEEE. 1997;16(2):26–27. doi: 10.1109/memb.1997.582172. March–April 1997. doi: 10.1109/MEMB.1997.582172. [DOI] [PubMed] [Google Scholar]
- Dufrêne YF, Pelling AE. Force nanoscopy of cell mechanics and cell adhesion. Nanoscale. 2013;5:4094–4104. doi: 10.1039/c3nr00340j. [DOI] [PubMed] [Google Scholar]
- Eggeling C, Ringemann C, Medda R, Schwarzmann G, Sandhoff K, Polyakova S, Belov VN, Hein B, Von Middendorff C, Schönle A, Hell SW. Direct observation of the nanoscale dynamics of membrane lipids in a living cell. Nature. 2008;457:1159–1162. doi: 10.1038/nature07596. [DOI] [PubMed] [Google Scholar]
- Fölling J, Bossi M, Bock H, Medda R, Wurm CA, Hein B, Jakobs S, Eggeling C, Hell SW. Fluorescence nanoscopy by ground-state depletion and single-molecule return. Nat methods. 2008;5:943–945. doi: 10.1038/nmeth.1257. [DOI] [PubMed] [Google Scholar]
- di Francia GT. Super-gain antennas and optical resolving power. Il Nuovo Cimento. 1952;9:426–438. [Google Scholar]
- di Francia GT. Resolving power and information. J Opt Soc Am. 1955;45:497–499. [Google Scholar]
- Galiani S, Harke B, Vicidomini G, Lignani G, Benfenati F, Diaspro A, Bianchini P. Strategies to maximize the performance of a STED microscope. Opt Express. 2012;20:7362–7374. doi: 10.1364/OE.20.007362. [DOI] [PubMed] [Google Scholar]
- Grigoriev I, Akhmanova A. Microtubule dynamics at the cell cortex probed by TIRF microscopy. In: Cassimeris L, Tran P, editors. Methods in Cell Biology. New York: Academic Press; 2010. pp. 91–109. Chapter 6. [DOI] [PubMed] [Google Scholar]
- Guo Q, Xia Y, Sandig M, Yang J. Characterization of cell elasticity correlated with cell morphology by atomic force microscope. J Biomech. 2012;45:304–309. doi: 10.1016/j.jbiomech.2011.10.031. [DOI] [PubMed] [Google Scholar]
- Gustafsson MGL. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J Microsc. 2000;198:82–87. doi: 10.1046/j.1365-2818.2000.00710.x. [DOI] [PubMed] [Google Scholar]
- Hamon L, Curmi PA, Pastré D. High-resolution imaging of microtubules and cytoskeleton structures by atomic force microscopy. In: Wilson L, Correia JJ, editors. Methods in Cell Biology. New York: Academic Press; 2010. pp. 157–174. Chapter 9. [DOI] [PubMed] [Google Scholar]
- Harke B, Keller J, Ullal CK, Westphal V, Sch\önle A, Hell SW. Resolution scaling in STED microscopy. Opt Express. 2008;16:4154–4162. doi: 10.1364/oe.16.004154. [DOI] [PubMed] [Google Scholar]
- Harke B, Chacko J, Canale C, Diaspro A. A novel nanoscopic tool by combining AFM with STED microscopy. Opt Nanosc. 2012;1:3. [Google Scholar]
- Heilemann M, van de Linde S, Schüttpelz M, Kasper R, Seefeldt B, Mukherjee A, Tinnefeld P, Sauer M. Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes. Angew Chem Int Ed. 2008;47:6172–6176. doi: 10.1002/anie.200802376. [DOI] [PubMed] [Google Scholar]
- Hein B, Willig KI, Hell SW. Stimulated emission depletion (STED) nanoscopy of a fluorescent protein-labeled organelle inside a living cell. Proc Natl Acad Sci USA. 2008;105:14271–14276. doi: 10.1073/pnas.0807705105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hell SW. Far-Field Optical Nanoscopy. Science. 2007;316:1153–1158. doi: 10.1126/science.1137395. [DOI] [PubMed] [Google Scholar]
- Hell SW, Wichmann J. Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt Lett. 1994;19:780–782. doi: 10.1364/ol.19.000780. [DOI] [PubMed] [Google Scholar]
- Hell SW, Dyba M, Jakobs S. Concepts for nanoscale resolution in fluorescence microscopy. Curr Opin Neurobiol. 2004;14:599–609. doi: 10.1016/j.conb.2004.08.015. [DOI] [PubMed] [Google Scholar]
