

Abstract
Calcific aortic valve disease (CAVD) affects 25% of people over 65, and the late-stage stenotic state can only be treated with total valve replacement, requiring 85,000 surgeries annually in the US alone [1]. As CAVD is an age-related disease, many of the affected patients are unable to undergo the open-chest surgery that is its only current cure. This challenge motivates the elucidation of the mechanisms involved in calcification, with the eventual goal of alternative preventative and therapeutic strategies. There is no sufficient animal model of CAVD, so we turn to potential in vitro models. In general, in vitro models have the advantages of shortened experiment time and better control over multiple variables compared to in vivo models. As with all models, the hypothesis being tested dictates the most important characteristics of the in vivo physiology to recapitulate. Here, we collate the relevant pieces of designing and evaluating aortic valve calcification so that investigators can more effectively draw significant conclusions from their results.
Keywords: Calcific aortic valve disease, Model, Quantification methods, in vitro
1. Introduction
Aortic stenosis, which has an estimated prevalence of 2% in patients between 70 and 80 years of age, is most often caused by calcific aortic stenosis, the late-stage presentation of calcific aortic valve disease (CAVD; note all acronyms and abbreviations used in this article can be found in Table 1) [2]. Prevalence of any aortic valve calcification was investigated in a randomized trial, and, for those aged 75–76, the prevalence was 48%; this further increased in the 80–81 and 85–86 year-old cohorts [3]. The incidence of this age-related disease is expected to grow dramatically as the US population over 65 nearly doubles over the next 25 years [4]. Calcific aortic valve stenosis is the main indication for valve replacement, which necessitates open chest surgery and is currently the only cure [5]. If the biological mechanism of valvular calcification was better understood, we could create more targeted, non-invasive therapeutics; a comprehensive review of CAVD therapeutic targets can be found in Hutcheson et al. [6]. To elucidate the important mechanisms that regulate the progression of CAVD, we first need to design models that recapitulate the in vivo human process.
Table 1.
Acronyms and Abbreviations
| αSMA | Alpha smooth muscle actin |
| AFM | Atomic force microscopy |
| ALP | Alkaline phosphatase |
| AVEC | Aortic valve endothelial cell |
| AVIC | Aortic VIC |
| aVIC | Activated VIC |
| BMP2 | Bone morphogenic protein 2 |
| BMP4 | Bone morphogenic protein 4 |
| β-catenin | Intracellular transducer of Wnt pathway |
| CAVD | Calcific aortic valve disease |
| CN | Calcific nodule |
| CNP | C-type natriuretic peptide |
| ECM | Extracellular matrix |
| EDS | Energy-dispersive X-ray spectroscopy |
| ELISA | Enzyme-linked immunosorbent assay |
| EMT | Endothelial to mesenchymal transformation |
| eNOS | Endothelial NO synthase |
| ESEM | Environmental SEM |
| FGF-2 | Fibroblast growth factor 2 |
| IHC | Immunohistochemistry |
| IL1-β | Interleukin 1-β |
| IL6 | Interleukin 6 |
| LRP5 | Low density lipoprotein receptor-related protein 5 |
| MMP | Matrix metalloproteinase |
| Msx2 | Msh homeobox 2 |
| NF-κB | Nuclear factor kappa-B |
| NO | Nitric oxide |
| Notch1 | Notch homolog 1 |
| obVIC | Osteoblastic VIC |
| Osteocalcin/BGLAP | Bone gamma-carboxyglutamic acid-containing protein |
| oxLDL | Oxidized low-density lipoprotein |
| PParγ | Peroxisome proliferator-activated receptor gamma |
| pVIC | Progenitor VIC |
| qVIC | Quiescent VIC |
| RANKL | Receptor activator of NF-κB ligand |
| RT-PCR | Reverse transcription polymerase chain reaction |
| ROS | Reactive oxygen species |
| Runx2/CBFα1 | Runt-related transcription factor 2/core-binding factor subunit alpha-1 |
| SEM | Scanning electron microscopy |
| Smad | Intracellular transducer of TGF-β pathway |
| Sox9 | Transcription factor Sox9 of the SoxE family |
| TEM | Transmission electron microscopy |
| TGF-β1 | Transforming growth factor β1 |
| TIMP | Tissue inhibitor of metalloproteinase |
| TLR2 | Toll-like receptor 2 |
| TLR4 | Toll-like receptor 4 |
| TNFα | Tumor necrosis factor alpha |
| VIC | Valve interstitial cell |
| Wnt3a | Signaling protein of the Wnt family |
| 5HT2B | 5-hydroxytryptamine receptor 2B |
In vivo models offer the complexity found in the human and can prevent overlooking an important variable. However, this complexity comes at the expense of confounding factors, especially because the experiments are performed in animals significantly different from humans. For example, leporine models must be fed very high cholesterol diets to induce the advanced disease observed in humans [7] or vitamin D2 to generate calcification [8]. Murine models require dietary and or genetic modification as well [9–11] to induce calcification; Ldlr−/− mice must be fed a high-cholesterol diet and while Apoe−/− mice develop hypercholesterolemia over time [12] it is unclear whether it progresses through the same mechanism as the human disease [13]. A full review of animal models of CAVD can be found in Sider et al. [13]. Since in vitro models allow better isolation and manipulation of variables and the in vivo models are far from perfect, we focus on in vitro models and their usefulness.
Once believed to be a passive process, aortic valve calcification is now thought to be an active process mediated largely by aortic valve interstitial cells (AVICs) [14]. AVICs are a heterogeneous population of fibroblast-like cells present in all three layers of the aortic valve and important in the structural maintenance of the valve, especially in maintenance of the extracellular matrix (ECM) [15, 16]. Progression of CAVD is marked by the formation of calcific nodules (CNs), which are cellular aggregates characterized in humans by a mixture of calcium phosphate phases [17]. Two well-established hypothetical mechanisms of CN formation exist: 1) transforming growth factor β1 (TGF-β1) mediates activation of myofibroblasts, causing calcification via apoptotic mechanisms [18], and 2) a population of myofibroblasts spontaneously transdifferentiate into osteoblast-like cells and these cells regulate mineralization (Figure 1) [19, 20]. In a study of human valves, 83% of the group demonstrated evidence of dystrophic calcification and 13% of those valves had mature lamellar bone and evidence of active bone remodeling [21]. It is unclear whether these processes occur simultaneously or sequentially [22]. Recent progress and the need for a robust in vitro system with which we can probe and clarify the mechanism of aortic valve calcification motivate this review.
Figure 1.

