

Abstract
Nanocarriers have recently emerged as an attractive platform for delivery of various types of therapeutics including anticancer agents. Previously, we developed an improved TPGS delivery system (PEG5K-VE2) which demonstrated improved colloidal stability and greater in vivo antitumor activity. Nevertheless, the application of this system is still limited by a relatively low drug loading capacity (DLC). In this study we report that incorporation of a fluorenylmethyloxycarbonyl (Fmoc) motif at the interfacial region of PEG5K-VE2 led to significant improvement of the system through the introduction of an additional mechanism of drug/carrier interaction. Doxorubicin (DOX) could be effectively loaded into PEG5K-Fmoc-VE2 micelles at a DLC of 39.9%, which compares favorably to most reported DOX nanoformulations. In addition, PEG5K-Fmoc-VE2/DOX mixed micelles showed more sustained release of DOX in comparison to the counterpart without Fmoc motif. MTT assay showed that PEG5K-Fmoc-VE2/DOX exerted significantly higher levels of cytotoxicity over DOX, Doxil as well as PEG5K-VE2/DOX in PC-3 and 4T1.2 cells. Cytotoxicity assay with NCI/ADR-RES, a drug resistant cell line, suggested that PEG5K-Fmoc-VE2 may have a potential to reverse the multidrug resistance, which was supported by its inhibition on P-gp ATPase. Pharmacokinetics (PK) and biodistribution studies showed an increased half-life in blood circulation and more effective tumor accumulation for DOX formulated in PEG5K-Fmoc-VE2 micelles. More importantly, DOX-loaded PEG5K-Fmoc-VE2 micelles showed an excellent safety profile with a MTD (~30 mg DOX/kg) that is about 3 times as much as that for free DOX. Finally, superior antitumor activity was demonstrated for PEG5K-Fmoc-VE2/DOX in both drug-sensitive (4T1.2 and PC-3) and drug-resistant (KB 8-5) tumor models compared to DOX, Doxil, and PEG5K-VE2/DOX.
Keywords: Fluorenylmethyloxycarbonyl, Drug-interactive Motif, Nanomedicine, Reversal of Multidrug Resistance, Chemotherapeutics
Introduction
Doxorubicin (DOX), one of the most potent anticancer agents, has been widely used for the treatment of ovarian, breast, prostate, cervix, brain, and lung cancers. It functions by intercalating between base pairs of the DNA helix, resulting in the suppression of DNA synthesis [1–3]. In addition, doxorubicin stabilizes the topoisomerase II-DNA complex after it has broken the DNA chain for replication, which prevents the DNA double helix from being resealed and thereby arrests the process of replication [1,4]. However, its clinical application has been compromised by its limited efficacy in vivo and systemic side effects [5,6]. Hence, there is a need to develop an effective drug carrier to specifically deliver DOX to tumors.
In the last two decades, D-α-tocopheryl polyethylene glycol succinate (TPGS) has gained increasing attention as an ideal biomaterial in developing various drug delivery systems such as micelles, liposomes, and other nanoparticles [7–13]. TPGS is able to function as a solubilizer, emulsifier, additive, permeability enhancer as well as absorption enhancer [14,15]. Additionally, TPGS is capable of overcoming multidrug resistance mediated by P-gp efflux pump [15–18]. As a stand-alone micellar formulation, the performance of TPGSs is affected by the molecular weight of PEG and the molar ratio of Vitamin E/PEG in the conjugates [19]. Previously, our lab developed PEG5K-VE2 nanomicelles comprising one molecule of polyethylene glycol 5000 and two molecules of Vitamin E succinate, which demonstrated improved performance in formulating and delivering paclitaxel (PTX) over other TPGS micellar formulations (TPGS5K, TPGS2K and PEG2K-Vitamin E2) [19]. Nonetheless, this improved system still has limited drug loading capacity. This is likely due to the fact that loading of drugs into these micellar formulations is largely driven by hydrophobic interaction. Such mechanism of carrier/drug interaction, while working well for highly hydrophobic drugs, may show limited effectiveness for many moderately hydrophobic drugs. This limitation is also shared by many other existing micellar systems [19–22].
Recently, Park’s group has demonstrated that inclusion of a hydrotropic molecule into the hydrophobic part of a block copolymer was able to improve the accommodation of drugs that are not entirely hydrophobic or lipophilic [23,24]. This was mainly due to the additional mechanism of carrier/drug interaction rendered by the hydrotropic molecules introduced. Hydrotropes, small amphiphilic molecules, are capable of solubilizing hydrophobic compounds in aqueous solutions through hydrogen bonding. Incorporation of the hydrotropic molecule into polymeric micelles led to improvement in both drug loading capacity and the colloidal stability of drug-formulated micelles.
We have recently shown that incorporation of a drug-interactive domain at the interfacial region of PEGylated lipopeptides resulted in a significant improvement in loading of hydrophobic drugs [25,26]. Among a number of functional motifs examined, fluorenylmethyloxycarbonyl (Fmoc), a commonly used amine protecting group in peptide chemistry, was found to be the most effective drug-interactive group in facilitating carrier/drug interaction [25]. A PEGylated lipopeptide with a built-in Fmoc at the interfacial region was effective in formulating various types of therapeutic agents of diverse structures [26]. Delivery of PTX via this formulation led to significant inhibition of tumor growth in a murine breast cancer model (4T1.2) [26].
This study is focused on the development of a new TPGS-based nanomicellar system with a built-in drug interactive motif, PEG5K-Fmoc-VE2. We hypothesized that incorporation of Fmoc at the interfacial region of PEG5K-VE2 shall greatly improve its performance in formulating and delivering hydrophobic anticancer agents. DOX was used as a model hydrophobic drug. Systematic comparison between PEG5K-VE2 and PEG5K-Fmoc-VE2 micelles was conducted with respect to the drug loading capacity and efficiency, stability, intracellular uptake, maximum tolerated dose, as well as in vitro and in vivo antitumor efficacy. Our data showed that PEG5K-Fmoc-VE2 was more effective than PEG5K-VE2 in forming stable mixed micelles with DOX. More importantly, DOX-loaded PEG5K-Fmoc-VE2 micelles exhibited significantly improved antitumor activity in vivo.
Materials and methods
Materials
Doxorubicin hydrochloride (98%) (DOX.HCl) was purchased from AK Scientific Inc. (CA, USA). Doxil was purchased from Avanti® Polar Lipids (AL, USA). Methoxy-PEG5,000-OH, succinate anhydride, Boc-lys-(Boc)-OH, Fmoc-lys-(Boc)-OH, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), aspartic acid, trypsin-EDTA solution, Triton X-100, and Dulbecco’s Modified Eagle’s Medium (DMEM) were all purchased from Sigma-Aldrich (MO, USA). D-alpha-tocopherol was purchased from Tokyo Chemical Industry (OR, USA). N,N′-dicyclohexylcarbodiimide (DCC) was purchased from Alfa Aesar (MA, USA). 4-Dimethylaminopyridine (DMAP) was purchased from Calbiochem-Novabiochem Corporation (CA, USA). Fetal bovine serum (FBS) and penicillin-streptomycin solution were from Invitrogen (NY, USA). All solvents used in this study were HPLC grade.
Synthesis of PEG5K-Fmoc-VE2 and PEG5K-VE2
PEG5K-VE2 was synthesized as reported previously [27]. PEG5K-Fmoc-VE2 was synthesized as follows: First, Fmoc-lys(Boc)-OH (4 eq.) was coupled to the terminal -OH of MeO-PEG5K-OH with the assistance of DCC (4 eq.) and DMAP (0.2 eq.) in DCM overnight. The resulting PEG5K-Fmoc-lys-(Boc) was washed and precipitated thrice by cold ethanol and ether, respectively. Boc group was then removed with 50% trifluoroacetic (TFA) acid in DCM for 2 h, and the resulting (PEG5K-Fmoc-lys-NH2) was precipitated by cold ethanol and ether thrice, respectively. Boc-lys(Boc)-OH (2 eq.) was then conjugated to the terminal -NH2 of PEG5K-Fmoc-lys via DCC (2 eq.) and DMAP (0.1 eq.) as coupling reagents in DCM overnight. The PEG5K-Fmoc-di-Boc obtained was precipitated and washed thrice with cold ethanol and ether, respectively. Boc groups in PEG5K-Fmoc-di-Boc were removed by 50% TFA in DCM for 2 h, and the resulting (PEG5K-Fmoc-di-NH2) was purified via precipitation in cold ethanol and ether, respectively. Finally, Vitamin E succinate (4 eq.) was coupled onto the deprotected amino groups of PEG5K-Fmoc-di-NH2 with DCC (4 eq.) and DMAP (0.2 eq.) as coupling reagents in DCM overnight to produce PEG5K-Fmoc-VE2. The resulting PEG5K-Fmoc-VE2 was further purified via precipitation in cold ethanol and ether, respectively. The purified PEG5K-Fmoc-VE2 was dried under vacuum prior to use. The identity and purity of PEG5K-Fmoc-VE2 were confirmed by 1NMR, MALDI-TOF and HPLC.
