

Abstract
The electrochemical pretreatment of diamond microelectrodes was investigated for the purpose of learning how an anodic, cathodic or a combined anodic + cathodic polarization affects the charge-transfer kinetics for two surface-sensitive redox systems: ferri/ferrocyanide and serotonin (5-hydroxytryptamine, 5-HT). The pretreatments were performed in 0.5 mol L−1 H2SO4. The anodic pretreatment was performed galvanically for 30 s at 250 mA cm−2. The 10 cathodic pretreatment was performed for 180 s at −250 mA cm−2. The combined pretreatment involved application of the anodic step first followed by the cathodic step. The results clearly demonstrate that the best performance for both redox systems is obtained after the cathodic polarization, which presumably activates the electrode by cleaning the surface and removing site-blocking surface carbon-oxygen functionalities. The cathodic pretreatment was found to be effective at activating a fouled microelectrode in situ. This observation has important implication for the measurement of 5-HT in the bowel.
1. Introduction
The development of surface pretreatments to create electrochemically-active (i.e., rapid electron-transfer 20 kinetics) and reproducibility-performing carbon electrodes has been the goal of many researchers over several decades (1). Generally for sp2 carbon electrodes, the pretreatment begins with a polishing step. This is often followed up with one of several activation steps including chemical (2), electrochemical (3-5), thermal (6), or radio frequency plasma (7) treatment. The resulting electrode activation has been attributed to (i) surface cleaning/removal of contaminants, (ii) increased in the coverage of surface carbon-oxygen functional groups that can act as redox mediators, (iii) increased electrode area due to surface roughening and/or (iv) exposure of clean edge plane or generation of new sites that support rapid rates of electron transfer. The extent to which a pretreatment activates an sp2 carbon electrode depends on the mechanism of the redox system being used to probe the electrode response (8-10).
Boron-doped diamond electrodes have emerged over the past 15+ years as a viable new carbon electrode for electroanalysis (11-13). While doped single crystal electrodes are now available, nearly all of the work thus far has utilized microcrystalline (14), nanocrystalline (15) and ultrananocrystalline (16) thin films. Generally in electroanalytical measurements, diamond offers significant improvements in linear dynamic range, limit of detection, response reproducibility and response stability as compared to sp2 carbon electrodes. A major benefit of diamond is that it exhibits a high level of electrochemical activity for some redox systems without the normal time-consuming pretreatment. In fact over the years, we have routinely used these electrodes without any pretreatment, or at most, a 20-min soak in ultraclean isopropanol (17). In cases where response loss is observed, diamond electrodes can often be reactivated by isopropanol cleaning or by hydrogen plasma treatment. The latter pretreatment (i) cleans the surface, (ii) replaces the surface oxygen termination (e.g., C-OH, C=O and C-O-C functionalities) with a hydrogen termination and (iii) adds subsurface hydrogen that increases the surface conductivity through an elevation in the carrier concentration (18,19). Given the fact that a microwave reactor for the hydrogen plasma treatment is not available to most users, work is needed to identify appropriate pretreatment methods that function to reactivate diamond electrodes and are convenient to apply.
To this end, several groups have investigated the effects of electrochemical pretreatment on the response of diamond electrodes. An electrochemical pretreatment can be either potentiostatic or galvanostatic in nature. Pioneering work was performed by the Rocha-Filho and Avaca groups who reported that an initial anodic polarization at +3.0 V vs. Ag/AgCl in 0.5 mol L−1 H2SO4 + 0.5 mol L−1 Na2SO4 (30 min) followed by a cathodic polarization at −3.0 V for an analogous time activated diamond electrodes for two surface-sensitive redox systems: ferri/ferrocyanide and pentachlorophenol (20).
Sluggish kinetics was observed for both redox systems after just the anodic pretreatment. However after the cathodic pretreatment, the ΔEp for ferri/ferrocyanide was significantly reduced and the Epox for pentachlorophenol was shifted to less positive potentials. In contrast, either pretreatment had little effect on the response for the surface-insensitive ferrocene. This result suggests the electrochemical pretreatments do not significantly alter the electronic properties of a more heavily boron-doped electrode. XPS results confirmed the cathodic pretreatment removed surface carbon-oxygen functionalities and presumably introduced surface hydrogen (21).