Cartoons depicting proposed mechanisms of valve calcification. The dystrophic pathway is mediated by a TGF-β1 mediated increase in αSMA and cadherin-11, which increases the cells’ contractility and strengthens their connections to each other. Under pathological strain, the increased and uneven tension tears cells apart, leading to calcification via apoptosis. The osteogenic pathway proceeds by osteogenic differentiation into obVICs, likely from qVICs. These obVICs actively form mineralized deposits.
2. Defining Aortic Valve Interstitial Cells
Human aortic valves consist of three layers: 1) the fibrosa faces the aorta and is composed mostly of type I fibrillar collagen arranged circumferentially in parallel bundles in a matrix of elastin, 2) the spongiosa is the middle layer composed of glycosaminoglycans that act as shock absorbers for the valve, and 3) the ventricularis faces the left ventricle and is primarily composed of elastin fibers oriented radially [23]. AVICs are present throughout the leaflets and are a heterogeneous population of myofibroblasts, fibroblasts, and smooth muscle like cells. Aortic valve endothelial cells (AVECs) sheath the surface of the leaflets and are oriented circumferentially and form single cell monolayers, expressing Von Willebrand factor and nitric oxide (NO) [24–26]. Circulating cells have recently been implicated in the progression of calcification as well; elevated levels of endothelial progenitor cells with an osteoblastic phenotype and osteogenic precursor cells have been associated with severe and early heterotopic ossification, respectively [27, 28]. Early stages of CAVD develop lesions similar to atherosclerotic lesions, which suggests a role for inflammatory cells and biochemical signals [29, 30]. Elevated levels of macrophages and T-lymphocytes have been found in human calcified aortic valves [21, 31–33]. These cell populations all contribute to CAVD progression, but it is likely that it is through secretion of factors that influence AVIC behavior.
As the AVIC population is heterogeneous, we should consider the characteristics of various subpopulations. Recently, AVICs were categorized into five groups based on their phenotypic behavior: embryonic progenitor endothelial/mesenchymal cells, quiescent VICs (qVICs), activated VICs (aVICs), progenitor VICs (pVICs), and osteoblastic VICs (obVICs) [34]. We will refer to these subtypes for ease of discussion. Embryonic progenitors are usually present in the cardiac cushions and give rise to qVICs via endothelial to mesenchymal transformation (EMT). While these are very important in valve development, there is also evidence that these progenitors participate in adult valve repair. qVICs are responsible for maintaining physiological valve structure and function. The exact activity of these cells is undefined, but they are believed to regulate low-level matrix degradation and synthesis and inhibition of angiogenesis. pVICs are considered valve stem cells and they are likely responsible for VIC proliferation in response to tissue injury. pVICs may originate from AVECs that undergo an EMT-like process [34–36]. These EMT-related events are likely directly mediated by the mechanical forces present in the valve. In a recent study using chick explanted atrioventricular canals, EMT was found to occur preferentially in higher regions of strain [37]. This developmental process is likely recapitulated in an unregulated fashion during CAVD progression. This suggests that as the valve stiffens, more AVECs are transformed into pVICs and qVICs, allowing subsequent activation.
aVICs are qVICs that have become myofibroblasts characterized by alpha smooth muscle actin (αSMA) and increased contraction [34]. This activation occurs under pathological injury cues or abnormal mechanical stress via cytokines and growth factors produced by activated AVECs and macrophages. aVICs are associated with increased ECM secretion and degradation, matrix metalloproteinase (MMP) and tissue inhibitor of metalloproteinase (TIMP) expression, proliferation and migration, and secretion of cytokines including TGF-β1. If apoptotic pathways become abnormal, aVICs can lead to calcification; this is referred to as the dystrophic pathway. obVICs are VICs that have undergone osteoblastic differentiation and promote calcification in vitro. This differentiation is induced by the addition of organic phosphate to culture media and subsequent calcification depends on the upregulation of alkaline phosphatase (ALP) activity. Adding bone morphogenic protein 2 (BMP2) and 25-hydroxycholesterol increased the rate of CN formation, as did TGF-β1, which induced calcification via an apoptotic mechanism [38]. BMP2 has been shown to be higher in stenotic human aortic valves [39] and upregulates osteogenic pathways involving Msx2 and Wnt signaling [40] and Runt-related transcription factor 2/core-binding factor subunit alpha-1 (Runx2/Cbfα-1) [41]. It is likely that AVECs are regulating aVIC or obVIC function and that, given the presence in vivo of both BMP2 and TGF-β1, a combination of osteogenic and dystrophic pathways is occurring. Therefore, we are most concerned with the transitions to and behavior of aVICs and obVICs.
AVIC-AVEC co-culture systems have provided some insight into the complex regulation of AVICs. When porcine AVICs were cultured with the same number of AVECs in a 3D model, they demonstrated decreased αSMA, cell number, and increased total protein and sulfated glycosaminogly can content compared to AVICs cultured alone in the 3D collagen gel [42]. Given osteogenic differentiation media, AVICs in 3D collagen hydrogels showed much higher levels of calcification via Alizarin Red stain, but the addition of AVECs brought calcification, osteocalcin, Runx2, and αSMA back down to at least control levels [43]. A more recent co-culture model utilizes magnetic nanoparticles to layer AVICs and AVECs and allow them to freely float in media [44]. While it would necessitate a system capable of flow, including relevant circulating cells, precursor and/or inflammatory, in a co-culture system would help elucidate their role in calcification.
3. The Appropriate Model Organism
The ideal in vitro model would use primary human AVICs, but availability is the chief limiter of using human-derived samples. The next best cell would retain all characteristics of the human cells important to CAVD. Since it is believed that the important mediators of calcification are AVICs, we can narrow our search to finding a species with AVICs comparable to human AVICs.
Non-human primates are a logical choice because of their genetic similarity. However, maintenance of these organisms requires more space, time, money, and permissions than other organisms. Likely for these reasons, non-human primate AVICs have not been isolated, though Macaca nemestrina aortic smooth muscle cells have been isolated to investigate proteoglycan expression [45]. Porcine hearts are both anatomically and physiologically similar to human hearts. The growth of the heart in swine from birth to four months is analogous to that in humans from birth to mid-teens [46] and remodeling in atherosclerosis of micropigs closely resembles human pathology [47]. Interestingly, their valves contain the same αSMA-positive population of cells in the ventricularis [15]. Swine can also develop spontaneous valvular atherosclerotic lesions, a precursor to calcification [20, 48]. The first isolation of porcine AVICs noted that they appear more homogenous than murine or leporine VICs and had a high recovery rate after being frozen, leading to the extensive use of porcine AVICs in in vitro studies [49]. Though these cells are widely used and multiple research groups have reported calcification and mineralization, Cloyd et al. reported that porcine AVICs cultured in osteogenic media with TGF-β1 (which should activate both dystrophic and osteogenic pathways) did not form mineral deposits. They used Raman spectroscopy to show that even Alizarin Red-positive nodules did not exhibit mineralization [17]. While pig anatomy is highly similar to human anatomy, porcine AVICs in vitro is still a limited model. One important limitation specific to in vitro cell culture systems is the age of the cells. In 20% of long-term cell culture, AVICs become contact-inhibited monolayers and behave unstably [50]. Also, metabolic activity of porcine AVICs was found to be passage number dependent [51]. Late-stage cultured AVICs demonstrated higher numbers of myofibroblasts [52–54]. Thus, porcine AVICs are generally used no later than passage 7. Though porcine AVICs have limitations, they are the best available model.