Preparation and characterization of DOX-loaded micelles
DOX·HCl was first neutralized by 3 molar eq. triethylamine in CHCl3/MeOH (1:1. v:v) to eliminate HCl. PEG5K-Fmoc-VE2/DOX mixed micelles were prepared as described before [27]. Briefly, PEG5K-Fmoc-VE2 (10 mM in CHCl3) was mixed with DOX (10 mM in CHCl3/MeOH) under different carrier/drug molar ratios. The mixture was first dried by nitrogen flow to form a thin dry film. The dry film was further dried under vacuum for 4 h to remove any traces of remaining solvent. The film was then reconstituted in saline without sonication. The micellar formulations were filtered through a 0.22 μm filter prior to characterization. The DOX-loaded PEG5K-VE2 micelles were similarly prepared as mentioned above. The mean diameter and size distribution of micelles with or without loaded drug were evaluated by dynamic light scattering (DLS). The morphology of DOX-free or DOX-loaded micelles was evaluated under TEM. The concentration of DOX in micelles was assessed by HPLC-Fluorescence detection (Excitation: 490 nm; Emission: 590 nm). The drug loading capacity (DLC) and drug loading efficiency (DLE) were calculated according to the following formula:
DOX release kinetics
The cumulative release kinetics of DOX in vitro was conducted by dialysis technique using DPBS (PH = 7.4) containing 0.5% (w/v) Tween 80 as the release medium. Free DOX was utilized as a control. Two mL of DOX-loaded PEG5K-VE2 or PEG5K-Fmoc-VE2 micelles (1 mg DOX/mL) were sealed in dialysis tubes (MWCO = 12 KDa, Spectrum Laboratories). The dialysis tubes were immersed in 500 mL release medium in a beaker capped with parafilm. The beakers were kept in an incubator shaker at 100 rpm and 37°C. At predetermined time points, aliquots of samples were collected, diluted, and filtered through a 0.22 μm filter prior to analysis by HPLC with the detector set at 490 nm. Values were reported as the means from triplicate samples.
Fluorescence quenching studies
PEG5K-VE2/DOX and PEG5K-Fmoc-VE2/DOX were prepared at varying carrier to drug molar ratio as mentioned above. To examine fluorescence quenching of the carriers, the molar concentrations of carriers were kept constant for comparison. In another set of experiment, DOX concentration was kept constant to examine the fluorescence quenching of DOX. The samples were placed into a 96-well plate, and the fluorescence intensity of carriers was recorded on a Synergy H1 Hybrid reader (BioTek), using an excitation wavelength of 270 nm and emission wavelength from 300–500 nm. The fluorescence intensity of DOX was examined using an excitation wavelength of 480 nm and emission wavelength from 510–650 nm.
UV absorbance spectroscopy of DOX
The absorption spectra were collected using Varian Cary 50 Bio UV-Visible Spectrophotometer over a wavelength ranging from 300 to 800 nm. PEG5K-VE2/DOX and PEG5K-Fmoc-VE2/DOX were prepared at a molar ratio of 2.5/1. All samples in distilled water were loaded into a quartz cell and measured against distilled water as the reference.
Fourier transform infrared spectroscopy (FT-IR)
FT-IR of DOX, DOX-loaded PEG5K-VE2, and DOX-loaded PEG5K-Fmoc-VE2 was evaluated using a VERTEX 70/70v FT-IR spectrometer (Bruker) to determine the hydrogen bonding of carrier/carrier and carrier/drug in the frequency of 4997-500 cm−1 (KBr pellet).
Hemolytic effect of PEG5K-Fmoc-VE2
The hemolytic activity of PEG5K-Fmoc-VE2 was examined using rat red blood cells (RBCs) following our published method [27].
Cell culture
Mouse breast cancer cell line, 4T1.2, human prostate cancer cell line, PC-3, and adriamycin (ADR)-resistant cell line, NCI/ADR-RES, were used in this work. All cell lines were cultured in DMEM containing 10% FBS and 1% penicillin-streptomycin in a humidified environment at 37 °C with 5% CO2.
Animals
Male and female and nude mice of 6–8 weeks of age were purchased from Harlan (Livermore, CA). Female BALB/c mice of 10–12 weeks of age were purchased from Charles River (Davis, CA). All animals were housed under pathogen-free conditions according to AAALAC (Association for Assessment and Accreditation of Laboratory Animal Care) guidelines. All animal-related experiments were performed in full compliance with institutional guidelines and approved by the Animal Use and Care Administrative Advisory Committee at the University of Pittsburgh.
In vitro cytotoxicity
The cytotoxicity of DOX formulated in PEG5K-VE2 or PEG5K-Fmoc-VE2 was evaluated in 4T1.2, PC-3, and NCI/ADR-RES cell lines, in comparison to free DOX and Doxil. Briefly, cells were seeded in 96-well plates followed by overnight attachment. Then cells were treated by different DOX formulations of varied concentrations. Free PEG5K-VE2 or PEG5K-Fmoc-VE2, at concentrations equivalent to those of carriers in the corresponding DOX formulation groups, was also added into cells. Cells were incubated for 72 h and cell viability was determined by MTT assay [19].
Intracellular uptake
3×105 NCI/ADR-RES cells/well were seeded into 6-well plates and incubated overnight prior to any treatment. Then cells were treated with free DOX, Doxil, and DOX-loaded PEG5K-VE2 and PEG5K-Fmoc-VE2, respectively at a DOX concentration of 6 μg/mL. Cells were then incubated for 1 h or 3 h at 37 °C. After that, cells were washed three times with cold PBS and fixed with 4% paraformaldehyde for 30 min. The nuclei were then stained by Hoechst33342 for 5 min. Subsequently, cells were washed thrice with cold PBS. The intracellular uptake of DOX in various formulations was observed under confocal laser scanning microscopy (CLSM, FluoView 1000, Olympus, Japan).
Quantitative cellular uptake of various DOX formulations was evaluated by flow cytometry. Briefly, NCI/ADR-RES cells were prepared as described above. Following treatment with various DOX formulations at 37 °C for 1 h or 3 h, cells were washed with cold PBS thrice, and resuspended in 500 μL PBS prior to the flow cytometry analysis with CyAn™ ADP Analyzer (Beckman Coulter, Inc.). Cell-associated DOX was excited with an argon laser (480 nm), and fluorescence was detected at 570 nm. Twenty-thousand events were collected for each sample.
P-gp ATPase assay
The effect of PEG5K-Fmoc-VE2 conjugate on P-gp was investigated through examining its impact on a verapamil-stimulated ATPase activity as reported previously [27].
Near infrared fluorescence (NIRF) imaging
The tumor-targeting efficiency and biodistribution of PEG5K-Fmoc-VE2 in vivo was examined using a near infrared fluorescence dye, DiD. Nude mice bearing PC-3 xenograft were employed in this study. Two hundred μL of DiD-loaded PEG5K-Fmoc-VE2 (DiD concentration in the formulation was 0.4 mg/mL) was i.v. injected into each mouse. At different time points (0.5 h, 6 h, 24 h, 48 h, 72 h and 96 h) post-injection, mice were imaged under Carestream Molecular Imaging System (Carestream Health, Inc.) with the excitation at 630 nm and the emission at 700 nm using an exposure time of 60 s. After 96 h, mice were euthanized and tumors and major organs were excised and imaged with Carestream Molecular Imaging System. The tissue distribution of DiD in tumors and other organs was quantified by measuring the signal intensity at the region of interest.
Pharmacokinetics and biodistribution
PEG5K-VE2/DOX, PEG5K-Fmoc-VE2/DOX, and free DOX were administered i.v. at a dose of 5 mg DOX/kg in 200 μL saline (n = 3). At predetermined time points (3 min, 8 min, 15 min, 30 min, 45 min, 1 h, 2 h, 4 h, 8 h and 12 h), blood was obtained from mice using a heparinized capillary tube. Plasma samples were isolated from the blood by centrifuging at 3000 rpm for 10 min. DOX in plasma was extracted via an extraction buffer (10% Triton X-100, deionized water, and isopropanol at a volumetric ratio of 1:2:15). The concentration of DOX at various time points was measured by HPLC with the detector set at 490 nm (Waters Alliance 2695 Separations Module combined with Waters 2998 Photodiode Array Detector, Waters Symmetry C18 5 μm 4.6*250 mm column, mobile phase: 80% MeOH:20% H2O isocrate, flow rate: 0.6 mL/min). Pharmacokinetic parameters such as t1/2, volume of distribution (Vd), area under the curve (AUC), and clearance (CL) were calculated by fitting the blood DOX concentrations to a non-compartment model using Phoenix WinNonlin.
For biodistribution study, free DOX and DOX-loaded micelles were i.v. injected into 4T1.2 tumor-bearing mice at a dose of 5 mg DOX/kg, respectively (n = 3). At 24 h post-injection, major organs (liver, spleen, lung, heart, and kidney), tumors, and blood were collected from the mice. Tissues were then homogenized utilizing a Power Gen 500 homogenizer (Fisher Scientific), in which 100 mg tissues were mixed with 900 μL extraction buffer, and DOX was extracted overnight at −20 °C. The samples were centrifuged at 3,000 rpm for 10 min, and the supernatant was then dried and dissolved in 400 μL 75% MeOH. Subsequently, the samples were further centrifuged at 14,500 rpm for 5 min to remove undissolved materials prior to HPLC measurement as mentioned above. The % injected dose and the % injected dose per gram (tissue) were calculated using the following equations:
Maximum tolerated dose (MTD)
Groups of 3 female BALB/c mice were treated intravenously with free DOX (5, 10, and 15 mg DOX/kg), DOX-loaded PEG5K-VE2 (5, 10, 15, 20, and 30 mg DOX/kg) or DOX-loaded PEG5K-Fmoc-VE2 (10, 15, 20, 25, 30, and 35 mg DOX/kg), respectively. Changes in body weight and general signs, and survival of mice were followed daily for two weeks. The MTD was determined as the maximal dose that causes neither mouse mortality nor greater than 15% loss in body weight and other significant changes in the general movement and signs within the entire period of the experiment.
In vivo antitumor efficacy
Both drug-sensitive (4T1.2 and PC-3) and drug-resistant (KB 8-5) tumor models were used to evaluate the therapeutic effect of DOX formulated in PEG5K-VE2 and PEG5K-Fmoc-VE2 micelles. 4T1.2 is an aggressive syngeneic murine breast cancer model and PC-3 and KB 8-5 are human prostate and cervical cancer xenograft model, respectively.