- Hell SW, Willig KI, Dyba M, Jakobs S, Kastrup L, Westphal V. Nanoscale resolution with focused light: Stimulated emission depletion and other reversible saturable optical fluorescence transitions microscopy concepts. Handbook Biol Confocal Microsc. 2006:571–579. [Google Scholar]
- Hess ST, Girirajan TPK, Mason MD. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys J. 2006;91:4258–4272. doi: 10.1529/biophysj.106.091116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann M, Eggeling C, Jakobs S, Hell SW. Breaking the diffraction barrier in fluorescence microscopy at low light intensities by using reversibly photoswitchable proteins. Proc Natl Acad Sci USA. 2005;102:17565. doi: 10.1073/pnas.0506010102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann UG, Rotsch C, Parak WJ, Radmacher M. Investigating the cytoskeleton of chicken cardiocytes with the atomic force microscope. J Struct Biol. 1997;119:84–91. doi: 10.1006/jsbi.1997.3868. [DOI] [PubMed] [Google Scholar]
- Huang B, Jones SA, Brandenburg B, Zhuang X. Whole-cell 3D STORM reveals interactions between cellular structures with nanometer-scale resolution. Nat Methods. 2008a;5:1047–1052. doi: 10.1038/nmeth.1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Wang W, Bates M, Zhuang X. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science. 2008b;319:810–813. doi: 10.1126/science.1153529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SA, Shim S-H, He J, Zhuang X. Fast, three-dimensional super-resolution imaging of live cells. Nat Methods. 2011;8:499–508. doi: 10.1038/nmeth.1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juette MF, Gould TJ, Lessard MD, Mlodzianoski MJ, Nagpure BS, Bennett BT, Hess ST, Bewersdorf J. Three-dimensional sub–100 nm resolution fluorescence microscopy of thick samples. Nat Methods. 2008;5:527–529. doi: 10.1038/nmeth.1211. [DOI] [PubMed] [Google Scholar]
- Klar TA, Jakobs S, Dyba M, Egner A, Hell SW. Fluorescence microscopy with diffraction resolution barrier broken by stimulated emission. Proc Natl Acad Sci. 2000;97:8206–8210. doi: 10.1073/pnas.97.15.8206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubitscheck U. Fluorescence Microscopy: From Principles to Biological Applications. Weinheim: Wiley; 2013. [Google Scholar]
- Lavagnino Z, Cella Zanacchi F, Ronzitti E, Diaspro A. Two-photon excitation selective plane illumination microscopy (2PE-SPIM) of highly scattering samples: characterization and application. Opt Express. 2013;21:5998–6008. doi: 10.1364/OE.21.005998. [DOI] [PubMed] [Google Scholar]
- Lee HD, Sahl SJ, Lew MD, Moerner WE. The double-helix microscope super-resolves extended biological structures by localizing single blinking molecules in three dimensions with nanoscale precision. Appl Phys Lett. 2012;100:153701–153701–3. doi: 10.1063/1.3700446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H, Centeno SP, Su L, Kenens B, Rocha S, Sliwa M, Hofkens J, Uji-i H. Mapping of surface-enhanced fluorescence on metal nanoparticles using super-resolution photoactivation localization microscopy. ChemPhysChem. 2012;13:973–981. doi: 10.1002/cphc.201100743. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Snapp E, Kenworthy A. Studying protein dynamics in living cells. Nat Rev Mol Cell Biol. 2001;2:444–456. doi: 10.1038/35073068. [DOI] [PubMed] [Google Scholar]