Ovine AVICs have been shown to form CNs when treated with TGF-β1 within 72 hours, and to calcify, assayed via Alizarin Red staining, within two weeks [18, 55]. Canine AVICs were also considered early in the development of CAVD research [38]. Specifically, beagles demonstrate age-related changes to aortic valves, including calcification; changes were especially apparent in the fibroblasts, suggesting a similar mechanism to human calcification [56]. In vitro, canine AVICs spontaneously formed CNs containing hydroxyapatite over two to three weeks, compared to human AVICs developing nodules in about six weeks under the same conditions [38]. Also, while an imperfect model, many similarities exist between canine and human myxomatous mitral valve disease, reinforcing the likeness between human and canine valves [57]. While canine AVICs were deemed very similar to humans’, they are not often used, likely as a function of convenience – dogs have longer life spans than small animal models and are not maintained at a large scale for another purpose, as pigs are for food. Rabbits are used for in vivo studies, but not as often in vitro, likely because they require high cholesterol diets to develop calcification [5, 58–60].
Mice are another popular model organism, perhaps because of their low cost, easy management, short life spans, and availability of genetic mutants. Murine cell lines can be easily immortalized, allowing for near indefinite expansion and use without regard for passage limitations. AVICs could be harvested from a variety of genetically-altered models such as ApoE−/−, Notch1+/−, and LDLr−/− [10, 20, 61–65]. Though some of these models are the only ones to exhibit the hemodynamic effects of aortic valve stenosis, murine valvular structure is significantly different from human [20, 66]. Specifically, human valves have trilaminar structure, but murine valves only have a fibrosa and spongiosa [66]. While non-ideal, murine AVICs would provide a convenient model that facilitates genetic manipulation allowing for further exploration of CAVD mechanisms. A summary of the advantages and limitations of the AVICs derived from each model organism can be found in Table 2.
Table 2.
Examination of advantages and disadvantages associated with AVICs derived from common model organisms.
| Organism | How are its AVICs useful? | Why are they imperfect? | Who has used these AVICs? |
|---|---|---|---|
| Human | Most appropriate | Difficult to obtain | [31, 38, 41, 95, 102, 105, 106, 108, 113, 121, 135, 138] |
| Porcine | Similar anatomy to human; easy to obtain; swine spontaneously develop calcification precursors | More homogenous than human | [16, 17, 22, 42–44, 48, 51–53, 69–71, 73, 82, 86, 87, 89, 93, 98, 110, 114, 115] |
| Ovine | CNs develop more quickly than human | More difficult to obtain than porcine | [18, 55, 117, 139, 140] |
| Canine | CNs develop more quickly than human; pathology naturally occurs | Difficult to obtain; require ageing | [38] |
| Leporine | Many osteogenic markers upregulated; easy to obtain | Require high cholesterol diets over time |
4. The Environment
4.1 Mechanical Characteristics
As a human ages, aortic valves remodel: AVIC density and proliferation decreases, elastin content increases, and collagen fibers become more aligned [67]. However, in CAVD, elastin content is fragmented and decreased, while collagen content increases and is disorganized, and the valve leaflet thickens. Remodeling of the ECM via MMP activity, and subsequent stiffening that is characteristic of CAVD has been shown to regulate cellular processes [31]. For example, AVICs cultured in the presence of TGF-β1 on type I fibrillar collagen gels or fibrin coated tissue culture plastic or hydrogels of ~25kPa formed osteogenic CNs, whereas nodules on ~120kPa formed through the dystrophic pathway via myofibroblastic differentiation [68, 69]. This suggests that after the initiation of disease, a positive feedback exacerbates the progression, at least in the dystrophic case.
Substrate composition has also been shown to affect calcification. AVICs cultured on fibrin or tissue culture polystyrene exhibited significantly more nodules than on collagen, fibronectin, or laminin [70]. In addition, the presentation of RGD to AVICs resulted in far more calcification than the presentation of YIGSR or DGEA. RGD, YIGSR, and DGEA are ECM-derived peptide sequences derived from fibronectin/fibrin/laminin/collagen, laminin, and collagen, respectively. Their receptors are αvβ3/α5β1/α1β1, 67-kDa laminin receptor, and α2β1, respectively. Further investigation showed that disruption of the α5β1 integrin or 67 kDa laminin receptor mediated binding between AVICs and ECM results in increased calcification [70]. Fibronectin coated tissue culture polystyrene suppressed calcification markers, while fibrin coated tissue culture plastic enhanced calcification as demonstrated by CN number, ALP activity, αSMA expression, Cbfα-1 expression, and calcium content via the o-cresolphthale in complex one method. However, both fibronectin and fibrin coating soft hydrogels suppressed calcification [71]. This suggests that substrate stiffness may be more important than specific ECM component interactions. However, the method in which stiffness is modulated (i.e. by increasing crosslinking) is often coupled to the presentation of ECM components, especially integrins.
Also important to consider are the effects of trying to recapitulate a 3-dimensional (3D) in vivo environment with a 2-dimensional (2D) environment in vitro. When porcine AVICs were encapsulated in peptide-modified polyethylene glycol hydrogels, results were consistent with 2D experiments [70]. However, in 3D spheroids, fibroblasts become much less sensitive to TGF-β1 than when arranged as a monolayer [72]. This suggests that the threshold for pathologic behavior induced by relevant biochemical cues may vary depending on the environment. Similarly, fewer isolated porcine AVICs express αSMA in 3D collagen type I gels when compared with those in 2D tissue culture flasks [73]. It appears that in a 3D environment, fewer aVICs are present and qVICs are less sensitive to TGF-β1 and thus more difficult to activate. This suggests that the concentration of TGF-β1 used to induce pathological behavior should vary dependent on the dimensionality of the system to best match in vivo levels. Similarly, porcine AVICs in a 3D collagen hydrogel were not susceptible to osteogenic media mediated calcification until mechanical stress was added [43]. It appears that a 3D environment may require more dramatic treatment to induce the same behaviors as a 2D environment.