In the breast cancer model, 1 × 105 4T1.2 cells in 200 μL PBS were inoculated s.c. at the right flank of female BALB/c mice. Various treatments were started when tumors reached a tumor volume around 50 mm3 and this day was named as day 1. On day 1, mice were randomly divided into six groups (n = 5) and received i.v. administration of free DOX (10 mg DOX/kg), Doxil (10 mg DOX/kg), DOX-loaded PEG5K-VE2 (10 mg DOX/kg), DOX-loaded PEG5K-Fmoc-VE2 (10 mg DOX/kg), DOX-loaded PEG5K-Fmoc-VE2 (20 mg DOX/kg) and saline, respectively on days 1, 4, and 7. Tumor sizes were monitored with a digital caliper on days 1, 4, 7, 10, 12,15, 18, 21, and 25 and calculated based on the formula: (L×W2)/2, where L and W are length and width of each tumor. To compare between groups, relative tumor volume (RTV) was calculated at each measurement time point (where RTV equals the tumor volume at a given time point divided by the tumor volume prior to first treatment). Mice were sacrificed if tumors reached 2000 mm3 or developed ulceration. Tumor growth inhibition rate (IR) was also calculated and defined as following:
Furthermore, to monitor the systemic toxicity, the body weights of mice were measured at the time of tumor size measurement.
In PC-3 xenograft tumor model, 2×106 PC-3 cells in 200 μL PBS were inoculated s.c. at the right flank in male nude mice. Treatments were initiated when tumors in the mice reached a volume around 50 mm3 and different groups (n = 6) were similarly treated as mentioned above on days 1, 4, and 7. Tumor size and body weight were monitored as described above.
In KB 8-5 xenograft tumor model, 5×106 cells were inoculated s.c at the right flank of female nude mice. When the tumor volume reached approximately 50 mm3, 6 groups of mice (n = 5) were treated with saline, DOX (5 mg/kg), DOX (7.5 mg/kg), PEG5K-VE2/DOX (5 mg/kg), PEG5K-Fmoc-VE2/DOX (5 mg/kg), and PEG5K-Fmoc-VE2/DOX (7.5 mg/kg), respectively, on days 1, 4, and 7. Tumor size and body weight were monitored very three days.
Statistical analysis
In all statistical analyses, the significance level was set at a probability of P < 0.05. All results were reported as the mean ± standard deviation (SD) unless otherwise indicated. Statistical analysis was performed by using the Student’s t-test for two groups, and one-way ANOVA for multiple groups, followed by Newman-Keuls test if P < 0.05.
Results and discussion
Coupling of Fmoc motif to PEG5K-VE2 at the interfacial region
In an effort to improve the TPGS-based micellar formulation, we have previously synthesized and characterized four PEG-Vitamin E conjugates that vary in the molecular weight of PEG (2 vs 5K) and the molar ratio of PEG/Vitamin E (1/1 vs 1/2) [27]. Our results showed that the conjugate with one molecule of PEG5K and two molecules of Vitamin E (PEG5K-VE2) was most effective in formulating and delivering PTX to tumor cells in vitro and in vivo. In this study, Fmoc was incorporated into PEG5K-VE2 at the interfacial region to further improve the performance of this carrier. Our recent study has shown that Fmoc is a highly effective drug-interactive motif and that incorporation of this motif into lipid-core micelles greatly facilitates the carrier/drug interaction. The synthesis route of PEG5K-Fmoc-VE2 was presented in Scheme 1. Briefly, MeO-PEG5K-OH was first reacted with Fmoc-lys-(Boc)-OH to yield PEG5K-Fmoc-lys-(Boc) followed by the removal of Boc to expose the terminal NH2. PEG5K-Fmoc-lys-NH2 was then coupled to Boc-lys-(Boc) to generate PEG5K-Fmoc-di-Boc followed by the removal of Boc groups. Finally, Vitamin E succinate was conjugated to PEG5K-Fmoc-di-NH2 to produce PEG5K-Fmoc-VE2. The chemical identity of the final product was verified by 1NMR (Fig. 2) and MALDI-TOF (Fig. S1). HPLC showed that the purity of the conjugate was 97.8% (Fig. S2).
Scheme 1.

Synthetic route of PEG5k-Fmoc-VE2 conjugate
Fig. 2.

1NMR spectra of free DOX in D2O, PEG5K-Fmoc-VE2 in D2O or DMSO and DOX formulated in PEG5K-Fmoc-VE2 in D2O or DMSO. Concentration of DOX was 1 mg/mL.
Physicochemical characterizations of DOX-free and DOX-loaded PEG5K-Fmoc-VE2 micelles
In aqueous solution, PEG5K-Fmoc-VE2 readily self-assembled into elongated worm-like nanoassemblies with a particle size around 55 nm as determined by DLS analysis (Fig. 1A, Tab. 1). This is in contrast to PEG5K-VE2 without Fmoc motif that formed typical spherical particles (Fig. S3A). A similar result was found in a recent study with an Fmoc-containing PEG-lipopeptide [26]. The formation of the filamentous structure is likely attributed to the Fmoc-mediated strong interaction among the carrier molecules since Fmoc-containing peptides are known to self-assemble into nanofibers or nanotubues [28–30]. However, in the case of Fmoc-containing PEG-lipopeptide, both lipid and Fmoc contributed to the formation of filamentous structure as the counterpart without lipid formed spherical structure [26]. It is likely that both the benzene ring and the alkane chain of Vitamin E are involved in the formation of filamentous structure.
Fig. 1.

Size distribution and TEM of PEG5K-Fmoc-VE2 (A), and PEG5K-Fmoc-VE2/DOX (0.1:1 m/m) (B).
Table 1.
Biophysical characterization of DOX-loaded PEG5K-VE2 or PEG5K-Fmoc-VE2
| Molar ratios | Size | PDI | DLC (%) | DLE (%) | |
|---|---|---|---|---|---|
| PEG5K-VE2 | N/A | 18.8 ± 0.12 | 0.09 | NA | N/A |
|
| |||||
| PEG5K-VE2:DOX | 2.5:1 | 23.2±0.91 | 0.15 | 2.9 | 84.3 |
| 24.5±0.17 (89 h later) | 0.24 | ||||
|
| |||||
| PEG5K-Fmoc-VE2 | N/A | 55.0±0.84 | 0.12 | ||
| 50.5±1.42 (One mouth later) | 0.23 | ||||
|
| |||||
| PEG5K-Fmoc-VE2:DOX | 0.1:1 | 61.4±2.36 | 0.21 | 39.9 | 79.5 |
| 62.4±1.83 (42 h later) | 0.25 | 86.4 | |||
| 0.2:1 | 53.6±1.54 | 0.18 | 26.5 | ||
| 56.8±1.87 (96 h later) | 0.24 | ||||
| 0.5:1 | 51.1±2.12 | 0.12 | 13.9 | 96.3 | |
| 52.7±1.89(290 h later) | 0.23 | ||||
Interestingly, following the incorporation of DOX into PEG5K-Fmoc-VE2, the morphology of the nanoparticles started to change drastically from filamentous (Fig. 1A) to spherical structure (Fig. 1B). This is likely due to the fact that the carrier/drug interaction impacted the interaction among the carrier molecules themselves following the incorporation of DOX, leading to the structural rearrangement. Both PEG5K-VE2 and DOX-loaded PEG5K-VE2 micelles were found to be spherical with a diameter around 20 nm (Fig. S3).
Tab. 1 compares the DOX loading capacity (DLC) and efficiency (DLE) of PEG5K-VE2 and PEG5K-Fmoc-VE2. For PEG5K-VE2, a minimal carrier/drug molar ratio of 2.5/1 was needed to form stable mixed micelles with DOX. In contrast, only a carrier/drug ratio of 0.1/1 was needed for PEG5K-Fmoc-VE2 to form stable mixed micelles with DOX. At this ratio, the DLC for PEG5K-Fmoc-VE2 was 39.9% which is about 13-fold higher than that (2.9%) for PEG5K-VE2. Increasing the carrier/drug ratio was associated with a further increase in both DLE and the colloidal stability of the DOX-loaded micelles.
To confirm whether DOX was indeed incorporated into the PEG5K-Fmoc-VE2 nanoassemblies, 1NMR studies were conducted for a number of samples including free DOX in D2O, PEG5K-Fmoc-VE2 in D2O or DMSO, and DOX formulated in PEG5K-Fmoc-VE2 in D2O. The concentration of DOX was kept at 1 mg/mL. As shown in Fig. 2, free DOX showed a 1H NMR spectrum in D2O that was consistent with data from literature [31]. A 1H NMR spectrum was discerned for PEG5K-Fmoc-VE2 in DMSO that matched to its structure (Fig. 2). However, when the 1H NMR spectrum of PEG5K-Fmoc-VE2 was collected in D2O, the VE signals (0.5–3 ppm) and Fmoc signals (7–8.5 ppm) were substantially suppressed (Fig. 2). This could be ascribed to the self-assembling process of PEG5K-Fmoc-VE2, as VE and Fmoc are hydrophobic and aggregated inward, while the highly hydrophilic PEG is oriented outward into the water phase, resulting in the shielding of the signals of VE and Fmoc by PEG. Similarly, a shielding of VE and Fmoc signals was observed when PEG5K-Fmoc-VE2/DOX was examined in D2O (Fig. 2). In addition, the typical spectrum of DOX was barely visible, indicating that DOX was effectively encapsulated into the interior core of PEG5K-Fmoc-VE2 micelles. Individual NMRs for DOX in DMSO or D2O, PEG5K-Fmoc-VE2 in DMSO or D2O, and PEG5K-Fmoc-VE2/DOX in DMSO or D2O are provided in Fig. S4, Fig. S5, Fig. S6, Fig. S7, Fig. S8, Fig. S9, respectively.
DOX release kinetics in vitro
To further understand the stability of DOX-loaded nanomicelles, the in vitro release kinetics of DOX was evaluated with dialysis method using DPBS (PH = 7.4) containing 0.5% (w/v) Tween 80 as the release medium. Free DOX was employed as a control. As shown in Fig. 3, PEG5K-VE2/DOX micelles exhibited significantly slower release kinetics over free DOX. During the first 9 h, 92.14% of DOX was released in the free DOX group, which was substantially higher than that in PEG5K-VE2 micelles (36.57%). Notably, after introducing Fmoc group into PEG5K-VE2, the release rate of DOX was further reduced. In addition, there was essentially no initial burst release for DOX-loaded PEG5K-Fmoc-VE2 nanoassemblies. Furthermore, DOX formulated in PEG5K-Fmoc-VE2 exhibited significantly slower DOX release over PEG5K-VE2/DOX during the entire period. The amount of released DOX after 74 h was only 36.45% for PEG5K-Fmoc-VE2/DOX mixed micelles, which is significantly less than that for free DOX (96.35%) and PEG5K-VE2/DOX (58.44%). The significantly slower and controlled release of DOX in DOX-loaded PEG5K-Fmoc-VE2 formulation may be attributed to the strong interactions between the carrier and DOX as well as among the carrier molecules themselves.