In other relevant work, Mahé et al. reported that alternating current pulses of ±250 mA cm−2 in 1 mol L−1 HNO3 can be used to activate diamond electrodes (22). Olivera et al. investigated the effects of anodic (+3.0 V) and cathodic (−3.0 V) potentiostatic polarization on the response of boron-doped diamond electrodes for ferri/ferrocyanide (23). They specifically investgated the influence of the supporting electrolyte and pH. Their results demonstrated that cathodic rather than anodic pretreatment provided a more active response. Kiran et al. recently reported that applying a train current pulses (±10 mA cm−2) of 100 msec duration in 0.5 mol L−1 LiClO4 activated boron-doped diamond films as evidenced by an increased heterogeneous electron-transfer rate constant, ko, for ferri/ferrocyanide (24). They also showed that the pretreatment enhanced the electrode response of a deactivated diamond electrode in urine. Cyclic voltammograms were presented for the irreversible oxidation of an unidentified species in urine, presumably uric acid, showing how the activation increased the oxidation current and shifted Epox to less positive potentials. Activation was also accomplished by pretreating the electrode directly in the biological medium.
In this paper, we report on how anodic and cathodic galvanostatic pretreatment affects the response of diamond microelectrodes toward two surface-sensitive redox systems: ferri/ferrocyanide and serotonin (5-HT). We have previously demonstrated that diamond microelectrodes provide stable and reproducible measurements of norepinephrine release from sympathetic nerves supplying blood vessels (25) and 5-HT release from enterochromaffin cells lining the mucosa of the small and large intestine in vitro (26). Generally, these microelectrodes can be used for these measurements from days to weeks with little response loss. However, from time-to-time, the microelectrodes can become deactivated in the 5-HT environment and the response cannot be regained with an isopropanol soak. Furthermore, insulated microelectrodes cannot be reactivated by the hydrogen plasma treatment. 5-HT may be the most notorious fouler of carbon electrodes. Wrona and Dryhurst have studied the 5-HT oxidation reaction mechanism on carbon paste electrodes (27). 5-HT is initially oxidized in a reversible one-electron reaction to form a radical cation (5-HT+•). In a rate determining step, the radical cation deprotonates to yield a 5-HT radical (5-HT•). These radicals can then react to form dimers and trimers that strongly adsorb on the electrode. The results presented herein clearly demonstrate that the cathodic, rather than the anodic, pretreatment effectively activates diamond microelectrodes for both redox systems.
2.1 Diamond deposition on Pt wires
The boron-doped diamond coating was deposited on a sharpened (76 μm diameter) Pt wire using microwave-assisted chemical vapor deposition (CVD). We use Pt as the substrate because it gives a very distinctive electrochemical signature if the diamond film is discontinuous or defective (28). The Pt wire (99.99%, Sigma-Aldrich Chemical) was cut (1.3 cm) and both ends of the wire were electrochemically sharpened in 1 mol L−1 KOH. The Pt wire was then ultrasonically cleaned in acetone for 20 min and then ultrasonically seeded for 30 min in a diamond powder suspension (3–6 nm diamond particles suspended in DMSO, 0.5 w/v% “Opal Seed”, ITC, Raleigh, NC). This served to enhance the initial nucleation of diamond growth on the surface. The wire was then rinsed with ultrapure water before being place in the CVD reactor, mounted horizontally, for pump down. Three wires were coated during a deposition run. A thin film of boron-doped diamond was deposited on the wire using a commercial reactor (1.5 kW, 2.54 GHz, Seki Diamond, San Jose, CA). At the beginning of the deposition, an amorphous carbon pre-growth layer was deposited from a 3% CH4/H2 source gas ratio (CH4 = 6.00 sccm, B2H6 = 2.00 sccm, H2 = 196 sccm) for 20 min at 600 90 W and 35 torr. This carbon layer further enhances the nucleation of diamond on the surface (29,30). At the end of this period, the CH4 flow was adjusted to give a 1% CH4/H2 ratio that was used for primary diamond growth (CH4 = 2.00 sccm, B2H6 = 2.00 sccm, H2 = 196 sccm). The microwave power was 600 W and the system pressure was 35 torr. Growth times ranged from 5 to 8 h. The boron-doping level was estimated to be ~1021 cm−3 based on studies of planar films grown under similar conditions. At the end of the growth, the CH4 and B2H6 flows were stopped and the diamond-coated wires remained exposed to a H2 plasma. The samples were then slowly cooled in the presence of atomic hydrogen by reducing the power and pressure over a 30-min period. The estimated substrate temperature (optical pyrometer) at the end of the period was ~400 °C or less. This step is critical for maintaining a hydrogen surface termination and keeping the near-surface carbon atoms in an sp3 bonding configuration.
2.2 Preparation of the diamond microelectrode
A diamond-coated Pt wire was cut in the middle to produce two electrodes. The cut end was affixed to a copper wire using conducting silver epoxy and super glue and then insulated with polypropylene from a heated pipette tip (28). The resulting microelectrode was a conically shaped with a tip diameter of about 10 μm and a cylinder diameter of about 80 μm. The exposed cone length was 300 - 700 μm.