4.2 Dynamics
Many traditional CAVD in vitro studies have been investigated in a static environment, but the valves exist in a dynamic environment; this likely affects calcification mechanisms. Interestingly, calcific lesions occur preferentially on the aortic side of the valve in the fibrosa, which is the stiffer side [74–76]. As the aorta stiffens with age, axial stiffening and circumferential compliance increase [77]; this results in higher mechanical loads placed on the circumferentially-aligned collagen fibers, along which AVICs reside [78]. Also, an increase in transvalvular flow greater than 0.3m/s per year is a clinical predictive marker for patients who might benefit from surgery, suggesting that increased flow contributes to pathological progression [79]. NO release by AVECs is regulated by flow; under laminar shear stress, NO is released and helps maintain valvular homeostasis via signaling to AVICs. However, low and oscillating shear stress, as would occur on the aortic side of a diseased valve, inhibits this release [80]. Also, while the AVICs themselves are not be directly exposed to fluid flow, it has been shown that flow alone can differentiate fibroblasts (the majority cell type of the AVIC population) into myofibroblasts [81]. This positive feedback of a stiffening valve that can no longer properly regulate its AVICs to maintain homeostasis is evidence of the importance of the dynamic environment on disease progression.
Several groups have begun probing CAVD progression in dynamic in vitro models. Fisher et al. showed that CN formation is strain dependent and that strain drastically reduced the time to nodule formation – 48 hours versus three days to three weeks [82]. At the tissue level, in a bioreactor under cyclic strain, porcine aortic valve cusps showed greater evidence of calcification under 15% (pathologic) strain than 10% strain (physiologic) [83]. In a related study of vascular calcification, 7% cyclic, equibiaxial strain yielded greater mineralization than unstrained calcifying vascular cells [84]. Strain alone was able to induce higher levels of myofibroblastic phenotype as measured by αSMA and collagen synthesis than untreated, unstrained cells, suggesting that strain exacerbates calcification via the dystrophic pathway [85]. In 3D culture of porcine AVICs, osteogenic media was unable to induce calcification, but the addition of mechanical stress via anchoring the gel led to significant calcification, as well as increases in αSMA, Runx2, and osteocalcin mRNA levels [43]. These studies demonstrate the critical role of the stress and strain placed on AVICs.
4.3 Biochemical Cues
Many cytokines are known to modulate AVIC behavior, including inducing disease progression in vitro. TGF-β1 is upregulated in diseased human valves, and when applied in vitro, exacerbates nodule formation [18]. TGF-β1 has been shown to activate myofibroblasts in valves leading to increased αSMA expression via Smads and p38 [86, 87]. As these myofibroblasts become more contractile, they likely activate latent TGF-β1 from the ECM [88]. This positive feedback loop provides a strong potential mechanism for dystrophic disease progression. Some experiments have shown that fibroblast growth factor 2 (FGF-2) treatment can block nodule formation and matrix contraction of AVICs, effectively counteracting TGF-β1 treatment [54]. In addition, antagonism of 5HT2B, a TGF-β1-dependent cardiopulmonary serotonin receptor, has been shown to prevent myofibroblast differentiation and CN formation in porcine AVICs [87]. Another recent strategy is to target cadherin-11, a protein believed to mediate cell-cell tension in CAVD and that has higher expression in calcified human valves; siRNA knockdown of cadherin-11 in vitro prevented TGF-β1-mediated CN formation [89].
Early aortic valve morphogenesis is regulated by many signaling factors that are also important in bone formation. Transcriptional factor Sox9 activity in AVICs promotes a chondrocytic phenotype (obVIC), but prevents progression to osteogenic mineralization; Msx2 inhibits Sox9 function [90]. Sox9fl/+; Col2a1-cre mice developed calcific lesions in their heart valves, supporting the important role of Sox9 in vivo [91]. Msx2 has also been shown to promote calcification via the Wnt signaling pathway, involving Wnt3a, Wnt7a, and nuclear translocation of β-catenin [92]. C-type natriuretic peptide (CNP) promotes endochondral bone formation and has been found in ventricular side AVICs, supporting its role as a protective factor. It also has been shown to prevent the differentiation of porcine AVICs to myofibroblasts or osteoblasts in vitro [93]. The combination of bone morphogenic protein 4 (BMP4), TGF-β1, and cyclic stretch can induce CN formation ex vivo in human leaflets and can be inhibited by noggin [83]. Wnt receptor low density lipoprotein receptor-related protein 5 (LRP5) and β-catenin, factors in canonical Wnt signaling, show increased expression in diseased human aortic valves [94]. Reactive oxygen species (ROS) signaling has also been shown to upregulate Runx2 via AKT and Msx2 [95, 96], whereas Notch suppresses Runx2 signaling and sustains Sox9 level in AVICs, thus inhibiting mineralization [11, 97, 98]. ROS have also induced in vitro calcification in vascular smooth muscle cells via BMP2 activity and the osteogenic pathway [99]. Many experimental models involve treatment of AVICs with osteogenic media, usually supplemented with β-glycerophosphate, dexamethasone, and ascorbic acid. It is understood that these factors work in concert to promote osteogenesis over time; dexamethasone activates and regulates Runx2 expression via Wnt/β-catenin signaling, ascorbic acid is required for collagen1 formation and subsequent ECM-mediated upregulation of Runx2, and β-glycerophosphate is a source of phosphate required to produce hydroxylapatite mineral and inorganic phosphate regulates BMP2 via the ERK pathway [100]. One confounding effect of β-glycerophosphate is that it can induce dystrophic calcification detectable by Alizarin Red and von Kossa, which leads to false positive results for osteogenic calcification [100]; this may explain the Alizarin Red positive nodules lacking mineralization observed by Cloyd et al. [17]. Also, β-glycerophosphate and high levels of inorganic phosphate have been shown to induce different calcification from in vivo mineralization [101], suggesting physiological levels of inorganic phosphate should be used to more accurately model in vivo processes.
Consideration of inflammatory pathways also provides insight since chronic inflammation often precedes calcification. Tumor necrosis factor-α (TNFα) has been shown to accelerate calcification, assayed via ALP activity, Alizarin Red, and von Kossa, in human AVICs via BMP2-Runx2 pathway [102]. Signals from the TNFα pathway also regulate Msx2-Wnt mediated calcification in LDLr−/− mice [103]. TNFα, interleukin 1-β (IL1-β), and IL6, have been shown to regulate the Notch signaling [104] and toll-like receptor 4 (TLR4) stimulation is enhanced by Notch1 in human AVICs via NF-κB [105]. Silencing TLR4 attenuates BMP2 expression, and stimulating TLR2 or TLR4 induces CN formation in human AVICs [106]. oxLDL increases Wnt3a, which drives osteogenic differentiation through LRP5 [107]. This links the early stage chronic inflammation to the osteogenic pathway of calcification. Receptor activator of nuclear factor kappa-B (NF-κB) ligand (RANKL), a surface bound molecule of the TNF family, and peroxisome proliferator-activated receptor gamma (PParγ) have also led to increased calcification in vitro [108, 109].