Fig. 3.

DOX cumulative release kinetics from free DOX, DOX-loaded PEG5K-VE2 and DOX-loaded PEG5K-Fmoc-VE2 micelles. DPBS (PH = 7.4) containing 0.5% (W/V) Tween 80 was used as the release medium. DOX concentration was kept at 1 mg/mL. Values reported are the means ± SD for triplicate samples.
Mechanism of interacrions between carrier and payload
The significantly improved DLC of PEG5K-Fmoc-VE2 over PEG5K-VE2 suggests a role of Fmoc/DOX interaction in the formation of PEG5K-Fmoc-VE2/DOX mixed micelles. To test this hypothesis, we examined the Fmoc/DOX interaction via fluorescence quenching assay. As shown in Fig. 4A, there is a distinct fluorescence peak at 310 nm, which is consistent with the fluorescence spectrum of Fmoc [32]. Incorporation of DOX into PEG5K-Fmoc-VE2 led to a significant quenching of the fluorescence in a dose-dependent manner. Fig. 4B shows the DOX fluorescence intensity at 595 nm before and after incorporation into PEG5K-Fmoc-VE2. Likewise, PEG5K-Fmoc-VE2 caused the quenching of DOX fluorescence in a dose-dependent manner. The quenching of either Fmoc or DOX fluorescence is likely due to the energy transfer triggered by the intermolecular π-π stacking interaction between the Fmoc motif and the aromatic rings of DOX.
Fig. 4.

A: fluorescence intensity of the carriers. B: Fluorescence change of DOX. C: UV-absorbance of DOX.
We also noticed a fluorescence emission at 300 nm from PEG5K-VE2 (Fig. 4A). This is likely attributed to the benzene ring in Vitamin E as no fluorescence emission was detected at this wavelength from a similar conjugate of PEG with two molecules of oleic acid (data not shown). Similarly, PEG5K-VE2 and DOX caused the quenching of the other’s fluorescence in a dose-dependent manner (Fig. 4A & B), suggesting that the benzene ring in Vitamin E is also involved in the carrier/DOX π-π stacking interaction, albeit to a lesser extent.
Fig. 4C shows the UV-absorbance of free DOX and DOX formulated in PEG5K-VE2 or PEG5K-Fmoc-VE2 micelles. A clear red shift of DOX UV-absorbance was detected in both micellar formulations, particularly in PEG5K-Fmoc-VE2/DOX mixed micelles, further supporting the notion that π-π stacking contributed to the carrier/DOX interactions.
In addition to hydrophobic interaction and π-π stacking, hydrogen bonding is likely to be involved in the carrier (PEG5K-VE2 or PEG5K-Fmoc-VE2)/DOX interaction. The carbamate group in Fmoc, the amide bond in lysine, as well as the ester linkage in between Vitamin E and lysine could interact with the hydroxyl groups in DOX through hydrogen bonding. To test this hypothesis, FT-IR was employed to compare the hydrogen bonding in DOX-loaded micelles and that in carrier alone. As shown in Fig. 5B, after subtracting the IR of PEG5K-Fmoc-VE2 from that of PEG5K-Fmoc-VE2/DOX, two peaks at 1637.1 cm−1 and 1754.3 cm−1 were detected. The reduction of absorbance at 1754.3 cm−1 and a simultaneous increase of absorbance at 1637.1 cm−1 suggest that incorporation of DOX led to disruption of the hydrogen bonding among carrier molecules themselves and, at the same time, the formation of new hydrogen bonding between carrier and DOX molecules. Similar results were found for PEG5K-VE2 micellar system (Fig. 5D).
Fig. 5.

A: FT-IR of DOX, PEG5K-Fmoc-VE2, and PEG5K-Fmoc-VE2/DOX. B: Difference of FT-IR between PEG5K-Fmoc-VE2 and PEG5K-Fmoc-VE2/DOX. C: FT-IR of DOX, PEG5K-VE2, and PEG5K-VE2/DOX. D: Difference of FT-IR between PEG5K-VE2 and PEG5K-VE2/DOX.
Evaluation of the hemolytic activity of PEG5K-Fmoc-VE2
As an intravenous formulation, the potential hemolytic activity of PEG5K-Fmoc-VE2 micellar formulation needs to be addressed. Fig. 6 shows the result of a hemolysis assay. PEI, a cationic polymer with potent cell surface activity, was included as a positive control. Treatment with PEI led to significant hemolysis in a dose-dependent manner. At a concentration of 1 mg/mL, almost all of the red blood cells were lysed by PEI. In contrast, only negligible level of hemolysis was observed for PEG5K-Fmoc-VE2 even at the concentration of 1 mg/mL. The very low level of hemolytic effect of PEG5K-Fmoc-VE2 indicates that PEG5K-Fmoc-VE2 is a mild surfactant that can be safely administered intravenously.
Fig. 6.

In vitro hemolysis assay of PEG5K-Fmoc-VE2 in comparison to PEI. PEG5K-Fmoc-VE2 and PEI of various concentrations were incubated with rat red blood cells (RBCs) for 4 h in an incubator shaker at 37 °C. The degree of RBCs lysis was determined spectrophotometrically (ƛ) according to the release of hemoglobin. 2% Triton X-100 and DPBS were used as a positive and negative control, respectively. Values reported are the means ± SD for triplicate samples.
In vitro cytotoxicity
Fig. 7 shows the cytotoxic effect of different DOX formulations on murine breast cancer cells, 4T1.2. Cells were challenged by DOX, Doxil, PEG5K-VE2/DOX and PEG5K-Fmoc-VE2/DOX at various concentrations of DOX, and the cytotoxicity was determined by MTT assay 72 h later. A time- and concentration-dependent cell-killing effect was shown for all DOX formulations in 4T1.2 cells. The IC50 was 186.70, 253.83, 89.21, and 77.57 ng/mL for DOX, Doxil, PEG5K-VE2/DOX, and PEG5K-Fmoc-VE2/DOX, respectively. The higher levels of cytotoxicity for the two micellar DOX formulations were likely due to an increased cellular uptake of DOX and effective release following intracellular delivery (Fig. 7). Doxil was slightly less active than free DOX, which is likely due to the ineffective release of DOX from the liposomes following intracellular delivery [33].
Fig. 7.

The cytotoxicity of PEG5K-VE2/DOX and PEG5K-Fmoc-VE2/DOX against mouse breast cancer cell line, 4T1.2, compared to DOX and Doxil. Cells were treated with indicated concentrations of different DOX formulations for 72 h and cytotoxicity was then examined by MTT assay. Values reported are the means ± SD for triplicate samples.
Reversal of multidrug resistance
Drug resistance is one of the major factors involved in the failure of chemotherapy [34]. Various mechanisms have been identified that are involved in the different types and/or stages of cancers [35,36]. One major mechanism involves the overexpression of P-glycoprotein (P-gp), which plays an important role in developing multiple drug resistance (MDR) [37]. P-gp, a member of ATP-binding cassette transporter, is one of the major drug efflux transporters and increased expression of P-gp leads to decreased drug accumulation in multidrug-resistant cells, and the development of resistance to anticancer drugs [38]. NCI/ADR-RES is a drug-resistant cell line that involves the overexpression of P-gp [39]. Thus, following the evaluation of cytotoxicity of the different DOX formulations in 4T1.2 cells, their cytotoxicity was further examined in NCI/ADR-RES cells. As shown in Fig. 8, the overall antiproliferative effects of all DOX formulations were significantly reduced in NCI/ADR-RES cells compared to drug-sensitive cell line, 4T1.2 cells. This is somewhat expected considering the drug resistant nature of NCI/ADR-RES cells. It is also apparent that PEG5K-Fmoc-VE2/DOX exhibited a higher level of cytotoxicity than the remaining DOX formulations in NCI/ADR-RES cells (Fig. 8). More importantly, PEG5K-Fmoc-VE2/DOX was 5.6-times as effective as free DOX in NCI/ADR-RES cells, which represents a more dramatic improvement of cytotoxicity over free DOX compared to that (2.4-times) in 4T1.2 cells. It is also worth noting that Doxil was more active than free DOX in NCI/ADR-RES cells, which is in contrast to what was shown in 4T1.2 cells. These data suggested that both PEG5K-Fmoc-VE2 and Doxil formulations were capable of partially reversing the drug resistance in NCI/ADR-RES cells.
Fig. 8.

The anti-proliferative effect of PEG5K-VE2/DOX and PEG5K-Fmoc-VE2/DOX in a drug resistant cell line, NCI/ADR-RES, in comparison to DOX and Doxil. Cells were challenged with indicated concentrations of different DOX formulations for 72 h and cytotoxicity was then determined by MTT assay. Values reported are the means ± SD for triplicate samples.
To examine whether the improved cytotoxicity of DOX-loaded PEG5K-Fmoc-VE2 was due to the enhanced intracellular delivery of DOX, DOX uptake was evaluated in NCI/ADR-RES cells following treatment of different DOX formulations. Fig. 9A shows the fluorescence images of cells 1 h following the different treatments. Least amounts of fluorescence signals were observed in the cells treated with free DOX, which could be attributed to the free accessibility of DOX by P-gp efflux transporter that is overexpressed in NCI/ADR-RES cells. Cells treated with Doxil showed increased fluorescence signals compared to free DOX group. This was consistent with the report that liposomal DOX was able to bypass the drug transport in MDR cells [40]. It is also apparent that cells treated with PEG5K-Fmoc-VE2/DOX showed the strongest fluorescence signals. Similar results were shown in cells treated with different DOX formulation for 3 h (Fig. 9B). The DOX uptake efficiency was further quantified by flow cytometry (Fig. 9C & 9D). The data were consistent with what was found in confocal imaging. Cells treated with PEG5K-Fmoc-VE2/DOX provided the highest level of cell-associated fluorescence signals. Endocytosis is likely to be involved in the cellular uptake of micellar DOX as punctuated distribution was visualized for both micellar formulations. More studies will be conducted in the future to further elucidate the mechanism of cellular uptake of PEG5K-Fmoc-VE2/DOX.