2.3 Anodic and cathodic pretreatment
Pretreatments were performed in 0.5 mol L−1 H2SO4. The anodic pretreatment was performed galvanically for 30 s at 250 mA cm−2. The cathodic pretreatment was performed for 180 s at −250 mA cm−2. The combined pretreatment involved application of the anodic step first followed by the cathodic step. Every conically-shaped electrode has a slightly different exposed length so the exposed area was somewhat variable from electrode to electrode. To calculate the current density for pretreatment, we assumed a constant area of 0.0048 cm2.
2.4 Electrochemical measurements
Cyclic voltammetry was performed using a commercial potentiostat/galvanostat (Model 660D, CH Instruments, Austin, TX). The measurements were made in a single-compartment, glass cell with a diamond working electrode, a Pt wire counter electrode and a Ag/AgCl (3 mol L−1 KCl) electrode as the reference. Before each measurement, the solution was degassed for 10 min with N2. The KCl, potassium ferricyanide and serotonin were supplied by Sigma-Aldrich. The ultrapure water was purified using a Barnstead E-Pure system (> 17 Mohm−cm). The protocol for the in vitro bowel measurements had been detailed elsewhere (26,31).
3. Results
Figure 1 shows background cyclic voltammograms in 0.1 30 mol L−1 KCl after the different pretreatments. In terms of the background current, capacitance and the onset potential for chlorine evolution, the anodic, cathode or combined potential for oxygen reduction to the least negative value. In contrast, the anodic pretreatment shifted the onset potential to the most negative value. The anodic pretreatment would appear to introduce surface oxygen functionalities that act as a partially blocking layer to inhibit oxygen reduction. The fact that the background current is not significantly different after any of the pretreatments indicates that the diamond coating is not roughened to increase the surface area, nor is the film delaminated to expose Pt.
Figure 1.
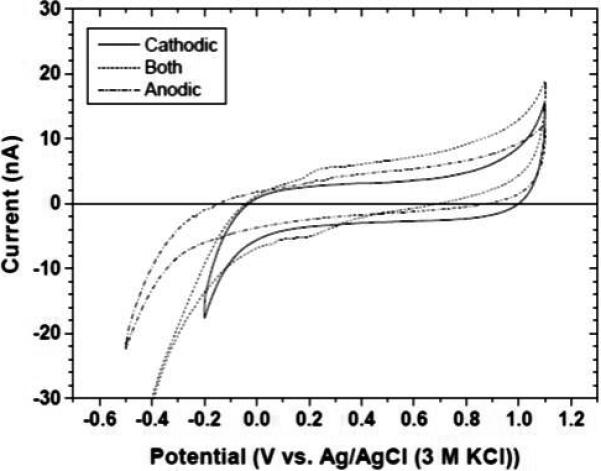
Cyclic voltammetric background curves for a diamond microelectrode after anodic (dash-dot), cathodic (solid) and combined anodic + cathodic (dash) pretreatment. Electrolyte = 0.1 mol L−1. Scan rate = 0.1 V s−1.
The effects of pretreatment on the response for surface-sensitive redox systems (5-HT and ferri/ferrocyanide) are shown in Figure 2. Figure 2A shows cyclic voltammograms for 10 μmol L-−1 5-HT in 0.1 mol L−1 KCl after anodic, cathodic and combined anodic and cathodic pretreatment. It can be see that the cathodic only or the combined anodic and cathodic pretreatment produced equivalent results in terms of Epox (~0.6 V) and ipox (~13 nA). The anodic pretreatment only deactivated the electrode causing Epox to shift positive by about 100 mV. The ipox was unaffected. Figure 2B shows cyclic voltammograms for 1 mmol L−1 ferricyanide in 0.1 mol L−1 KCl after the different pretreatments. As was the case for 5-HT, the cathodic only and the combined anodic and cathodic pretreatment produced similar levels of activity with ΔEp values of ~75 mV. In contrast after the anodic pretreatment, ΔEp increased to ~400 mV indicative of sluggish kinetics. The reduction peak current also was affected by the pretreatment. ipred was 850 nA for the cathodically pretreated microelectrode and decreased to ~700 nA after the combined anodic and cathodic pretreatment. After the anodic pretreatment, ipred decreased further to ~600 nA. Clearly, the surface condition created by the pretreatment has a significant effect on the interfacial electron-transfer kinetics for these two systems. The oxygenated surface was the least active and the low oxygen surface was the most active.
Figure 2.