It has been thought that statins may provide a protective effect against calcification, although this is controversial; in vitro experiments showed promise, but randomized human trials did not demonstrate the benefit of statin therapy [5]. NO donors have also been shown to reduce nodule formation, likely via soluble guanylyl cyclase activation [110]. Increasing the expression and activity of endothelial NO synthase (eNOS) in hypercholesterolaemic leporine aortic valves led to decreased calcification [111]. Porcine AVECs were able to inhibit AVIC calcification via NO secretion inhibiting the differentiation to obVICs. Additionally, blocking NO led to increased calcification even in 3D AVEC-AVIC co-culture [43]. Ex vivo culture of porcine aortic valve cusps in osteogenic media demonstrated significantly more CN formation on the fibrosa side than the ventricularis, which was exacerbated with NO inhibition. In healthy human valves, eNOS levels were much higher on the ventricularis than the fibrosa, further supporting the important protective effect of NO [43].
5. Evaluation of Assays
5.1 Calcium Assays
Evaluation of valve calcification can be separated into two categories: direct, in which the level of calcium or mineralization is directly measured, and indirect, in which markers of the proposed dystrophic and/or osteogenic pathways toward calcification are measured. Direct evaluation has the advantage of determining whether the assay leads to a pathological outcome functionally, whereas the indirect measurements yield more mechanistic information (Table 3).
Table 3.
Summary of direct techniques for evaluating calcification in vitro including advantages, limitations, and expected results in the normal and pathological states of human or porcine aortic valves (or rat, for atomic absorption spectroscopy).
| Techniques for Evaluation of Calcification | ||||
|---|---|---|---|---|
| Technique | Advantages | Limitations | Normal/Pathological Results | Notes on Images |
| Alizarin Red | Easy to stain; relatively easy to quantify with large range; inexpensive | Other elements, like magnesium, iron, and manganese also stain red |
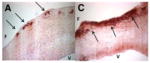
|
Tissue sections from porcine aortic valves; F=fibrosa; V=ventricularis; Balachandran 2010. |
| Arsenazo III | No interference from cations commonly found in plasma; easy to quantify; more stable and accurate than o-cresolphthalein complexone | Cannot differentiate between intracellular and extracellular calcium |

|
Porcine aortic valves; 10% strain is physiologic; 15% is pathologic; Balachandran 2010. |
| Atomic Absorption | Gold standard to determine sample composition | Requires vaporization of sample; expensive |

|
Calcium in porcine cusp or bovine pericardium after glutaraldehyde or triglycidylamine crosslinking in transplant rat model; Connolly 2005. |
| Energy-Dispersive X-ray Spectroscopy | Easily quantifiable; can perform during SEM or ESEM; ESEM yields more authentic data (no coating interference) | Expensive |

|
Human aortic valves; region with and without calcific lesions; Bertazzo 2013. |
| O-Cresolphthalein Complexone | Easily quantifiable | Not as stable and accurate as Arsenazo III |

|
Porcine AVICs on various coated tissue culture polystyrene; withTGF-β1 is pathologic (black); Benton 2008. |
| Raman Spectroscopy | Can be performed on live cells; algorithms can use data to accurately diagnose valve calcification | Expensive |
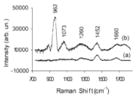
|
Human aortic valves; a is physiologic; b is pathologic; Otero 2004. |
| Scanning Electron Microscopy | Topographical and compositional information; resolution ~nm; can be performed on hydrated samples (ESEM) | Difficult to quantify without EDS; expensive |

|
Human aortic valves; scale bar is 3μm; green to orange represents increasing intensity; Bertazzo 2013. |
| Transmission Electron Microscopy | Chemical composition information; resolution ~pm | Expensive; difficult to perform on hydrated tissue |

|
Human aortic valves; scale bar is 2μm; S=spherical particles; OM=organic matter; Pt=platinum; Bertazzo 2013. |
| von Kossa | Easy to stain; inexpensive | Melanocytes in valves of a black or brown mouse will appear as false positive stain; not specific for calcium phosphate |

|
Tissue sections from porcine aortic valves; black is calcification; Balachandran 2010. |
Direct evaluation techniques include von Kossa staining [19, 83, 89, 96, 98, 99, 102, 108, 112, 113], Alizarin Red staining [17, 43, 55, 60, 70, 71, 82, 83, 87, 89, 93, 95, 96, 98, 102, 103, 106, 110, 114–117], energy-dispersive X-ray spectroscopy (EDS) [19, 118], Raman spectroscopy [17, 119, 120], scanning electron microscopy (SEM) [17, 60, 118], transmission electron microscopy (TEM) [17, 118, 121], atomic absorption [117, 122], arsenazo III [60, 83, 112], and o-cresolphthalein complexone [71, 108, 117] measurements. While these are all used as measures of calcification, not all are perfectly specific and thus are often used in concert. The gold standard for calcium detection is atomic absorption spectroscopy. Atomic absorption spectroscopy is based on the principle that different elements absorb different wavelengths of light and it works by atomizing the sample, sending light usually from a hollow cathode lamp of a specific wavelength through the vaporized sample, and measuring the amount absorbed [123]. Samples with increased mineralization content exhibit higher absorbance levels compared to controls.
Probably the most common measure of calcification, Alizarin Red, or 1,2-dihydroxyanthraquinone, stains hydroxyapatite mineralized matrix red-orange. Calcium, but also magnesium, manganese, barium, strontium, and iron, forms complexes with the dye in a chelation process, and results in a birefringent stain. Calcium is usually in much higher concentration than the other elements, allowing the inference that the areas stained have calcium present. Alizarin Red is often used to stain CNs to verify their mineralization and to help quantify the nodule assay, either by making the nodules easier to count or by extracting the dye for more rigorous quantification. Typical methods for quantifying the amount of dye involve staining of the cells or tissue, washing extensively, extracting via acetic acid or cetylpyridinium chloride, neutralization with ammonium hydroxide, and colorimetric detection at 405nm or 550nm. The acetic acid-ammonium hydroxide method is three times more sensitive than the cetylpyridinium method and results in better signal to noise ratio, especially for weakly stained samples [70, 124]. This method is also advantageous over Arsenazo III quantification because it has a higher and wider linear range of detection [124].