Fig. 9.

Confocal laser scanning microscopy (CLSM) images of NCI/ADR-RES cells incubated with free DOX, Doxil, PEG5K-VE2/DOX and PEG5K-Fmoc-VE2/DOX for 1 h (A) and 3 h (B); Quantitative analysis of uptake of different DOX formulations in NCI/ADR-RES cells after 1 h (C) and 3 h (D) treatment using flow cytometry. Arrows indicated the area that was co-localized by DOX.
The more effective DOX accumulation in cells treated with PEG5K-Fmoc-VE2/DOX is likely attributed to the stable mixed micelles as a result of strong carrier/DOX interaction as discussed earlier. This will minimize the release of DOX from the micelles before they are taken up by the tumor cells. On the other hand, PEG5K-Fmoc-VE2 may facilitate the intracellular accumulation of DOX via inhibiting the function of P-gp. TPGS1K is a well-known inhibitor of P-gp via directly inhibiting the activity of P-gp ATPase. We hypothesized that PEG5K-Fmoc-VE2 shall possess a similar biological activity considering the structural similarity between TPGS and PEG5K-Fmoc-VE2. To test this hypothesis, the effect of PEG5K-Fmoc-VE2 on P-gp activity was evaluated via examining its impact on verapamil-stimulated P-gp ATPase activity (Fig. 10). TPGS1k was utilized as a positive control. ΔRLU represents the consumption of ATP in the system. As expected, TPGS1k treatment led to a decrease of ΔRLU in a dose-dependent fashion. Importantly, PEG5K-Fmoc-VE2 showed a level of inhibition on P-gp ATPase activity that was comparable to that of TPGS1K. The mechanism for the inhibition of P-gp ATPase by PEG5K-Fmoc-VE2 is not clearly understood at present. PEG5K-Fmoc-VE2 may be a substrate for ATPase and compete directly with other substrates for the binding to ATPase. In addition, PEG5K-Fmoc-VE2 may bind to the ATPase-substrate complex, limiting the effectiveness of ATPase to hydrolyze ATP. More studies are underway to fully unveil the details on how PEG5K-Fmoc-VE2 inhibits P-gp ATPase activity. It should be noted that, in addition to DOX, there are many other potent chemotherapeutics that are the substrates of P-gp, such as camptothecin and paclitaxel. Therefore, PEG5K-Fmoc-VE2 may hold a promise to improve the effectiveness of these therapeutic agents as well.
Fig. 10.

Inhibitory effect of TPGS and PEG5K-Fmoc-VE2 on verapamil-stimulated P-gp ATPase activity. Test samples containing verapamil (50 μM) and PEG5K-Fmoc-VE2 (final concentrations at 10 and 100 μM, respectively) or Na3VO4 (a selective inhibitor of P-gp) were incubated with P-gp membrane for 5 min at 37 °C. The reaction was initiated by the addition of Mg/ATP followed by another 40 minutes’ incubation at 37 °C. ATP detection reagent was then added and luminescence was examined. Values reported are the means ± SD for triplicate samples.
Near infrared fluorescence imaging
The ability of PEG5K-Fmoc-VE2 for targeting tumors was investigated in nude mice bearing PC-3 xenograft, using a hydrophobic near infrared fluorescence (NIRF) dye, DiD. Two hundred μL of PEG5K-Fmoc-VE2 nanoparticles containing DiD was intravenously injected into a mouse bearing bilateral PC-3 tumors. Fluorescence signals were observed in tumor areas as early as 6 h post-injection, which peaked around 24 h and retained at a significant level for 96 h (Fig. 11A). Following the last imaging at 96 h post-injection, mice were sacrificed and tumors and major organs were excised, imaged and quantified using a Carestream Molecular Imaging System (Fig. 11B). Intense fluorescence signals were observed in tumors for DiD-loaded PEG5K-Fmoc-VE2. Moderate levels of fluorescence signals were also discerned in liver and lungs, which was due to the nonspecific clearance of foreign particles via the RES. Fig. 11C showed the quantified fluorescence intensity for different organs, which was consistent with the observation in Fig. 11B. The effective tumor accumulation of PEG5K-Fmoc-VE2/DiD could be ascribed to its excellent stability endowed by the strong carrier/DiD and carrier/carrier interactions. PEG on the surface of the carrier can also provide shielding effect against opsonins. Furthermore, the very small sizes of PEG5K-Fmoc-VE2/DiD nanoparticles shall facilitate the extravasation and deep penetration into the tumor tissues [41,42].
Fig. 11.

In vivo (A) and ex vivo (B) NIRF optical images of PC-3 tumor-bearing nude mice administered intravenously with DiD-loaded PEG5K-Fmoc-VE2 nanoparticles. Tumors and major organs were excised for ex vivo imaging at 96 h post-injection and the quantitated DiD fluorescence intensity from different organs were presented (C).
In vivo DOX pharmacokinetics and biodistribution
The DOX blood kinetics as a function of time following i.v. bolus administration of DOX, DOX-loaded PEG5K-VE2 and DOX-loaded PEG5K-Fmoc-VE2 was illustrated in Fig. 12A. It is apparent that the DOX-loaded PEG5K-Fmoc-VE2 showed the highest level of DOX retention in circulation in comparison to free DOX and DOX-loaded PEG5K-VE2. Meanwhile, the pharmacokinetic parameters, obtained by fitting the data to a non-compartment model, were outlined in Tab. 2. Incorporation of DOX into PEG5K-VE2 micelles resulted in a significantly greater t1/2, AUC, and Cmax over free DOX. However, these parameters were further improved in DOX-loaded PEG5K-Fmoc-VE2 nanomicelles.
Fig. 12.
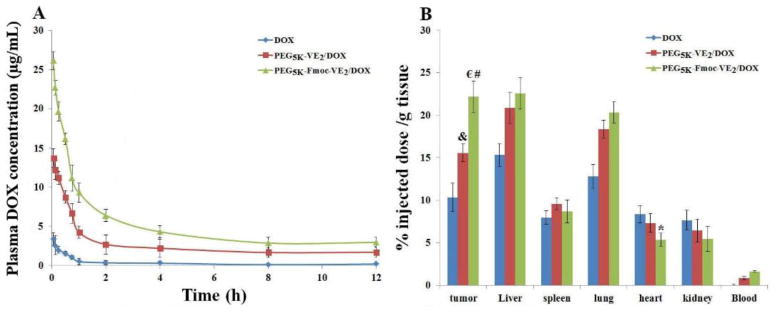
DOX pharmacokinetics (A) and biodistribution profiles (B) in 4T1.2-tumor bearing mice receiving intravenous administration of different DOX formulations at the dose of 5mg/kg. *p < 0.05, compared to DOX and PEG5K-VE2/DOX; € p < 0.005, compared to DOX; # p < 0.01, compared to PEG5K-VE2/DOX; & p < 0.01, compared to DOX. Values reported are the means ± SD for triplicate samples.
Table 2.
Pharmacokinetic parameters of DOX in different formulations
| t1/2 (h) | AUC (μg/mL×h) | CL (mL/h) | Cmax (μg/mL) | Vd (mL) | |
|---|---|---|---|---|---|
| DOX | 4.20±0.35 | 5.43±0.11 | 18.40±2.39 | 3.64 t0.30 | 25.32±2.15 |
| PEG5K-VE2/DOX | 14.89±2.08 | 70.58f±6.68 | 1.47±0.15 | 14.51±1.39 | 6.26±0.43 |
| PEG5K-Fmoc-VE2/DOX | 19.41±1.76 | 104.36±11.52 | 0.72±0.055 | 27.62±2.60 | 3.29±0.24 |
The t1/2, AUC, and Cmax of DOX in PEG5K-Fmoc-VE2/DOX were 3.62, 18.22, and 6.59-folds higher, respectively, than those of free DOX. In contrast, Vd and CL for PEG5K-Fmoc-VE2/DOX were significantly lower than those for free DOX and PEG5K-VE2/DOX. Taken together, these data demonstrated that DOX formulated in PEG5K-Fmoc-VE2 micelles was able to circulate for a significantly longer period of time in blood.
Next we went on to investigate whether our PEG5K-Fmoc-VE2/DOX formulation can improve the DOX biodistribution profile in tumor-bearing mice. Different DOX formulations were i.v. administered to 4T1.2 tumor-bearing mice at a DOX dosage of 5 mg/kg. Twenty-four h following the injection, blood, tumors, and major organs were collected for the quantification of DOX. Compared to free DOX, there were significantly greater amounts of DOX accumulated in tumors for both types of DOX nanomicelles (Fig. 12B), which was attributed to the EPR effect of micellar formulations. We also noticed that DOX formulated in PEG5K-Fmoc-VE2 micelles was more effective in tumor accumulation compared to the counterpart without Fmoc motif (Fig. 12B). This is likely attributed to a better stability of PEG5K-Fmoc-VE2/DOX mixed micelles in the blood due to the enhanced carrier/drug interaction as discussed before. In addition, the presence of Fmoc may help improve the carrier stability via imposing steric hindrance against the degrading enzymes in the blood. Indeed, PEG5K-Fmoc-VE2 is less sensitive to esterase-mediated cleavage than the counterpart without an Fmoc motif (Fig. S10) in an in vitro study. The improved stability of PEG5K-Fmoc-VE2/DOX mixed micelles may contribute significantly to a longer t1/2 in the blood (Tab. 2) and thus increased chance for passive accumulation at tumor tissues via EPR effect.