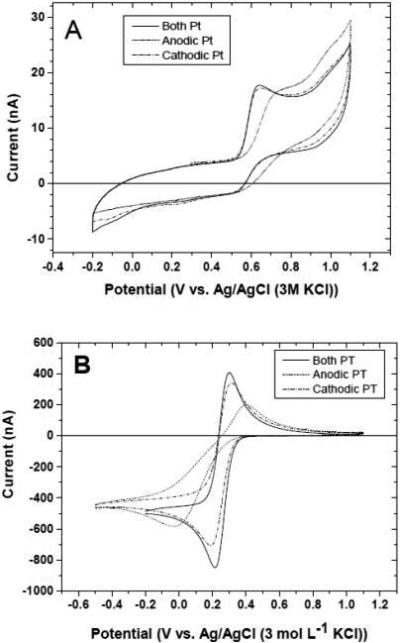
(A) Cyclic voltammograms for 10 μmol L−1 5-HT + 0.1 mol L−1 KCl after anodic (dash-dot), cathodic (solid) and 55 combined anodic and cathodic (dash) pretreatment. (B) Cyclic voltammograms for 1 mmol L-1 ferricyanide + 0.1 mol L-1 KCl. Scan rate = 0.1 V s−1.
Figure 3 shows the effect of a combined anodic +cathodic pretreatment on the response of a fouled diamond microelectrode. Fouling was accomplished by exposing a new microelectrode to 50 cycles in 10 μmol L−1 5-HT + 0.1 mol L−1 KCl solution An ill-defined oxidation curve is evident for the deactivated electrode with no clear Epox or ipox. In contrast, a new microelectrode is quite active with an Epox of ~630 mV and an ipox of 14 nA. The deactivated microelectrode was returned to its native state of activity after the combined pretreatment. The Epox and ipox values are identical to those of the microelectrode in its pristine state.
Figure 3.
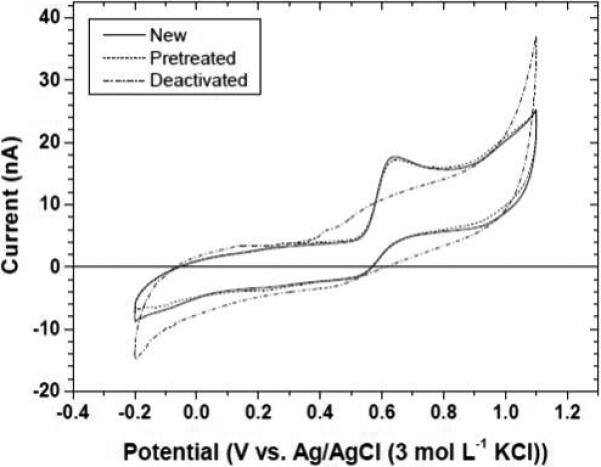
Cyclic voltammograms for 10 μmol L−1 5-HT in 0.1 mol L−1 KCl for a new diamond microelectrode (solid), after deactivating the electrode by extended cycling in a 5-HT solution (dash-dot) and after a combination anodic + cathodic pretreatment (dash). Scan rate = 0.1 V s−1.
Figure 4 shows the effect of repetitive pretreatments on the diamond microelectrode response for 1 mmol L−1 ferricyanide in 0.1 mol L−1 KCl. Curves are shown for a new microelectrode and the same microelectrode after the tenth pretreatment cycle (anodic + cathodic). Multiple pretreatments do not alter the electrode in any way as the ΔEp remains unchanged at ~80 mV and ipred is unaltered at ~700 nA. There is no increase in the background current indicating that the diamond film is stably covering the Pt and the electrode microstructure/morphology is unaltered.
Figure 4.
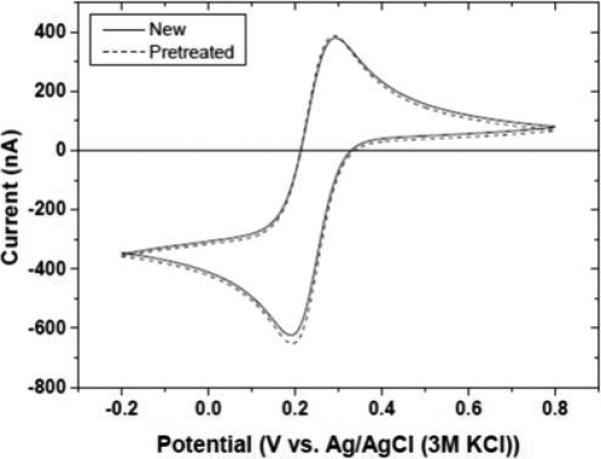
Cyclic voltammograms for 1 mmol L−1 ferricyanide in 0.1 mol L−1 KCl before and after ten combined anodic + cathodic pretreatments. Scan rate = 0.1 V s−1.
To demonstrate that the pretreated microelectrodes exhibit an analytically useful response, square wave voltammetry was performed to measure the 5-HT oxidation current as a function of the solution concentration. Figure 5 shows response curves for three different microelectrodes activated by the combined anodic + cathodic pretreatment. Clearly, all three microelectrodes exhibited a linear increase in the oxidation peak current with increasing solution concentration. The regression statistics were as follows: (electrode #1: slope = 0.694 nA−L μmol−1, y-intercept = 2.28 nA, R2 = 0.9965; electrode #2: slope = 0.639 nA−L μmol−1, y-intercept = 2.62 nA, R2 = 0.9912; 70 electrode #3: slope = 0.708 nA−L μmol−1, y-intercept = 2.94 nA, R2 = 0.9783).
Figure 5.
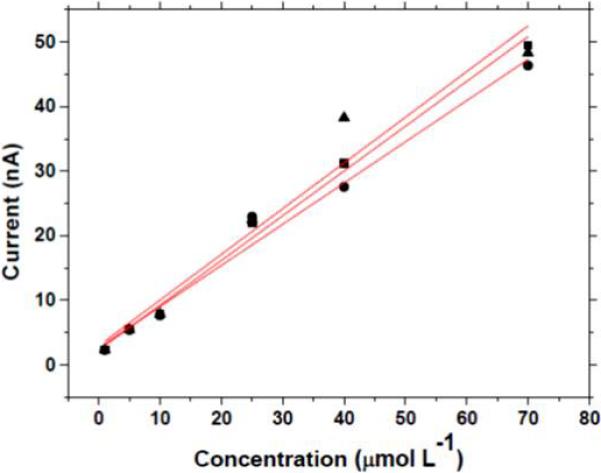
Response curves for three different pretreated diamond microelectrodes as a function of the 5-HT concentration. The square wave voltammetric conditions were: increment: 7 mV; frequency: 15 Hz; amplitude: 50 mV. Electrolyte = 0.1 mol L−1 KCl. The electrodes were activated by the combined anodic and cathodic pretreatment.
The ability to reactivate a poorly performing diamond microelectrode has important implications for in vitro peripheral tissue measurement of 5-HT (30,31). A major source of bioavailable 5-HT in the body is in the bowel, specifically in enterochromaffin cells that are part of the intestinal epithelium (32). These cells synthesize 5-HT from tryptophan, store it in granules and secrete it in response to a luminal stimulus (e.g., mechanical force). The release of this signaling molecule activates various motor, sensory and secretory reflexes. Using continuous amperometry with a diamond microelectrode, we have measured 5-HT secretion in the gut mucosa (guinea pigs, rats, mice and humans) (30,31). In these measurements, the microelectrode is placed near the mucosa of the small or large intestine. Stimulation of the enterochromaffin cells evokes release of 5-HT that can be measured in the nearby extracellular space as an oxidation current. The relatively high concentrations present (10's μmol L−1) along with the complex environment of the bowel present measurement challenges for any electrode material. From time to time, the diamond microelectrode response deteriorates with time during the in vitro measurement. We conducted tests to determine if the galvanostatic pretreatment could reactivate a fouled microelectrode in situ.
Figure 6 shows typical approach curves for the measured 5-HT oxidation current versus the distance between the diamond microelectrode and the mucosa surface. These measurements were made in vitro in the mouse ileum. The perfusion buffer provides a sufficient mechanical stimulus to the cells to evoke secretion. As the microelectrode approaches the surface (2000 to 10 μm), the current progressively increases. This is shown in the left-most data for a “clean electrode”. The center data shows the response for a “deactivated” diamond microelectrode in the same tissue. The microelectrode was deactivated by cycling it in a solution of 100 μmol L−1 5-HT + Krebs buffer (pH 7.4) for an extended period at 0.10 V s−1 prior to the tissue measurement. It can be seen that the 5-HT oxidation current at all distances is about half the magnitude of that for the “clean” electrode, reflecting the microelectrode's unclean surface condition. After this measurement, the microelectrode was retracted from the tissue but left in the perfusion bath. It was then pretreated in situ in the flowing Krebs buffer. An anodic pretreatment was performed galvanically for 30 s at 250 mA cm−2. A cathodic pretreatment was performed for 180 s at −250 mA cm−2. The right-most data in the figure show that the original current magnitude was regained for the “reactivated electrode”. This result shows that reactivation of the diamond microelectrode can be conveniently accomplished in situ. Figure 7 shows plots of the 5-HT oxidation current measured as a function of distance during the approach. Approach curves were recorded from 2000 to 50 μm for a clean electrode, after the electrode was deactivated and after reactivation by galvanostatic pretreatment. Clearly, the galvanostatic pretreatment effectively restored the current response of a “deactivated” diamond microelectrode.
Figure 6.
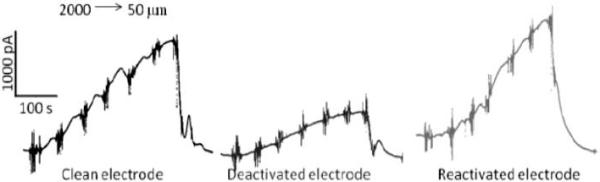
Oxidation current versus the diamond microelectrode-mucosa spacing. The measurements were made by continuous amperometry in the mouse ileum at 0.8 V vs. Ag/AgCl. The tissue was mounted in a bath with perfusing Krebs buffer (pH 7.4). Approach curves were recorded from 2000 to 10 μm for a clean electrode, after the electrode was deactivated and after reactivation by galvanostatic pretreatment. Error bars represent standard deviations for three approach curves measured for each electrode in the same tissue.
Figure 7.
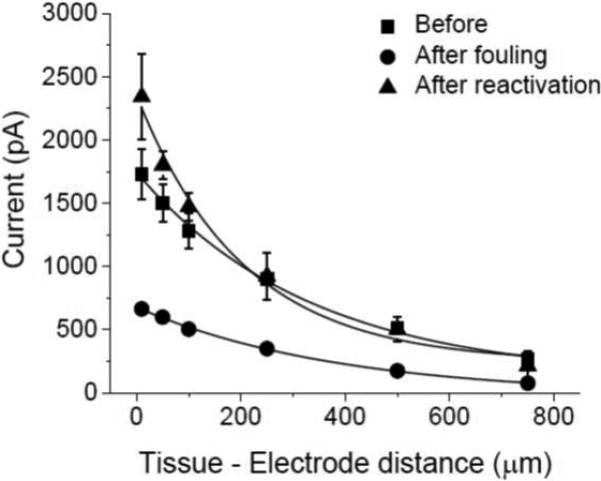
Oxidation current versus the diamond microelectrode-mucosa spacing. The measurements were made by continuous amperometry in the mouse ileum at 0.8 V vs. Ag/AgCl. The tissue was mounted in a bath with Krebs buffer (pH 7.4) perfusing. Approach curves were recorded from 2000 to 10 μm for a clean electrode, after the electrode was deactivated and after reactivation by galvanostatic pretreatment.
Finally studies were performed to determine if the degree of activation by the two-part galvanostatic pretreatment depends on the electrolyte medium in which the pretreatment is performed. To this end, we tested three media: 0.1 mol L-1 H2SO4, 0.1 mol L phosphate buffer (pH 7.2) and 0.1 mol L−1 NaOH. Figure 8A-C shows cyclic voltammetric i-E curves for 100 μmol L−1 5-HT in 1 mol L−1 KCl (0.05 V s−1) recorded before, after the anodic pretreatment and after the cathodic pretreatment in each of the three media. It can be seen that the degree of activation is similar regardless of the electrolyte solution. In all media, the anodic pretreatment deactivates the diamond electrode for 5-HT oxidation as evidenced by the positive shift in Epox by ~100 mV or so as compared to the value for the clean, unused electrode (so-called background), ~0.45 V. In contrast, in all three media the cathodic pretreatment activates the electrode as evidenced by the more negative Epox.
Figure 8.
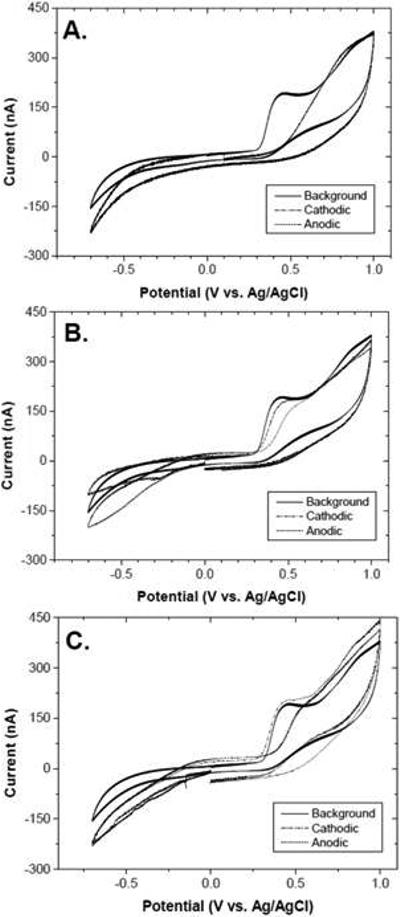
Cyclic voltammograms for 100 μmol L−1 5-HT in 1 mol L−1 KCl before and after the anodic and cathodic pretreatment in (A) 0.1 mol L−1 H2SO4, (B) 0.1 mol−1 L phosphate buffer (pH 7.2) and (C) 0.1 mol L−1 NaOH. Scan rate = 0.1 V s−1.