Von Kossa is another common stain for mineralization, especially in tissue sections. The stain works by reducing the calcium ions with light and replacing them with silver deposits that appear dark grey or black in tissue [125]. This method is not specific for calcium phosphates [126], though it has been suggested that the yellow precipitates are specific [127]. Von Kossa can be further confused if performed on a C57BL/6 mouse, which has melanocytes that appear black in the aortic valve. Thus, von Kossa is performed frequently in combination with Alizarin Red staining.
Calcium content can be measured more directly by various methods, but it is important to note that these methods all require lysing of the sample, meaning that calcium from mineralized areas or calcific lesions is not differentiated from intracellular calcium. Arsenazo III is a metallochromogen that complexes with calcium at pH 6.75 without interference from any other cations commonly present in serum or plasma, and is measured at 650nm [128]. When compared with the o-cresolphthalein complexone method, accuracy and calibration stability increased [129]. The o-cresolphthalein complexone method involves a reaction of Ca2+ ions with o-cresolphthalein complexone in an alkaline solution (8-Hydroxyquinoline at pH 10.6) and reading the sample absorbance at 660nm [130]. While these methods do not have a range of detection as large as Alizarin Red quantified via the acetic acid-ammonium hydroxide method, they are still useful for samples with low levels of calcium.
Other elemental methods include SEM, TEM, and Raman spectroscopy. SEM yields topographical and compositional information about the sample’s surface with a resolution on the order of nanometers. It can be performed on fixed, dehydrated, and gold-/platinum-/or carbon-sputter-coated samples or in wet conditions via environmental SEM (ESEM) [131]. TEM yields information about the sample’s chemical identity based on how it absorbs electrons and has a resolution on the order of picometers [132]. It can be performed on fixed, dehydrated, and stained samples. EDS analysis allows one to determine particular elements and their proportions in the sample. It functions on the principle that different elements will absorb different energy x-rays and the amount absorbed corresponds to the amount of element present [133]. EDS can be performed during SEM and ESEM; the advantage to using ESEM is that the samples do not have to be coated and high accelerating voltages can be used. EDS performed during ESEM is better because of the lack of interference from the coating and because the lack of sample preparation yields more authentic data. EDS coupled with ESEM yields quantitative data as well as qualitative [131, 133]. Raman spectroscopy is unique in that it can be performed on live cells. This allows calcification to be measured over time. Raman has also been shown to be an effective diagnostic for human heart valve calcification. Given the appropriate training data, an algorithm based on spectral shifts could predict whether the tissue was calcified with 100% sensitivity and specificity [119, 120].
5.2 Indirect Assays
In addition to quantifying mineralization, there are assays commonly employed to assess the progression of calcification by investigating mechanistic markers in the context of CAVD. For example, characterizing the phenotypic changes of AVICs toward myofibroblasts is commonly accomplished via immunofluorescence staining, western blotting, or ELISA for αSMA, collagen gel contraction assays, and wound assays. While ELISA is the most quantitative method for detecting changes in αSMA, immunofluorescence provides information about the protein’s localization and both immunofluorescence staining and western blots provide a high enough resolution to see changes in expression level. Collagen gel contraction assays indirectly quantify the myofibroblastic differentiation of AVICs based on the principle that higher levels of αSMA will result in higher contractility, measured by the change in size of the collagen gel after being seeded with cells. The wound assay involves disruption of a cell monolayer with a pipette tip and it measures the tension between cells via the wound area. The larger the wound, the more neighboring cells there are pulling on those that were disrupted [89].
Alternatively, the osteogenic process of calcification is often evaluated via ALP activity, RT-PCR, immunofluorescence staining, ELISA, and/or western blotting for Runx2 and osteocalcin. ALP activity is measured by how much p-nitrophenyl phosphate is dephosphorylated by ALP, which turns the solution yellow and can be quantified by absorbance at 405nm [134]. Runx2/CBFα-1 is a transcription factor associated with osteoblast differentiation and osteocalcin/BGLAP is a protein secreted only by osteoblasts. Runx2 is often used as an early stage marker of osteoblast activity, and osteocalcin and ALP are later stage indicators of osteoblast activity. MMPs have also been investigated via zymography, collagenase activity, immunofluorescence staining, and western blots to determine which were most important for pathological matrix remodeling [135].
Atomic force microscopy (AFM) has also been used to characterize the composition of calcified valves ex vivo in an effort to better understand the mechanism of formation. The ultra-fine structure of calcified regions of a human aortic valve was examined on a nanometer scale and found to contain 30–70nm diameter closely connected crystals. They suggest the mechanism of formation is deposit from supersaturated interstitial fluid and the crystals then grow on the organic substrate regulated by volume diffusion of interstitial fluid [136]. Recently, an AFM technique for evaluating the mechanical stiffness of valves has also been developed. This technique allows researchers to characterize mechanical properties of small animal models of CAVD, which can be extended to larger animal models and other diseases as well, while leaving enough tissue for concurrent histological studies [75]. Also, AFM comparison of human aortic valves with current valve replacement materials can yield insight into development of better prosthetics and a possible mechanism of the calcification that is common in prosthetics [137].
6. Conclusions
Utilizing a combination of CAVD models to investigate the factors important to its progression will likely yield information critical to the development of new therapeutic strategies. In vitro models are useful because they allow the quick and controlled manipulation of a large set of variables. While this simplifies the task of clarifying each factor’s contribution to the disease, it presents the challenge of determining which other variables it interacts with and how oversimplification of the model may lead to results that are not relevant in vivo. Similarly, the proper evaluation of the chosen assays is necessary to yield significant insight into CAVD mechanism. Combining evaluation methods also increases the significance of results. For example, Alizarin Red staining may yield interesting and quantitative results, but when paired with von Kossa its specificity increases. Raman spectroscopy should be used more frequently as it allows evaluation of live cells. Indirect evaluation of calcification can yield important mechanistic and functional data via western blot, immunofluorescence, PCR, and ELISA or ALP activity, collagen gel contraction, wound assay, and zymography, respectively. Figure 2 presents a summary of the important variables to consider when designing and evaluating a relevant in vitro model of aortic valve calcification. From choosing an appropriate origin of cells to the combination of evaluation techniques, any in vitro assay should recapitulate the conditions of the normal and disease state in an efficient and informative manner. Thoughtful choices should lead to novel and more promising targets to prevent and reduce CAVD.
Figure 2.

Both the design and the evaluation of in vitro models of calcification must be considered. During design, it is important to consider the type and origin of the cell or cells and the composition, cues, and mechanical environment of these cells. During evaluation, a coupling of mechanistic and functional evaluation leads to powerful conclusions. Examples of calcification-related assays that fall under each category are listed.
Highlights.
Heart valve calcification is an idiopathic disease
Suitable animal models for valve calcification are lacking
In vitro models may provide mechanistic insights for calcification in vivo
This review highlights in vitro valve calcification models
Acknowledgments
Funding
MAB was supported by the National Science Foundation (Graduate Research Fellowship) (DGE-0909667). WDM was supported by the National Institutes of Health (HL094707 and HL115103) and the National Science Foundation (1055384).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Center, U.o.M.M. US Aortic Stenosis Disease Prevalence & Treatment Statistics. 2013 Available from: http://umm.edu/programs/heart/services/programs/surgery/valve-surgery/facts.
- 2.Roger VL, et al. Heart disease and stroke statistics--2012 update: a report from the American Heart Association. Circulation. 2012;125(1):e2–e220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lindroos M, et al. Prevalence of aortic valve abnormalities in the elderly: an echocardiographic study of a random population sample. J Am Coll Cardiol. 1993;21(5):1220–5. doi: 10.1016/0735-1097(93)90249-z. [DOI] [PubMed] [Google Scholar]
- 4.Aging, A.o; D.o.H.H. Services, editor. Projected Future Growth of the Older Population. 2000. [Google Scholar]
- 5.Hermans H, et al. Statins for calcific aortic valve stenosis: into oblivion after SALTIRE and SEAS? An extensive review from bench to bedside. Curr Probl Cardiol. 2010;35(6):284–306. doi: 10.1016/j.cpcardiol.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 6.Hutcheson JD, Aikawa E, Merryman WD. Potential drug targets for calcific aortic valve disease. Nature Reviews Cardiology. 2014 doi: 10.1038/nrcardio.2014.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rajamannan NM, et al. Atorvastatin inhibits hypercholesterolemia-induced cellular proliferation and bone matrix production in the rabbit aortic valve. Circulation. 2002;105(22):2660–5. doi: 10.1161/01.cir.0000017435.87463.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Drolet MC, Couet J, Arsenault M. Development of aortic valve sclerosis or stenosis in rabbits: role of cholesterol and calcium. J Heart Valve Dis. 2008;17(4):381–7. [PubMed] [Google Scholar]
- 9.Drolet MC, et al. A high fat/high carbohydrate diet induces aortic valve disease in C57BL/6J mice. J Am Coll Cardiol. 2006;47(4):850–5. doi: 10.1016/j.jacc.2005.09.049. [DOI] [PubMed] [Google Scholar]
- 10.Awan Z, et al. The LDLR deficient mouse as a model for aortic calcification and quantification by micro-computed tomography. Atherosclerosis. 2011;219(2):455–62. doi: 10.1016/j.atherosclerosis.2011.08.035. [DOI] [PubMed] [Google Scholar]
- 11.Garg V, et al. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005;437(7056):270–4. doi: 10.1038/nature03940. [DOI] [PubMed] [Google Scholar]
- 12.Plump AS, et al. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell. 1992;71(2):343–53. doi: 10.1016/0092-8674(92)90362-g. [DOI] [PubMed] [Google Scholar]
- 13.Sider KL, Blaser MC, Simmons CA. Animal models of calcific aortic valve disease. Int J Inflam. 2011;2011:364310. doi: 10.4061/2011/364310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li C, Xu S, Gotlieb AI. The progression of calcific aortic valve disease through injury, cell dysfunction, and disruptive biologic and physical force feedback loops. Cardiovasc Pathol. 2013;22(1):1–8. doi: 10.1016/j.carpath.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 15.Chester AH, Taylor PM. Molecular and functional characteristics of heart-valve interstitial cells. Philos Trans R Soc Lond B Biol Sci. 2007;362(1484):1437–43. doi: 10.1098/rstb.2007.2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ku CH, et al. Collagen synthesis by mesenchymal stem cells and aortic valve interstitial cells in response to mechanical stretch. Cardiovasc Res. 2006;71(3):548–56. doi: 10.1016/j.cardiores.2006.03.022. [DOI] [PubMed] [Google Scholar]
- 17.Cloyd KL, et al. Characterization of porcine aortic valvular interstitial cell ‘calcified’ nodules. PLoS One. 2012;7(10):e48154. doi: 10.1371/journal.pone.0048154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jian B, et al. Progression of aortic valve stenosis: TGF-beta1 is present in calcified aortic valve cusps and promotes aortic valve interstitial cell calcification via apoptosis. Ann Thorac Surg. 2003;75(2):457–65. doi: 10.1016/s0003-4975(02)04312-6. discussion 465–6. [DOI] [PubMed] [Google Scholar]
- 19.Rajamannan NM, et al. Human aortic valve calcification is associated with an osteoblast phenotype. Circulation. 2003;107(17):2181–4. doi: 10.1161/01.CIR.0000070591.21548.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guerraty M, Mohler ER., III Models of aortic valve calcification. J Investig Med. 2007;55(6):278–83. doi: 10.2310/6650.2007.00012. [DOI] [PubMed] [Google Scholar]
- 21.Mohler ER, 3rd, et al. Bone formation and inflammation in cardiac valves. Circulation. 2001;103(11):1522–8. doi: 10.1161/01.cir.103.11.1522. [DOI] [PubMed] [Google Scholar]
- 22.Monzack EL, Masters KS. Can valvular interstitial cells become true osteoblasts? A side-by-side comparison. J Heart Valve Dis. 2011;20(4):449–63. [PMC free article] [PubMed] [Google Scholar]
- 23.Arjunon S, et al. Aortic valve: mechanical environment and mechanobiology. Ann Biomed Eng. 2013;41(7):1331–46. doi: 10.1007/s10439-013-0785-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deck JD. Endothelial cell orientation on aortic valve leaflets. Cardiovasc Res. 1986;20(10):760–7. doi: 10.1093/cvr/20.10.760. [DOI] [PubMed] [Google Scholar]
- 25.Manduteanu I, et al. Calf Cardiac Valvular Endothelial-Cells in Culture - Production of Glycosaminoglycans, Prostacyclin and Fibronectin. Journal of Molecular and Cellular Cardiology. 1988;20(2):103–118. doi: 10.1016/s0022-2828(88)80024-5. [DOI] [PubMed] [Google Scholar]
- 26.Nathan C. Nitric-Oxide as a Secretory Product of Mammalian-Cells. Faseb Journal. 1992;6(12):3051–3064. [PubMed] [Google Scholar]