Relatively high levels of DOX uptake were also observed in liver and lung, which was due to the nonspecific uptake of particles by these tissues. Importantly, heart distribution of DOX was significantly reduced in PEG5K-Fmoc-VE2/DOX compared with free DOX and PEG5K-VE2/DOX. This is significant considering that cardiotoxicity is a major adverse effect associated with the application of DOX in clinic. This will allow higher dose of DOX to be used to maximize the therapeutic effect.
Maximum tolerated dose (MTD)
To examine whether the significantly improved stability of PEG5K-Fmoc-VE2/DOX will lead to reduced systemic toxicity in vivo, MTD of various DOX formulations was assessed in tumor-free mice. Mice were treated with i.v. bolus injection of different doses of free DOX, PEG5K-VE2/DOX and PEG5K-Fmoc-VE2/DOX followed by observation of changes in body weight and other general signs of toxicity. As shown in Tab. 3, no significant toxicity was noticed for free DOX group at a dosage of 10 mg DOX/kg. Increasing the dosage to 15 mg/kg led to the death of 2 out of 3 treated mice. Therefore, the MTD for free DOX at a single injection was around 10 mg/kg, which was consistent with the literature [43]. PEG5K-VE2/DOX was well tolerated at a dosage of 20 mg/kg: no significant changes were observed in the general appearance or activity other than 6.2% weight loss. Increasing the DOX dose to 30 mg/kg resulted in the death of all 3 treated mice, suggesting that the single i.v. MTD for DOX-loaded PEG5K-VE2 micelles was around 20 mg DOX/kg, which was a 2-fold increase over free DOX. PEG5K-Fmoc-VE2/DOX showed a lowest level of toxicity with a MTD around 30 mg/kg, which was 3-fold and 1.5-fold increase over free DOX and PEG5K-VE2/DOX, respectively. The improved MTD of DOX-loaded PEG5K-Fmoc-VE2 is likely attributed to a slower and sustained DOX release kinetics (Fig. 3) due to the enhanced stability, which will liberate its payload gradually instead of a bursting release; as such, the toxicity will be better tolerated. In addition, it has been shown that Vitamin E and Fmoc both have anti-inflammatory activities which may also contribute to the reduced toxicity of PEG5K-Fmoc-VE2/DOX [44–46]. More studies are still needed to fully elucidate the underlying mechanisms for the reduced toxicity of PEG5K-Fmoc-VE2/DOX.
Table 3.
MTD of DOX and DOX-loaded nanomicellar formulations
| Formulations | Does (mg/kg) | Animal death | Weight loss (%) |
|---|---|---|---|
| DOX | 5 | 0/3 | 2.2 |
| 10 | 0/3 | 7.8 | |
| 15 | 2/3 | N/A | |
| DOX-loaded PEG5K-VE2 | 5 | 0/3 | 0.9 |
| 10 | 0/3 | −1.8 | |
| 15 | 0/3 | 3.9 | |
| 20 | 0/3 | 6.2 | |
| 30 | 3/3 | N/A | |
| DOX-loaded PEG5K-Fmoc-VE2 | 10 | 0/3 | −2.2 |
| 15 | 0/3 | 0.2 | |
| 20 | 0/3 | −1.3 | |
| 25 | 0/3 | 3.5 | |
| 30 | 0/3 | 7.4 | |
| 35 | 2/3 | N/A |
In vivo anti-tumor efficacy
Both drug-sensitive (4T1.2 and PC-3) and drug-resistant (KB 8-5) tumor models were employed to evaluate the in vivo anti-tumor activity of DOX-loaded PEG5K-Fmoc-VE2. 4T1.2 is an aggressive syngeneic murine breast cancer model. As depicted in Fig. 13A, a rapid and unrestrained growth of tumors was observed in mice treated with saline and mice had to be sacrificed early due to the development of severe ulceration. After three consecutive injections, all of the mice treated with free DOX died from systemic toxicity on day 7. PEGylated liposomal DOX (Doxil) was well tolerated and showed a moderate level of activity in delaying the tumor growth, which is consistent with previous reports [40, 47–50]. It is also apparent that incorporation of Fmoc motif into PEG5K-VE2/DOX led to a significant improvement in antitumor activity. The IRs were 82.63, 67.73, and 62.24% for PEG5K-Fmoc-VE2/DOX, PEG5K-VE2/DOX, and Doxil, respectively (Tab. 4). In addition, increasing DOX dosage to 20 mg/kg resulted in further enhancement in the antitumor activity of DOX-loaded PEG5K-Fmoc-VE2 micelles. No obvious toxicity was noticed at both dosages (Fig. 13B).
Fig. 13.
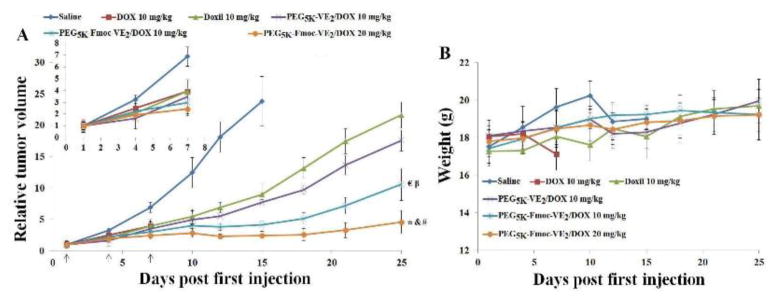
In vivo antitumor activity of different DOX formulations in 4T1.2 syngeneic mouse model. Solid arrows indicate the i.v. administration. A: relative tumor volume. B: body weight. *p < 0.0005, compared to Doxil. & p < 0.001, compared to PEG5K-VE2/DOX; # p < 0.05, compared to PEG5K-Fmoc-VE2/DOX (10 mg/kg); € p < 0.05, compared to PEG5K-VE2/DOX; β p < 0.005, compared to Doxil.
Table 4.
Tumor growth inhibition rate (IR) in 4T1.2 tumor bearing mice.
| Different DOX formulations | IR (%) |
|---|---|
| Doxil 10 mg/kg | 62.24 |
| PEG5K-VE2/DOX 10 mg/kg | 67.73 |
| PEG5K-Fmoc-VE2/DOX 10 mg/kg | 82.63 |
| PFG5K-Fmoc-VE2/DOX 20 mg/kg | 89.77 |
IR (%) = (1 − relative tumor volume in the treated group ÷ relative tumor volume in the saline group) × 100%
IR (%) was calculated based on the RTV on day 15 (Mice in saline group died after day 15. Mice in DOX group died after third injection due to severe toxicity)
The improved performance of the two micellar formulations (PEG5K-VE2 and PEG5K-Fmoc-VE2) over Doxil in inhibiting the tumor growth may be attributed to their more effective accumulation at tumors based on their relatively small size (~20 nm and ~60 nm for PEG5K-VE2/DOX and PEG5K-Fmoc-VE2/DOX, respectively). It has been generally regarded that a drug carrier needs to have a size range of ~200 nm in order to capitalize on the EPR effect. However, recent studies have shown that sub-100 nm is required for particles to effectively penetrate the tumor tissues, particularly poorly vascularized tumors [41,42]. The size advantage of the two micellar DOX formulations over Doxil (~100 nm) may contribute to their enhanced antitumor activity. In addition, the carriers may contribute to the overall antitumor activity via delaying or reversing the drug resistance through inhibiting the activity of P-gp. The further improved performance of PEG5K-Fmoc-VE2 over PEG5K-VE2 is likely due to the more effective drug/carrier interaction as well as the interaction among the carrier molecules themselves, leading to improved formulation stability and more effective DOX delivery to tumors (Fig. 12).
After demonstrating effective inhibition of tumor growth in the syngeneic murine breast cancer model, the therapeutic effect of DOX-loaded PEG5K-Fmoc-VE2 micelles was further examined in nude mice bearing human prostate cancer xenograft (PC-3) (Fig. 14). In this study, a lower DOX dosage (5 mg/kg) was used to avoid the death of animals in free DOX group, which allowed the comparison of all treatment groups over a relatively long period of time. Similarly, PEG5K-Fmoc-VE2/DOX gave the highest level of tumor growth inhibition among all DOX formulations examined. Doubling the DOX dosage to 10 mg/kg led to a further improvement of the antitumor efficacy (Fig. 14A). Tab. 5 shows the IR of different DOX formulations on day 19. Mice in both saline and free DOX groups showed significant loss of body weight after day 19 (Fig. 14B), which was likely attributed to the cachexia that is caused by the overgrowth of tumor. No significant changes were noticed in other treatment groups in either body weight (Fig. 14B) or the general appearance.
Fig. 14.
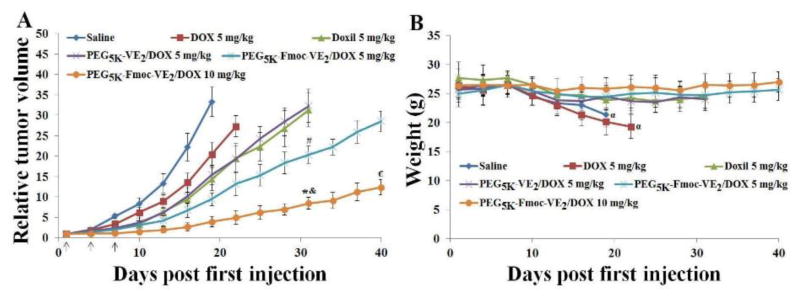
In vivo antitumor activity of various DOX formulations in PC-3 tumor-bearing mice. Solid arrows indicate the i.v. administration. A: relative tumor volume. B: body weight. *p < 0.001, compared to PEG5K-VE2/DOX or Doxil on day 31; & p < 0.01, compared to PEG5K-Fmoc-VE2/DOX (5 mg/kg) on day 31; # p < 0.01, compared to PEG5K-VE2/DOX or Doxil on day 31; € p < 0.001, compared to PEG5K-Fmoc-VE2/DOX (5 mg/kg) on day 40; α p < 0.05, compared to day 1.
Table 5.
Tumor growth inhibition rate (IR) in PC-3 tumor bearing mice.
| Different DOX formulations | IR (%) |
|---|---|
| DOX 5 mg/kg | 38.72 |
| Doxil 5 mg/kg | 56.98 |
| PEG5K-VE2/DOX 5 mg/kg | 53.71 |
| PEG5K-Fmoc-VE2/DOX 5 mg/kg | 71.64 |
| PEG5K-Fmoc-VE2/DOX 10 mg/kg | 88.10 |
IR (%) = (1 − relative tumor volume in the treated group ÷ relative tumor volume in the saline group) × 100%
IR (%) was calculated based on the RTV on day 19 (Mice in saline group died after day 19)
Fig. 15 shows the in vivo antitumor activity of DOX-loaded PEG5K-Fmoc-VE2 micelles in a drug-resistant tumor model, KB 8-5. At a dosage of 5 mg/kg, free DOX barely showed any effect in inhibiting the tumor growth. Increasing the dose to 7.5 mg/kg only led to a slight improvement in antitumor activity. These are in consistence with the drug-resistant nature of this tumor model [51]. In contrast, incorporation of DOX into PEG5K-VE2 led to a significant improvement in antitumor activity: PEG5K-VE2/DOX at 5 mg/kg was even more effective than free DOX at 7.5 mg/kg. In agreement with studies in other tumor models (Figs. 13 and 14), PEG5K-Fmoc-VE2/DOX was even more effective than PEG5K-VE2/DOX in inhibiting the tumor growth at a same dosage (5 mg/kg). Increasing the dose to 7.5 mg/kg led to almost complete inhibition of tumor growth with an IR of 89.21% (Fig. 15 and Tab. 6). Both micellar formulations were well tolerated. In contrast, significant weight loss was observed in free DOX-treated mice at both dosages (Fig. 15B). Data from Fig. 15 strongly suggest that our improved formulation is capable of reversing the drug-resistance, at least partially, although more studies are needed to better understand the underlying mechanism.
Fig. 15.

In vivo antitumor activity of varying DOX formulations in a drug-resistant xenograft tumor model, KB 8-5. Solid arrows indicate the i.v. injection. A: relative tumor volume. *p < 0.002, compared to PEG5K-Fmoc-VE2/DOX (5 mg/kg); & p < 0.01, compared to PEG5K-VE2/DOX (5 mg/kg); # p < 0.05, compared to DOX (7.5 mg/kg); € p < 0.05, compared to DOX (5 mg/kg) or saline. B: body weight, α p < 0.0005, compared to day 1; β p < 0.005, compared to day 1; £ p < 0.05, compared to day 1. C: tumor images. D: tumor weight, *p < 0.005, compared to PEG5K-Fmoc-VE2/DOX (5 mg/kg); & p < 0.01, compared to PEG5K-VE2/DOX (5 mg/kg); # p < 0.001, compared to DOX (7.5 mg/kg); € p < 0.05, compared to DOX (5 mg/kg).
Table 6.
Tumor growth inhibition rate (IR) in KB 8-5 tumor bearing mice.
| Different DOX formulations | IR (%) |
|---|---|
| DOX 5 mg/kg | 6.13 |
| DOX 7.5 mg/kg | 20.91 |
| PEG5K-VE2/DOX 5 mg/kg | 39.15 |
| PEG5K-Fmoc-VE2/DOX 5 mg/kg | 62.15 |
| PEG5K-Fmoc-VE2/DOX 7.5 mg/kg | 89.21 |
IR (%) = (1 − relative tumor volume in the treated group ÷ relative tumor volume in the saline group) × 100%
IR (%) was calculated based on the RTV on day 22
Conclusions
Incorporation of an Fmoc motif at the interfacial region of PEG5K-VE2 led to significant improvement of the system through the introduction of an additional mechanism of drug/carrier interaction. Doxorubicin (DOX) could be effectively loaded into PEG5K-Fmoc-VE2 micelles at a DLC of 39.9%, which compares favorably to most reported DOX nanoformulations. PEG5K-Fmoc-VE2/DOX exerted significantly higher levels of cytotoxicity over DOX, Doxil as well as PEG5K-VE2/DOX in 4T1.2 cells and demonstrated a potential to reverse the DOX resistance in NCI/ADR-RES cells. PK and biodistribution studies showed an increased half-life in blood circulation and more effective tumor accumulation for DOX formulated in PEG5K-Fmoc-VE2 micelles. More importantly, DOX-loaded PEG5K-Fmoc-VE2 micelles showed an excellent safety profile with a MTD (~30 mg DOX/kg) that is about 3 times as much as that for free DOX. Finally, superior antitumor activity was demonstrated for PEG5K-Fmoc-VE2/DOX in both drug-sensitive (4T1.2 and PC-3) and drug-resistant (KB 8-5) tumor models.
Supplementary Material
Acknowledgments
This work was supported in part by NIH grants RO1CA174305, RO1GM102989, and R21CA173887. We would like to thank Dr. Robert Gibbs for critical reading of this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tacar O, Sriamornsak P, Dass CR. Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J Pharm Pharmacol. 2013;65:157–170. doi: 10.1111/j.2042-7158.2012.01567.x. [DOI] [PubMed] [Google Scholar]
- 2.Fornari FA, Randolph JK, Yalowich JC, Ritke MK, Gewirtz DA. Interference by doxorubicin with DNA unwinding in MCF-7 breast tumor cells. Mol Pharmacol. 1994;45:649– 656. [PubMed] [Google Scholar]
- 3.Momparler RL, Karon M, Siegel SE, Avila F. Effect of adriamycin on DNA, RNA, and protein synthesis in cell-free systems and intact cells. Cancer Res. 1976;36:2891–2895. [PubMed] [Google Scholar]
- 4.Pommier Y, Leo E, Zhang H, Marchand C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem Biol. 2010;17:421–433. doi: 10.1016/j.chembiol.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takemura G, Fujiwara H. Doxorubicin-induced cardiomyopathy from the cardiotoxic mechanisms to management. Prog Cardiovasc Dis. 2007;49:330–352. doi: 10.1016/j.pcad.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 6.Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev. 2004;56:185–229. doi: 10.1124/pr.56.2.6. [DOI] [PubMed] [Google Scholar]
- 7.Mi Y, Liu Y, Feng SS. Formulation of docetaxel by folic acid-conjugated D-α-tocopheryl polyethylene glycol succinate 2000 (Vitamin E TPGS2k) micelles for targeted and synergistic chemotherapy. Biomaterials. 2011;32:4058–4066. doi: 10.1016/j.biomaterials.2011.02.022. [DOI] [PubMed] [Google Scholar]
- 8.Win KY, Feng SS. In vitro and in vivo studies on vitamin E TPGS-emulsified poly(D,L-lactic-co-glycolic acid)nanoparticles for paclitaxel formulation. Biomaterials. 2006;27:2285–2291. doi: 10.1016/j.biomaterials.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 9.Ma Y, Zheng Y, Liu K, Tian G, Tian Y, Xu L, et al. Nanoparticles of poly(lactide-co-glycolide)-d-α-tocopheryl polyethylene glycol 1000 succinate random copolymer for cancer treatment. Nanoscale Res Lett. 2010;6:1161–1169. doi: 10.1007/s11671-010-9620-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang J, Sun J, Chen Q, Gao Y, Li L, Li H, et al. Star-shape copolymer of lysine-linked di-tocopherol polyethylene glycol 2000 succinate for doxorubicin delivery with reversal of multidrug resistance. Biomaterials. 2012;28:6877–6888. doi: 10.1016/j.biomaterials.2012.06.019. [DOI] [PubMed] [Google Scholar]
- 11.Muthu MS, Kulkarni SA, Xiong J, Feng SS. Vitamin E TPGS coated liposomes enhanced cellular uptake and cytotoxicity of docetaxel in brain cancer cells. Int J Pharm. 2011;421:332–340. doi: 10.1016/j.ijpharm.2011.09.045. [DOI] [PubMed] [Google Scholar]
- 12.Cao N, Feng SS. Doxorubicin conjugated to D-alpha-tocopheryl polyethylene glycol 1000 succinate (TPGS): conjugation chemistry, characterization, in vitro and in vivo evaluation. Biomaterials. 2008;29:3856–3865. doi: 10.1016/j.biomaterials.2008.05.016. [DOI] [PubMed] [Google Scholar]
- 13.Anbharasi V, Cao N, Feng SS. Doxorubicin conjugated to D-alpha-tocopheryl polyethylene glycol succinate and folic acid as a prodrug for targeted chemotherapy. J Biomed Mater Res A. 2010;94:730–743. doi: 10.1002/jbm.a.32734. [DOI] [PubMed] [Google Scholar]
- 14.Yu L, Bridgers A, Polli J, Vickers A, Long S, Roy A, et al. Vitamin E-TPGS increases absorption flux of an HIV protease inhibitor by enhancing its solubility and permeability. Pharm Res. 1999;16:1812–1817. doi: 10.1023/a:1018939006780. [DOI] [PubMed] [Google Scholar]
- 15.Dintaman JM, Silverman JA. Inhibition of P-glycoprotein by D-alpha-tocopheryl polyethylene glycol 1000 succinate (TPGS) Pharm Res. 1999;16:1550–1556. doi: 10.1023/a:1015000503629. [DOI] [PubMed] [Google Scholar]
- 16.Varma MVS, Panchagnula R. Enhanced oral paclitaxel absorption with vitamin E-TPGS: effect on solubility and permeability in vitro, in situ and in vivo. Eur J Pharm Sci. 2005;25:445–453. doi: 10.1016/j.ejps.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 17.Collnot EM, Baldes C, Schaefer UF, Edgar KJ, Wempe MF, Lehr CM. Vitamin E TPGS p-glycoprotein inhibition mechanism: influence on conformational flexibility, intracellular ATP levels, and role of time and site of access. Mol Pharm. 2010;7:642–651. doi: 10.1021/mp900191s. [DOI] [PubMed] [Google Scholar]
- 18.Constantinides PP, Han JH, Davis SS. Advances in the use of tocols as drug delivery vehicles. Pharm Res. 2006;23:243–255. doi: 10.1007/s11095-005-9262-9. [DOI] [PubMed] [Google Scholar]
- 19.Huang Y, Lu J, Gao X, Li J, Zhao W, Sun M, et al. PEG-derivatized embelin as a dual functional carrier for the delivery of paclitaxel. Bioconjug Chem. 2012;23:1443–1451. doi: 10.1021/bc3000468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu J, Huang Y, Zhao W, Marquez RT, Meng X, Li J, et al. PEG-Derivatized Embelin as a Nanomicellar Carrier for Delivery of Paclitaxel to Breast and Prostate Cancers. Biomaterials. 2013;34:1591–1600. doi: 10.1016/j.biomaterials.2012.10.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang X, Lu J, Huang Y, Zhao W, Chen Y, Li J, et al. PEG-farnesylthiosalicylate conjugate as a nanomicellar carrier for delivery of paclitaxel. Bioconjug Chem. 2013;24:464–472. doi: 10.1021/bc300608h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Y, Zhang X, Lu J, Huang Y, Li J, Li S. Targeted Delivery of Curcumin to Tumors via PEG-Derivatized FTS-Based Micellar System. AAPS J. 2014;16:600–608. doi: 10.1208/s12248-014-9595-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim JY, Kim S, Papp M, Park K, Pinal R. Hydrotropic solubilization of poorly water-soluble drugs. J Phar Sci. 2010;99:3953–3965. doi: 10.1002/jps.22241. [DOI] [PubMed] [Google Scholar]
- 24.Kim JY, Kim S, Pinal R, Park K. Hydrotropic polymer micelles as versatile vehicles for delivery of poorly water-soluble drugs. J Control Release. 2011;152:13–20. doi: 10.1016/j.jconrel.2011.02.014. [DOI] [PubMed] [Google Scholar]
- 25.Gao X, Huang Y, Makhov AM, Epperly M, Lu J, Grab S, et al. Nanoassembly of surfactants with interfacial drug-interactive motifs as tailor-designed drug carriers. Mol Pharm. 2013;10:187–98. doi: 10.1021/mp300319m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang P, Lu J, Huang Y, Zhao W, Zhang Y, Zhang X, et al. Design and evaluation of a PEGylated lipopeptide equipped with drug-interactive motifs as an improved drug carrier. AAPS J. 2014;16:114–124. doi: 10.1208/s12248-013-9536-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu J, Huang Y, Zhao W, Chen Y, Li J, Gao X, et al. Design and characterization of PEG-derivatized vitamin E as a nanomicellar formulation for delivery of paclitaxel. Mol Pharm. 2013;10:2880–2890. doi: 10.1021/mp300729y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Y, Gu H, Yang Z, Xu B. Supramolecular hydrogels respond to ligand-receptor interaction. J Am Chem Soc. 2003;125:13680–13681. doi: 10.1021/ja036817k. [DOI] [PubMed] [Google Scholar]
- 29.Jayawarna V, Ali M, Jowitt TA, Miller AF, Saiani A, Gough JE, et al. Nanostructured Hydrogels for Three-Dimensional Cell Culture Through Self-Assembly of Fluorenylmethoxycarbonyl–Dipeptides. Adv Mater. 2006;18:611–614. [Google Scholar]
- 30.Mahler A, Reches M, Rechter M, Cohen S, Gazit E. Rigid, Self-Assembled Hydrogel Composed of a Modified Aromatic Dipeptide. Adv Mater. 2006;18:1365–1370. [Google Scholar]
- 31.Wang Y, Wang R, Lu X, Lu W, Zhang C, Liang W. Pegylated phospholipids-based self-assembly with water-soluble drugs. Pharm Res. 2010;27:361–370. doi: 10.1007/s11095-009-0029-6. [DOI] [PubMed] [Google Scholar]
- 32.Mao J, Zhang H, Luo J, Li L, Zhao R, Zhang R, Liu G. New method for HPLC separation and fluorescence detection of malonaldehyde in normal human plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 2006;832:103–108. doi: 10.1016/j.jchromb.2005.12.041. [DOI] [PubMed] [Google Scholar]
- 33.Zhao Y, Alakhova DY, Kim JO, Bronich TK, Kabanov AV. A simple way to enhance Doxil® therapy: drug release from liposomes at the tumor site by amphiphilic block copolymer. J Control Release. 2013;168:61–69. doi: 10.1016/j.jconrel.2013.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu CM, Zhang L. Therapeutic nanoparticles to combat cancer drug resistance. Curr Drug Metab. 2009;10:836–841. doi: 10.2174/138920009790274540. [DOI] [PubMed] [Google Scholar]
- 35.Gottesman MM. Mechanisms of cancer drug resistance. Annu Rev Med. 2002;53:615–627. doi: 10.1146/annurev.med.53.082901.103929. [DOI] [PubMed] [Google Scholar]
- 36.Yuan H, Li X, Wu J, Li J, Qu X, Xu W, et al. Strategies to overcome or circumvent P-glycoprotein mediated multidrug resistance. Curr Med Chem. 2008;15:470–476. doi: 10.2174/092986708783503258. [DOI] [PubMed] [Google Scholar]
- 37.Tijerina M, Fowers KD, Kopecková P, Kopecek J. Chronic exposure of human ovarian carcinoma cells to free or HPMA copolymer-bound mesochlorin e6 does not induce P-glycoprotein-mediated multidrug resistance. Biomaterials. 2000;21:2203–2210. doi: 10.1016/s0142-9612(00)00161-7. [DOI] [PubMed] [Google Scholar]
- 38.Desai PV, Sawada GA, Watson IA, Raub TJ. Integration of in silico and in vitro tools for scaffold optimization during drug discovery: predicting P-glycoprotein efflux. Mol Pharm. 2013;10:1249–1261. doi: 10.1021/mp300555n. [DOI] [PubMed] [Google Scholar]
- 39.Xu D, Kang H, Fisher M, Juliano RL. Strategies for inhibition of MDR1 gene expression. Mol Pharmacol. 2004;66:268–275. doi: 10.1124/mol.66.2.268. [DOI] [PubMed] [Google Scholar]
- 40.Ogawara K, Un K, Tanaka K, Higaki K, Kimura T. In vivo anti-tumour effect of PEG liposomal doxorubicin (DOX) in DOX resistant tumour-bearing mice: Involvement of cytotoxic effect on vascular endothelial cells. J Control Release. 2009;133:4–10. doi: 10.1016/j.jconrel.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 41.Luo J, Xiao K, Li Y, Lee JS, Shi L, Tan YH, et al. Well-defined, size-tunable, multifunctional micelles for efficient paclitaxel delivery for cancer treatment. Bioconjug Chem. 2010;21:1216–1224. doi: 10.1021/bc1000033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li Y, Xiao K, Luo J, Lee J, Pan S, Lam KS. A novel size-tunable nanocarrier system for targeted anticancer drug delivery. J Control Release. 2010;144:314–323. doi: 10.1016/j.jconrel.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xiao K, Luo J, Li Y, Lee JS, Fung G, Lam KS. PEG-oligocholic acid telodendrimer micelles for the targeted delivery of doxorubicin to B-cell lymphoma. J Control Release. 2011;155:272–281. doi: 10.1016/j.jconrel.2011.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shirpoor A, Norouzi L, Khadem Ansari MH, Ilkhanizadeh B, Gharaaghaji R. Vasoprotective effect of vitamin E: rescue of ethanol-induced atherosclerosis and inflammatory stress in rat vascular wall. Int Immunopharmacol. 2013;16:498–504. doi: 10.1016/j.intimp.2013.04.024. [DOI] [PubMed] [Google Scholar]
- 45.Buse J, El-Aneed A. Properties, engineering and applications of lipid-based nanoparticle drug-delivery systems: current research and advances. Nanomedicine (Lond) 2010;5:1237–1260. doi: 10.2217/nnm.10.107. [DOI] [PubMed] [Google Scholar]
- 46.Yen CT, Hwang TL, Wu YC, Hsieh PW. Design and synthesis of new N-(fluorenyl-9-methoxycarbonyl) (Fmoc)-dipeptides as anti-inflammatory agents. Eur J Med Chem. 2009;44:1933–1940. doi: 10.1016/j.ejmech.2008.11.008. [DOI] [PubMed] [Google Scholar]
- 47.Chen X, Wang X, Wang Y, Yang L, Hu J, Xiao W, et al. Improved tumor-targeting drug delivery and therapeutic efficacy by cationic liposome modified with truncated bFGF peptide. J Control Release. 2010;145:17–25. doi: 10.1016/j.jconrel.2010.03.007. [DOI] [PubMed] [Google Scholar]
- 48.Ishida T, Shiraga E, Kiwada H. Synergistic antitumor activity of metronomic dosing of cyclophosphamide in combination with doxorubicin-containing PEGylated liposomes in a murine solid tumor model. J Control Release. 2009;134:194–200. doi: 10.1016/j.jconrel.2008.11.019. [DOI] [PubMed] [Google Scholar]
- 49.Schiffelers RM, Koning GA, ten Hagen TL, Fens MH, Schraa AJ, Janssen AP, et al. Anti-tumor efficacy of tumor vasculature-targeted liposomal doxorubicin. J Control Release. 2003;91:115–122. doi: 10.1016/s0168-3659(03)00240-2. [DOI] [PubMed] [Google Scholar]
- 50.Maeng JH, Lee DH, Jung KH, Bae YH, Park IS, Jeong S, et al. Multifunctional doxorubicin loaded superparamagnetic iron oxide nanoparticles for chemotherapy and magnetic resonance imaging in liver cancer. Biomaterials. 2010;31:4995–5006. doi: 10.1016/j.biomaterials.2010.02.068. [DOI] [PubMed] [Google Scholar]
- 51.Ma Y, Liu D, Wang D, Wang Y, Fu Q, Fallon JK, Yang X, He Z, Liu F. Combinational Delivery of Hydrophobic and Hydrophilic Anticancer Drugs in Single Nanoemulsions To Treat MDR in Cancer. Mol Pharm. 2014;11:2623–2630. doi: 10.1021/mp400778r. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.