4. Discussion
The results demonstrate that diamond microelectrodes can be activated by cathodic and/or a combined anodic and cathodic pretreatment in agreement with published work for planar diamond films (20-24,34). This was demonstrated using two surface-sensitive redox systems: ferri/ferrocyanide and 5-HT. The anodic nor the cathodic pretreatment, at least as performed herein, causes no major increase in surface roughness or detachment of the diamond coating from the Pt wire substrate. Background voltammetric currents and capacitance were largely unchanged by either pretreatment. One thing the cathodic treatment does do is lower the surface carbon-oxygen functionality coverage, presumably by introducing surface hydrogen (21,34). Since our data were consistent with these findings, we did not perform a surface analysis to learn how the diamond microelectrode surface chemistry was changed by the pretreatment.
At the current densities used in this work, the electrode potential is well into the oxygen evolution and hydrogen evolution regimes. While the anodic pretreatment might be effective at cleaning the electrode surface through the generation of reactive oxygen species at the electrode surface (oxidizing adsorbed organics to CO2), it inhibits the interfacial electron-transfer kinetics for these two surface-sensitive redox systems. The evidence at hand is consistent with a mechanism whereby the introduced carbon-oxygen functionalities block sites involved in the electron transfer of these two redox systems. The exact site and the types of blocking functional groups remain to be determined. For the highly boron-doped diamond films used in this work, the anodic pretreatment does not introduce changes in the surface electronic properties. This was confirmed in prior published work for the surface-insensitive ferrocene (20) and in our work by use of surface-insensitive redox systems like Ru(NH3)6+3/+2 (data not presented herein). Relatively rapid electron transfer was observed for these two redox systems on electrodes pretreated by both methods. If the anodic pretreatment altered the electronic properties, then inhibited electron transfer for all redox systems would be expected (more positive Epox and or larger ΔEp) and this is not the finding. The site blocking effects of surface carbon-oxygen functionalities is specific for certain redox systems and was first demonstrated by us for ferri/ferrocyanide by comparing the response of oxidized versus hydrogen plasma-treated diamond films (35). A similar finding was made by the Fujishima group (36). In more recent work, Actis et al. reported more sluggish electron transfer for ferri/ferrocyanide at oxidized diamond as compared to hydrogen- or amine-terminated diamond (37). Their work involved the use of more semiconducting diamond electrodes. The cathodic pretreatment appears essential mainly because of surface cleaning and its effect on lowering the coverage of site blocking carbon-oxygen functionalities. It should be emphasized that these effects are specific to the more heavily doped diamond. Greater effects on the electronic properties are certainly possible for lower doped diamond.
Conclusions
The results presented herein demonstrate that electrochemical pretreatment is a convenient and reproducible method for activating diamond microelectrodes. The pretreatment is especially useful for activating diamond microelectrodes in situ during measurements of 5-HT in the bowel. Unlike the effects on sp2 carbon fibers, anodic and cathodic pretreatment, at least as applied here, produced no diamond surface roughening or caused no changes in the voltammetric background current or capacitance. Additionally, the pretreatment did not cause any diamond film delamination from the Pt substrate. The most active electrodes were produced by a final cathodic pretreatment which served to clean the surface and lower the site-blocking surface carbon-oxygen functional group coverage. While we have 15 not performed any systematic studies of how the pretreatment affects the microelectrode response for other bioanalytes, in principle, it should be as effective. We have recently shown how the pretreatment activates diamond thin films for the oxidation of estrogenic compounds (38,39).
Acknowledgements
The work was also supported in part by a grant from the National Science Foundation (CHE-0911383). B.D 25 acknowledges the doctoral fellowships provided by Conicyt (N° 21100047), Becas Chile (N° 75120025) and VRI of Pontificia Universidad Católica de Chile. R.B. PDSE scholarship (for R.F. Brocenschi) from the Brazilian funding agency CAPES
References
- 1.Hu I-F, Karweik DH, Kuwana T. J. Electroanal. Chem. 1985;188:59. [Google Scholar]
- 2.Taylor RJ, Humffray AA. J. Electroanal. Chem. 1973;42(347):50. [Google Scholar]
- 3.Bladel WJ, Jenkins RA. Anal. Chem. 1974;46:1952. [Google Scholar]
- 4.Engstrom RC. Anal. Chem. 1982;54:2310. [Google Scholar]
- 5.Engstrom RC, Strasser VA. Anal. Chem. 1984;56(136):55. [Google Scholar]
- 6.Fagan DT, Hu I-F, Kuwana T. Anal. Chem. 1985;57:2759. [Google Scholar]
- 7.Evans JF, Kuwana T. Anal. Chem. 1979;51:358. [Google Scholar]
- 8.Einaga Y. J. Appl. Electrochem. 2010;40(1807):60. [Google Scholar]
- 9.Luong JHT, Male KB, Glennon Analyst JD. 2009;134:1865. doi: 10.1039/b910206j. [DOI] [PubMed] [Google Scholar]
- 10.Compton RG, Foord JS, Marken F. Electroanalysis. 2003;14:1349. [Google Scholar]
- 11.Kristen-Cline K, McDermott MT, McCreery RL. J. Phys. Chem. 1994;98(65):5314. [Google Scholar]
- 12.Chen P, Fryling MA, McCreery RL. Anal. Chem. 1995;67:3115. [Google Scholar]
- 13.Chen P, McCreery RL. Anal. Chem. 1996;68(3958):70. [Google Scholar]
- 14.Fischer AE, Show Y, Swain GM. Anal. Chem. 2004;76:2553. doi: 10.1021/ac035214o. [DOI] [PubMed] [Google Scholar]
- 15.Wang S, Swope VM, Butler JE, Swain GM. Diam. Rel. Mater. 2009;18:669. [Google Scholar]
- 16.Show Y, Witek MA, Sonthalia P, Swain GM. Chem. Mater. 2003;15(75):879. [Google Scholar]
- 17.Ranganathan S, Kuo T-C, McCreery RL. Anal. Chem. 1999;71:3574. [Google Scholar]
- 18.Looi HJ, Jackman RB, Foord JS. Appl. Phys. Lett. 1998;72(353):80. [Google Scholar]
- 19.Looi HJ, Pang LYS, Molloy AB, Jones F, Foord JS, Jackman RB. Diam. Rel. Mater. 1998;7:550. [Google Scholar]
- 20.Suffredini HB, Pedrosa VA, Codognoto L, Machado SAS, Rocha-Filho RC, Avaca LA. Electrochim. Acta. 2004;49(85):4021. [Google Scholar]
- 21.Salazar-Barda GR, Andrade LS, P Nascente PA, Dizani PS, Rocha-Filho RC, Avaca LA. Electrochim. Acta. 2006;51:4612. [Google Scholar]
- 22.Mahé E, Devilliers D, Comminellis Ch. Electrochim. Acta. 2005;50(90):2263. [Google Scholar]
- 23.Oliveira SCB, Oliveira-Brett AM. Electrochim Acta. 2010;55:4599. [Google Scholar]
- 24.Kiran R, Scorsone E, De Sanoit J, Arnault J-C, Mailley P, Bergonzo P. J. Electrochem. Soc. 2013;95(160):H67. [Google Scholar]
- 25.Park J, Galligan JJ, Fink GD, Swain GM. Anal. Chem. 2006;78:6756. doi: 10.1021/ac060440u. [DOI] [PubMed] [Google Scholar]
- 26.Patel BA, Bian X, Quaiserova-Mocko V, Swain GM. Analyst. 2007;132(41):100. doi: 10.1039/b611920d. [DOI] [PubMed] [Google Scholar]
- 27.Wrona MZ, Dryhurst G. J. Pharm. Sciences. 1988;77:911. doi: 10.1002/jps.2600771102. [DOI] [PubMed] [Google Scholar]
- 28.Cvacká, J J, Quaiserová V, Park J, Swain GM. Anal. Chem. 2003;75:2678. doi: 10.1021/ac030024z. [DOI] [PubMed] [Google Scholar]
- 29.Rotter SZ, Madaleno JC. Chem. Vap. Dep. 2009;15(105):206. [Google Scholar]
- 30.Fhaner M, Zhao H, Bian X, Galligan JJ, Swain GM. Diam. Rel. Mater. 2011;20:75. doi: 10.1016/j.diamond.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao H, Bian X, Galligan JJ, Swain GM. Diam. Rel. Mater. 2010;19(182):110. doi: 10.1016/j.diamond.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bertrand PP, Bertrand RL. Auton. Neurosci. 2010;153:47. doi: 10.1016/j.autneu.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 33.Marcelli G, Patel BA. Analyst. 2010;135:2340. doi: 10.1039/c0an00260g. [DOI] [PubMed] [Google Scholar]
- 34.Hoffman R, Kriele A, Obloh H, Hees J, Wolfer M, Smirnov W, Yang N, Nebel CE. Appl. Phys. Lett. 2010;97(115):052103. [Google Scholar]
- 35.Granger MC, Swain GM. J. Electrochem. Soc. 1999;146:4551. [Google Scholar]
- 36.Duo I, Lévy-Clément C, Fujishima A, Comninellis C. J. Appl. Electrochem. 2004;34(935):120. [Google Scholar]
- 37.Actis P, Denoyelle A, Boukherroub R, Szunerits S. Electrochem. Comm. 2008;10:402. [Google Scholar]
- 38.Brocenschi RF, Rocha-Filho RC, Li L, Swain GM. J. Electroanal. Chem. 2014;712:207. [Google Scholar]
- 39.Brocenschi RF, Rocha-Filho RC, Duran B, Swain GM. Talanta. 2014 doi: 10.1016/j.talanta.2014.02.047. available online at http://dx.doi.org/10.1016/j.talanta.2014.02.047. [DOI] [PubMed]