- Mueller V, Ringemann C, Honigmann A, Schwarzmann G, Medda R, Leutenegger M, Polyakova S, Belov V, Hell S, Eggeling C. STED nanoscopy reveals molecular details of cholesterol-and cytoskeleton-modulated lipid interactions in living cells. Biophys J. 2011;101:1651–1660. doi: 10.1016/j.bpj.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronzitti E, Harke B, Diaspro A. Frequency dependent detection in a STED microscope using modulated excitation light. Opt Express. 2013;21:210–219. doi: 10.1364/OE.21.000210. [DOI] [PubMed] [Google Scholar]
- Rust MJ, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nat methods. 2006;3:793–796. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schermelleh L, Heintzmann R, Leonhardt H. A guide to super-resolution fluorescence microscopy. J Cell Biol. 2010;190:165–175. doi: 10.1083/jcb.201002018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard CJR. Super-resolution in confocal imaging. Optik. 1988;80:53–54. (Stuttg.) [Google Scholar]
- Subach FV, Patterson GH, Renz M, Lippincott-Schwartz J, Verkhusha VV. Bright monomeric photoactivatable red fluorescent protein for two-color super-resolution sptPALM of live cells. J Am Chem Soc. 2010;132:6481–6491. doi: 10.1021/ja100906g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai E, Costa KD, Shaheen A, Hung CT, Guo XE. Osteoblast elastic modulus measured by atomic force microscopy is substrate dependent. Ann Biomed Eng. 2005;33:963–971. doi: 10.1007/s10439-005-3555-3. [DOI] [PubMed] [Google Scholar]
- Tamarit B, Bugault F, Pillet A-H, Lavergne V, Bochet P, Garin N, Schwarz U, Thèze J, Rose T. Membrane microdomains and cytoskeleton organization shape and regulate the IL-7 receptor Signalosome in human CD4 T-cells. J Biol Chem. 2013;288:8691–8701. doi: 10.1074/jbc.M113.449918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trache A, Lim S-M. Live cell response to mechanical stimulation studied by integrated optical and atomic force microscopy. J Vis Exp. 2010;44:e2072. doi: 10.3791/2072. doi: 10.3791/2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner RD, Hurd AF, Cadby A, Hobbs JK, Foster SJ. Cell wall elongation mode in Gram-negative bacteria is determined by peptidoglycan architecture. Nat Commun. 2013;4:1496. doi: 10.1038/ncomms2503. doi: 10.1038/ncomms2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban NT, Willig KI, Hell SW, Nägerl UV. STED nanoscopy of actin dynamics in synapses deep inside living brain slices. Biophys J. 2011;101:1277–1284. doi: 10.1016/j.bpj.2011.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicidomini G, Moneron G, Han KY, Westphal V, Ta H, Reuss M, Engelhardt J, Eggeling C, Hell SW. Sharper low-power STED nanoscopy by time gating. Nat Methods. 2011;8:571–573. doi: 10.1038/nmeth.1624. [DOI] [PubMed] [Google Scholar]
- Westphal V, Hell SW. Nanoscale resolution in the focal plane of an optical microscope. Phys Rev Lett. 2005;94:143903. doi: 10.1103/PhysRevLett.94.143903. [DOI] [PubMed] [Google Scholar]
- Xu K, Zhong G, Zhuang X. Actin, spectrin, and associated proteins form a periodic cytoskeletal structure in axons. Science. 2013;339:452–456. doi: 10.1126/science.1232251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang TT, Hampilos PJ, Nathwani B, Miller CH, Sutaria ND, Liao J-C. Superresolution STED microscopy reveals differential localization in primary cilia. Cytoskeleton. 2013;70:54–65. doi: 10.1002/cm.21090. [DOI] [PubMed] [Google Scholar]
- Zobel T, Bogdan S. A high resolution view of the fly actin cytoskeleton lacking a functional WAVE complex. J Microsc. 2013;251:224–231. doi: 10.1111/jmi.12020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary