

Abstract
Group A streptococci (GAS) (Streptococcus pyogenes) are common causes of infections in humans for which there is no licensed vaccine. Decades of work has focused on the role of the surface M protein in eliciting type-specific protective immunity. Recent studies have identified additional surface proteins of GAS that contain opsonic epitopes. In the present study, we describe a serotype M65 GAS originally isolated during an epidemiologic study in Bamako, Mali, which simultaneously expressed M, M-related protein (Mrp), and streptococcal protective antigen (Spa) on the bacterial surface. The emm, mrp, and spa genes were sequenced from PCR amplicons derived from the M65 chromosome. Rabbit antisera raised against synthetic peptides copying the N-terminal regions of M, Mrp, and Spa were highly specific for each peptide, reacted with the surface of M65 GAS, and promoted bactericidal activity against the organism. A mixture of antisera against all three peptides was most effective in the bactericidal assays. Immunofluorescence microscopy revealed that the M, Mrp, and Spa antisera bound to the bacterial surface in the presence of human plasma proteins and resulted in the deposition of complement. Five additional spa genes were identified in the Mrp-positive GAS serotypes, and their sequences were determined. Our results indicate that there are multiple antigens on the surface of GAS that evoke antibodies that promote bacterial killing. A more complete understanding of the relative contributions of M, Mrp, and Spa in eliciting protective immunity may aid in the development of GAS vaccines with enhanced coverage and efficacy.
INTRODUCTION
Group A streptococci (GAS) (Streptococcus pyogenes) are common causes of infections in humans that range from uncomplicated pharyngitis and pyoderma to more serious infections, including sepsis, necrotizing fasciitis, pneumonia, and toxic shock syndrome. The nonsuppurative complications include poststreptococcal glomerulonephritis and acute rheumatic fever. The main driving force behind the development of safe and effective vaccines designed to prevent GAS infections has been the morbidity and mortality associated with invasive infections and rheumatic heart disease (1), which together account for an estimated excess mortality of 500,000 annually (2). Although there are several GAS candidate vaccines in development (3), most efforts have focused on the surface M protein, which contains epitopes that evoke opsonic (bactericidal) antibodies that protect against infection (4).
The seminal work of Lancefield (5) defined the central role of the M protein in protective immunity and resulted in the classical view of serotype-specific protection following natural infection. Since these early studies, additional cell wall proteins have been described that confer virulence and also contain protective epitopes (6–8). M18 and M36 GAS were shown to express the M protein and streptococcal protective antigen (Spa), both of which evoked antibodies that promoted bactericidal activity. M4 GAS expressed M4 and M-related protein (Mrp), and opsonic antibodies were directed against both surface antigens. To our knowledge, the expression of all three protective antigens by a single serotype has not been described. In the present study, we identified a serotype M65 GAS recovered during an epidemiologic study of pharyngitis in Bamako, Mali (9), which simultaneously expressed M, Mrp, and Spa on its surface. We show that antibodies against each surface protein opsonized the organism and promoted bactericidal activity in whole human blood. We also identified and sequenced five additional spa genes from different serotypes of GAS, all of which have been predicted to be Mrp positive (10), suggesting that the expression of these three distinct protective proteins by GAS may not be limited to the M65 serotype.
MATERIALS AND METHODS
Synthesis of M, Mrp, and Spa peptides and immunization of rabbits.
The M65emm and M65mrp sequences were determined from the PCR products that were generated using genomic DNA from the Malian isolate of M65 GAS as the template, as previously described (6). The M65spa sequence was determined using the methods described below. The peptides synthesized (Bio-Synthesis, Lewisville, TX) were those that copied the N-terminal regions of the deduced amino acid sequences of each protein using peptide-antigen algorithms that predict antigenic epitopes (Peptide Antigen Database; Protein Lounge). Each peptide contained a C-terminal cysteine to facilitate coupling to keyhole limpet hemocyanin (KLH), and the peptides were designated sM65, sM65Mrp, and sM65Spa for M, Mrp, and Spa, respectively. The sequence of each peptide was Y17SKLLNENDILRDKQDDYL35C (sM65), L13PGKEANKVFEERKALEKQA32C (sM65Mrp), and K16DSSELIKLITDRNRNRNKM35C (sM65Spa).
New Zealand White rabbits were immunized with 100 μg of each peptide adsorbed to an equal amount of alum at 0, 4, and 8 weeks (11). Serum samples were obtained prior to the first injection and 2 weeks after the final booster injection.
ELISA.
Enzyme-linked immunosorbent assay (ELISA) was performed using the synthetic peptides or whole bacteria as solid-phase antigens, as previously described (6). Nonspecific immunoglobulin binding to the surface of whole streptococci was blocked by the addition of a mixture of 3% pig and 2% goat serum prior to the addition of the test antiserum.
Bactericidal assays.
The bactericidal activities of antisera against sM65, sM65Mrp, and sM65Spa were assessed individually and in various combinations, using previously described assays (12), with some modifications. Briefly, each reaction mixture consisted of 50 μl of M65 GAS grown to early log phase (inoculum of ∼25 CFU), test serum, and 350 μl of fresh nonimmune human blood. For the preparation of the test sera, 33 μl of each of the three antisera was added in various combinations, and sterile phosphate-buffered saline (PBS) was added to bring the final volume added to the rotation mixture to 100 μl. After end-over-end rotation for 3 h at 37°C, an aliquot was removed, and pour plates were made with melted sheep's blood agar. The surviving colonies were enumerated, and the percent killing was expressed as the fraction of the growth of the organism in immune serum compared to that in preimmune serum. The results were expressed as the average killing ± the standard deviation (SD) observed from four experiments, and statistical analyses were performed using a one-way analysis of variance (ANOVA) of the independent samples, followed by Tukey's honestly significant difference (HSD) test.
Immunofluorescence assays.
To detect the binding of M, Mrp, and Spa antibodies to the surface of M65 GAS, bacteria were grown at 37°C in Todd-Hewitt broth (THB) plus 0.5% yeast extract to an optical density (OD) at 546 nm of ∼0.200. Fifteen microliters of 10 mg/ml hyaluronidase (Sigma, St. Louis, MO) was added, and the bacteria were allowed to incubate for approximately 30 min at 37°C. The bacteria were then washed with cold PBS plus 0.05% Tween (PBS-Tween) and resuspended in rabbit antiserum diluted 1:20 in nonimmune human plasma. The bacteria, plasma, and antiserum mixture was rotated at 37°C for 15 min and then washed with PBS-Tween. Fluorescein-conjugated goat anti-rabbit IgG (IgG fraction; MP Biomedicals, Solon, OH) was diluted 1:100 in PBS-Tween and used to resuspend the bacteria. The mixture was again rotated at 37°C for 15 min, washed twice, and resuspended in sterile water. Twenty microliters of the final mixture was dropped on a slide, air-dried, and then mounted in Gelvatol (EMD Chemicals, San Diego, CA) with a coverslip. Fluorescent images were captured using a Zeiss fluorescence microscope (Carl Zeiss Microscopy, Thornwood, NY).
Complement binding to the bacterial surface in the presence and absence of M, Mrp, and Spa antibodies was also detected by fluorescence microscopy. The bacteria were grown and washed as described above and resuspended in nonimmune human plasma that had been heat-inactivated at 56°C for 25 min. The bacteria-and-plasma mixture was incubated at 37°C for 15 min, antiserum diluted 1:20 in PBS was added, and the mixture was incubated at 37°C for 15 min. The bacteria were pelleted, fresh human plasma was added as a source of complement, and the mixture was incubated again at 37°C for 15 min. The bacteria were washed twice with cold PBS-Tween and resuspended in fluorescein-conjugated goat anti-human complement C3 (MP Biomedicals, Solon, OH) diluted 1:50 in cold PBS-Tween, incubated for 15 min at 37°C, washed twice, and resuspended in sterile water. The slides were prepared for fluorescence microscopy as described above.
Identification and sequencing of spa genes.
Our previous studies indicated that the spa genes of M18 and M36 GAS had identical leader sequences, highly conserved 3′ sequences, and shared locations in the chromosome between RelA and NRDI (8). In the current study, the PCR primers used to amplify the M18 and M36 spa genes were unsuccessful, presumably because they contained different 3′ sequences and/or occupied different chromosomal locations. From an ongoing and unrelated genome sequencing project of multiple GAS serotypes, we identified a putative open reading frame in the M15 chromosome that showed homology with M18Spa and M36Spa by BLAST analysis. Thus, we synthesized new PCR primer pairs complementing the putative leader sequence and the 3′ end of the M15 spa gene (Sigma, St. Louis, MO), TGTCTTTTTCGTCTTTTAGGAATAGGA and AAAAGGAGAATAAACAATGCCTAAAAC. These primers amplified putative spa genes using chromosomal templates from M15, M65, M67, M74, and M78. The DNA sequences were determined directly from the purified PCR products at the University of Tennessee Health Science Center Molecular Resource Center. A phylogenetic tree showing the sequence relatedness of the N-terminal 50 amino acids of the Spa proteins was constructed using the Jukes-Cantor tree builder within Geneious (version 6.0.6; Biomatters, Auckland, New Zealand).
RESULTS
Specificities of M, Mrp, and Spa antibodies and binding to M65 streptococci.
Rabbit antisera raised against the synthetic peptides copying the N-terminal regions of M65, M65Mrp, and M65Spa were assayed by ELISA using each of the synthetic peptides as solid-phase antigens (Table 1). Each antiserum contained antibodies with a titer of 12,800 against the respective immunogen. There was no reactivity with the heterologous antigens, indicating that the antibodies were highly specific. The antisera were then tested in ELISA using the whole M65 bacteria as solid-phase antigens. All three antisera reacted with the surface of M65 streptococci (Table 1), indicating that the N-terminal epitopes of the respective proteins were exposed and available for antibody binding. The experiments were performed three times, with similar results (data not shown).
TABLE 1.
Specificity of rabbit antisera against sM65, sM65Mrp, and sM65Spa and reactivity with whole M65 streptococci
| Antiserum | ELISA titer against: |
|||
|---|---|---|---|---|
| sM65 | sM65Mrp | sM65Spa | M65 GAS | |
| Anti-sM65 | 12,800 | <200 | <200 | 1,600 |
| Anti-sM65Mrp | <200 | 12,800 | <200 | 3,200 |
| Anti-sM65Spa | <200 | <200 | 12,800 | 3,200 |
Bactericidal activities of M, Mrp, and Spa antibodies against M65 streptococci.
The functional activities of the antisera against the three surface proteins of M65 GAS were assessed by in vitro bactericidal assays, in which nonimmune human blood served as the source of phagocytes and complement. To determine the relative contributions of the M, Mrp, and Spa antibodies to the promotion of opsonization and phagocytic killing, the assays were performed using the antisera singly or in various combinations (Fig. 1). All three individual antisera promoted bactericidal activity, ranging from 46 to 65%. The bactericidal activity observed with a combination of all three antisera (96%) was significantly greater than that observed with antisera against Mrp or Spa or that with a combination of antisera against M and Mrp (Fig. 1). Taken together, these results indicate that highly specific synthetic peptide antisera against M, Mrp, and Spa reacted with the surface proteins of M65 streptococci, and each antiserum promoted bactericidal activity. A combination of all three antisera resulted in the highest level of bactericidal activity observed.
FIG 1.

Bactericidal activity of M, Mrp, and Spa antisera against M65 GAS. The assays were performed with 33 μl of each of the three antisera, which were added in various combinations, as indicated. Sterile PBS was added to bring the final volume in the test mixture to 100 μl. The percent bacterial killing was calculated based on the reduction in the number of CFU that survived the rotation in the presence of immune serum compared to the number of CFU recovered from tubes containing preimmune serum. The results are expressed as the average killing ± SD observed in four experiments. *, P < 0.05 (ANOVA with Tukey's HSD test).
Patterns of antibody binding and C3 deposition on the surface of M65 streptococci.
Previous studies have demonstrated the importance of the location of antibody binding to the M protein (13) and the pattern of C3 deposition on the bacterial surface (14) in promoting optimal phagocytic uptake of opsonized streptococci. To visualize the binding patterns of M, Mrp, and Spa antibodies and the resultant deposition of complement, immunofluorescence microscopy was performed (Fig. 2). In order to mimic the potential in vivo conditions, antibody binding assays were performed in the presence of human plasma, which contains multiple proteins that bind to the bacterial surface (15–17) that can alter the binding of functional antibodies (18). The M, Mrp, and Spa peptide antisera each reacted with the surface of M65 streptococci in a semicircular pattern at the pole of each coccus, a location that represents the mature cell wall with fully inserted integral proteins (Fig. 2, top panels). Preimmune rabbit serum produced weak fluorescence, which likely resulted from nonspecific IgG binding that was incompletely blocked by human plasma proteins.
FIG 2.
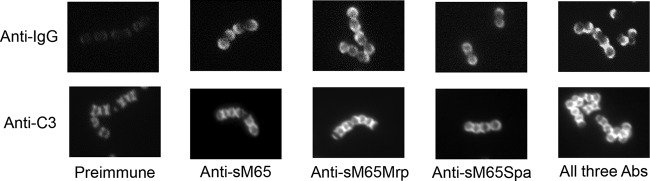
Fluorescence microscopy of M65 streptococci showing the patterns of M, Mrp, and Spa antibody binding to the surface of the organism in the presence of human plasma proteins (top panels) and C3b deposition after antibody binding to the respective antigens (bottom panels). Abs, antibodies.
Although antibody binding to the surface proteins of GAS is necessary for immune-mediated phagocytosis of the organisms, it is the activation and deposition of C3b that constitute the most potent opsonin (14). To visualize the pattern of C3b binding, M65 streptococci were first incubated with M, Mrp, or Spa antisera and then incubated with fluorescent antibody against human C3 (Fig. 2, bottom panels). The antibody binding was performed in the presence of heat-inactivated human plasma, followed by incubation with fresh plasma as a complement source. As shown in previous studies (19), C3 binding to the streptococcal surface in the absence of specific antibodies but in the presence of plasma proteins resulted in complement deposition in a linear or “X” pattern (Fig. 2, bottom left panel). This pattern of binding has been interpreted to represent C3 deposition at the sites of new cross-wall formation that do not contain fully inserted integral proteins. Once these proteins are fully expressed, they are capable of binding plasma proteins that otherwise block the nonimmune recognition of the exposed cell wall. After first reacting the organisms with M, Mrp, or Spa antibodies, followed by fresh human plasma and anti-C3 antibodies, the complement deposition was observed to follow a more circumferential pattern, which included the nonspecific and antibody-mediated deposition of C3. Circumferential C3 deposition has been shown to result in optimal phagocytic uptake because the C3 receptor interacts with C3b on the bacterial surface, functioning as a “zipper” to fully engulf the organisms (14).
Sequences of spa genes from additional emm types of GAS.
The observation that at least one serotype of GAS expressed three distinct integral cell wall proteins that contained opsonic epitopes prompted us to perform additional studies to identify spa genes in organisms that also contain the emm and mrp genes. Genomic DNA was used as a PCR template to amplify potential spa genes from M15, M67, M74, M78, and another M65 strain that was isolated during an epidemiologic study in North America (20). The sequences of the Spa proteins from M18 and M36 GAS were determined previously (7, 8). An analysis of the predicted N-terminal amino acid sequences of the six newly sequenced Spa proteins (GenBank accession no. KJ794096 to KJ794101) and the previously sequenced Spa18 and Spa36 (Fig. 3A) indicated that the Spa sequence from the M65 Malian strain was highly homologous to the Spa sequence from serotype M67 (Fig. 3B). Interestingly, the Spa sequence from the M65 North American strain was completely different from the Spa sequence of the M65 Malian strain. In addition, the predicted sequences of the Spa proteins from M15, M74, and M78 were almost identical, with the exception of a single amino acid substitution in M15Spa.
FIG 3.

Deduced amino acid sequences of the N-terminal 50 amino acids of different Spa proteins (A) and their sequence relatedness depicted by a phylogenetic tree (B).
DISCUSSION
It has been known for decades that M antibodies have the capacity to neutralize the antiphagocytic effect of M protein. Virulent GAS multiply in nonimmune human blood because the M protein specifically binds numerous plasma proteins that block or interfere with the activation and deposition of complement (15). Antibodies against the N-terminal regions of the M protein are opsonic and are able to bind to the M epitopes that extend beyond the layer of human plasma proteins covering the cell surface (18). In the current study, we have shown that serotype M65 GAS expressed three distinct cell wall proteins that contained bactericidal epitopes. Highly specific rabbit antisera against synthetic peptides copying the N-terminal regions of M, Mrp, and Spa reacted with the surface of the organism, activated complement, and promoted phagocytic uptake and killing. A combination of all three antisera against M, Mrp, or Spa resulted in greater bactericidal activity than that with any one antiserum alone. Thus, all three proteins functioned in concert as potential protective antigens.
The seminal studies of Lancefield (5) resulted in the classical view of type-specific immunity to GAS infections. It was thought that each serotype of GAS expressed one M protein with unique opsonic epitopes, and once infected, individuals were protected against subsequent infection by that serotype only. We now understand that GAS, depending on the serotype, display an array of surface proteins that have multiple functions related to protective immunity and/or virulence (15). Previous studies showed that when M and Mrp of serotype 4 streptococci are coexpressed, they function in concert as determinants of virulence, and both contain opsonic epitopes (6). Likewise, M and Spa of type 18 and type 36 streptococci were both required for optimal virulence, and both contained protective epitopes (8, 19). In the current study, type 65 streptococci expressed all three proteins that contained opsonic epitopes. It is of interest that the highest level of bactericidal activity was observed when all three antisera were present in the reaction mixture. This result suggests that because M, Mrp, and Spa were expressed simultaneously in a mosaic pattern on the bacterial surface, antibodies against all three antigens may be required for optimal opsonization and phagocytic killing. In addition, the apparent enhanced functional effect of the antisera against the three distinct surface proteins suggests that competition for antibody binding sites on the bacterial surface did not occur, at least with the antiserum concentrations used in the assays.
Although the roles of M and Mrp in virulence have been studied (6, 16, 21), the precise function of Spa in mediating resistance to phagocytosis is under investigation. Our previous studies showed that the function of Spa in the Mrp-negative GAS serotypes M18 and M36 resembled that of the M protein. Indeed, the 3′ sequences of these spa genes were shown to be highly homologous to those of the seM gene of Streptococcus equi, a group C streptococcus (8), suggesting that GAS acquired the gene via horizontal transfer, and the 5′ region subsequently mutated under immunological pressure from the human host. Using antisera against the conserved C-terminal region of the M18Spa protein, we previously reported that 25/70 GAS serotypes expressed Spa-like proteins on their surface (8), and 24/25 are potentially Mrp positive based on the structure of the mga regulon (10). Analysis of the emm gene cluster of GAS indicates that 61% of all serotypes may express Mrp. The coexpression of M, Mrp, and Spa, coupled with our finding that all three contain opsonic epitopes, has potential implications for vaccine development and design. The addition of dominant protective epitopes, in particular those that are shared among different serotypes of GAS, may broaden the potential efficacy of vaccines designed to prevent these infections.
ACKNOWLEDGMENTS
This work was supported by U.S. Public Health Service grants AI-010085 and AI-060592 (both to J.B.D.) from the National Institute of Allergy and Infectious Diseases. S.E.N., a UTHSC medical student, was the recipient of a Medical Research Scholar Award from the UTHSC Department of Medicine.
We thank Harry Courtney for helpful suggestions and critical review of the manuscript.
Footnotes
Published ahead of print 30 July 2014
REFERENCES
- 1.Dale JB, Fischetti VA, Carapetis JR, Steer AC, Sow S, Kumar R, Mayosi BM, Rubin FA, Mulholland K, Hombach JM, Schödel F, Henao-Restrepo AM. 2013. Group A streptococcal vaccines: paving a path for accelerated development. Vaccine 31(Suppl 2):B216–B222. 10.1016/j.vaccine.2012.09.045. [DOI] [PubMed] [Google Scholar]
- 2.Carapetis JR, Steer AC, Mulholland EK, Weber M. 2005. The global burden of group A streptococcal diseases. Lancet Infect. Dis. 5:685–694. 10.1016/S1473-3099(05)70267-X. [DOI] [PubMed] [Google Scholar]
- 3.Bisno AL, Rubin FA, Cleary PP, Dale JB, National Institute of Allergy and Infectious Diseases 2005. Prospects for a group A streptococcal vaccine: rationale, feasibility, and obstacles—report of a National Institute of Allergy and Infectious Diseases workshop. Clin. Infect. Dis. 41:1150–1156. 10.1086/444505. [DOI] [PubMed] [Google Scholar]
- 4.Dale JB. 2008. Current status of group A streptococcal vaccine development. Adv. Exp. Med. Biol. 609:53–63. 10.1007/978-0-387-73960-1_5. [DOI] [PubMed] [Google Scholar]
- 5.Lancefield RC. 1962. Current knowledge of the type-specific M antigens of group A streptococci. J. Immunol. 89:307–313. [PubMed] [Google Scholar]
- 6.Courtney HS, Hasty DL, Dale JB. 2006. Anti-phagocytic mechanisms of Streptococcus pyogenes: binding of fibrinogen to M-related protein. Mol. Microbiol. 59:936–947. 10.1111/j.1365-2958.2005.04977.x. [DOI] [PubMed] [Google Scholar]
- 7.Dale JB, Chiang EY, Liu SY, Courtney HS, Hasty DL. 1999. New protective antigen of group A streptococci. J. Clin. Invest. 103:1261–1268. 10.1172/JCI5118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahmed EA, Penfound TA, Brewer SC, Tennant PA, Chiang EY, Dale JB. 2010. Streptococcal protective antigens (Spa): a new family of type-specific proteins of group A streptococci. Eur. J. Clin. Microbiol. Infect. Dis. 29:51–57. 10.1007/s10096-009-0819-0. [DOI] [PubMed] [Google Scholar]
- 9.Dale JB, Penfound TA, Tamboura B, Sow SO, Nataro JP, Tapia M, Kotloff KL. 2013. Potential coverage of a multivalent M protein-based group A streptococcal vaccine. Vaccine 31:1576–1581. 10.1016/j.vaccine.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bessen DE, Sotir CM, Readdy TL, Hollingshead SK. 1996. Genetic correlates of throat and skin isolates of group A streptococci. J. Infect. Dis. 173:896–900. 10.1093/infdis/173.4.896. [DOI] [PubMed] [Google Scholar]
- 11.Dale JB. 1999. Multivalent group A streptococcal vaccine designed to optimize the immunogenicity of six tandem M protein fragments. Vaccine 17:193–200. [DOI] [PubMed] [Google Scholar]
- 12.Hu MC, Walls MA, Stroop SD, Reddish MA, Beall B, Dale JB. 2002. Immunogenicity of a 26-valent group A streptococcal vaccine. Infect. Immun. 70:2171–2177. 10.1128/IAI.70.4.2171-2177.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones KF, Fischetti VA. 1988. The importance of the location of antibody binding on the M6 protein for opsonization and phagocytosis of group A M6 streptococci. J. Exp. Med. 167:1114–1123. 10.1084/jem.167.3.1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jacks-Weis J, Kim Y, Cleary PP. 1982. Restricted deposition of C3 on M+ group A streptococci: correlation with resistance to phagocytosis. J. Immunol. 128:1897–1902. [PubMed] [Google Scholar]
- 15.Walker MJ, Barnett TC, McArthur JD, Cole JN, Gillen CM, Henningham A, Sriprakash KS, Sanderson-Smith ML, Nizet V. 2014. Disease manifestations and pathogenic mechanisms of group A streptococcus. Clin. Microbiol. Rev. 27:264–301. 10.1128/CMR.00101-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Courtney HS, Li Y. 2013. Non-immune binding of human IgG to M-related proteins confers resistance to phagocytosis of group A streptococci in blood. PLoS One 8:e78719. 10.1371/journal.pone.0078719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y, Courtney HS. 2011. Promotion of phagocytosis of Streptococcus pyogenes in human blood by a fibrinogen-binding peptide. Microbes Infect. 13:413–418. 10.1016/j.micinf.2010.12.008. [DOI] [PubMed] [Google Scholar]
- 18.Whitnack E, Dale JB, Beachey EH. 1984. Common protective antigens of group A streptococcal M proteins masked by fibrinogen. J. Exp. Med. 159:1201–1212. 10.1084/jem.159.4.1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McLellan DG, Chiang EY, Courtney HS, Hasty DL, Wei SC, Hu MC, Walls MA, Bloom JJ, Dale JB. 2001. Spa contributes to the virulence of type 18 group A streptococci. Infect. Immun. 69:2943–2949. 10.1128/IAI.69.5.2943-2949.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shulman ST, Tanz RR, Dale JB, Beall B, Kabat W, Kabat K, Cederlund E, Patel D, Rippe J, Li Z, Sakota V, North American Streptococcal Pharyngitis Surveillance Group 2009. Seven-year surveillance of North American pediatric group A streptococcal pharyngitis isolates. Clin. Infect. Dis. 49:78–84. 10.1086/599344. [DOI] [PubMed] [Google Scholar]
- 21.Podbielski A, Schnitzler N, Beyhs P, Boyle MDP. 1996. M-related protein (Mrp) contributes to group A streptococcal resistance to phagocytosis by human granulocytes. Mol. Microbiol. 19:429. 10.1046/j.1365-2958.1996.377910.x. [DOI] [PubMed] [Google Scholar]