- 27.Gossl M, et al. Role of Circulating Osteogenic Progenitor Cells in Calcific Aortic Stenosis (vol 60, pg 1945, 2012) Journal of the American College of Cardiology. 2012;60(24):2606–2606. doi: 10.1016/j.jacc.2012.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Egan KP, et al. Role for Circulating Osteogenic Precursor Cells in Aortic Valvular Disease. Arteriosclerosis Thrombosis and Vascular Biology. 2011;31(12):2965–U534. doi: 10.1161/ATVBAHA.111.234724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Freeman RV, Otto CM. Spectrum of calcific aortic valve disease - Pathogenesis, disease progression, and treatment strategies. Circulation. 2005;111(24):3316–3326. doi: 10.1161/CIRCULATIONAHA.104.486738. [DOI] [PubMed] [Google Scholar]
- 30.Cote N, et al. Inflammation Is Associated with the Remodeling of Calcific Aortic Valve Disease. Inflammation. 2013;36(3):573–581. doi: 10.1007/s10753-012-9579-6. [DOI] [PubMed] [Google Scholar]
- 31.Kaden JJ, et al. Inflammatory regulation of extracellular matrix remodeling in calcific aortic valve stenosis. Cardiovasc Pathol. 2005;14(2):80–7. doi: 10.1016/j.carpath.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 32.New SEP, Aikawa E. Molecular Imaging Insights Into Early Inflammatory Stages of Arterial and Aortic Valve Calcification. Circulation Research. 2011;108(11):1381–1391. doi: 10.1161/CIRCRESAHA.110.234146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wallby L, et al. Inflammatory Characteristics of Stenotic Aortic Valves: A Comparison between Rheumatic and Nonrheumatic Aortic Stenosis. Cardiol Res Pract. 2013;2013:895215. doi: 10.1155/2013/895215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu AC, Joag VR, Gotlieb AI. The emerging role of valve interstitial cell phenotypes in regulating heart valve pathobiology. Am J Pathol. 2007;171(5):1407–18. doi: 10.2353/ajpath.2007.070251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paruchuri S, et al. Human pulmonary valve progenitor cells exhibit endothelial/mesenchymal plasticity in response to vascular endothelial growth factor-A and transforming growth factor-beta2. Circ Res. 2006;99(8):861–9. doi: 10.1161/01.RES.0000245188.41002.2c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Balachandran K, et al. Cyclic strain induces dual-mode endothelial-mesenchymal transformation of the cardiac valve. Proc Natl Acad Sci U S A. 2011;108(50):19943–8. doi: 10.1073/pnas.1106954108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sewell-Loftin MK, et al. Myocardial contraction and hyaluronic acid mechanotransduction in epithelial-to-mesenchymal transformation of endocardial cells. Biomaterials. 2014;35(9):2809–15. doi: 10.1016/j.biomaterials.2013.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mohler ER, 3rd, et al. Identification and characterization of calcifying valve cells from human and canine aortic valves. J Heart Valve Dis. 1999;8(3):254–60. [PubMed] [Google Scholar]
- 39.Kaden JJ, et al. Expression of bone sialoprotein and bone morphogenetic protein-2 in calcific aortic stenosis. J Heart Valve Dis. 2004;13(4):560–6. [PubMed] [Google Scholar]
- 40.Cheng SL, et al. Msx2 promotes osteogenesis and suppresses adipogenic differentiation of multipotent mesenchymal progenitors. Journal of Biological Chemistry. 2003;278(46):45969–45977. doi: 10.1074/jbc.M306972200. [DOI] [PubMed] [Google Scholar]
- 41.Yang XP, et al. Bone morphogenic protein 2 induces Runx2 and osteopontin expression in human aortic valve interstitial cells: Role of Smad1 and extracellular signal-regulated kinase 1/2. Journal of Thoracic and Cardiovascular Surgery. 2009;138(4):1008–U241. doi: 10.1016/j.jtcvs.2009.06.024. [DOI] [PubMed] [Google Scholar]
- 42.Butcher JT, Nerem RM. Valvular endothelial cells regulate the phenotype of interstitial cells in co-culture: effects of steady shear stress. Tissue Eng. 2006;12(4):905–15. doi: 10.1089/ten.2006.12.905. [DOI] [PubMed] [Google Scholar]
- 43.Richards J, et al. Side-specific endothelial-dependent regulation of aortic valve calcification: interplay of hemodynamics and nitric oxide signaling. Am J Pathol. 2013;182(5):1922–31. doi: 10.1016/j.ajpath.2013.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tseng H, et al. A three-dimensional co-culture model of the aortic valve using magnetic levitation. Acta Biomater. 2014;10(1):173–82. doi: 10.1016/j.actbio.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chang MY, et al. Oxidized low density lipoproteins regulate synthesis of monkey aortic smooth muscle cell proteoglycans that have enhanced native low density lipoprotein binding properties. Journal of Biological Chemistry. 2000;275(7):4766–4773. doi: 10.1074/jbc.275.7.4766. [DOI] [PubMed] [Google Scholar]
- 46.Swindle MM, et al. Swine as models in biomedical research and toxicology testing. Vet Pathol. 2012;49(2):344–56. doi: 10.1177/0300985811402846. [DOI] [PubMed] [Google Scholar]
- 47.de Smet BJ, et al. The atherosclerotic Yucatan animal model to study the arterial response after balloon angioplasty: the natural history of remodeling. Cardiovasc Res. 1998;39(1):224–32. doi: 10.1016/s0008-6363(98)00085-6. [DOI] [PubMed] [Google Scholar]
- 48.Simmons CA, et al. Spatial heterogeneity of endothelial phenotypes correlates with side-specific vulnerability to calcification in normal porcine aortic valves. Circ Res. 2005;96(7):792–9. doi: 10.1161/01.RES.0000161998.92009.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Johnson CM, Hanson MN, Helgeson SC. Porcine cardiac valvular subendothelial cells in culture: cell isolation and growth characteristics. J Mol Cell Cardiol. 1987;19(12):1185–93. doi: 10.1016/s0022-2828(87)80529-1. [DOI] [PubMed] [Google Scholar]
- 50.Mulholland DL, Gotlieb AI. Cell biology of valvular interstitial cells. Can J Cardiol. 1996;12(3):231–6. [PubMed] [Google Scholar]
- 51.Wang L, et al. Factors influencing the oxygen consumption rate of aortic valve interstitial cells: application to tissue engineering. Tissue Eng Part C Methods. 2009;15(3):355–63. doi: 10.1089/ten.tec.2008.0415. [DOI] [PubMed] [Google Scholar]
- 52.Cushing MC, Liao JT, Anseth KS. Activation of valvular interstitial cells is mediated by transforming growth factor-beta 1 interactions with matrix molecules. Matrix Biology. 2005;24(6):428–437. doi: 10.1016/j.matbio.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 53.Pho M, et al. Cofilin is a marker of myofibroblast differentiation in cells from porcine aortic cardiac valves. Am J Physiol Heart Circ Physiol. 2008;294(4):H1767–78. doi: 10.1152/ajpheart.01305.2007. [DOI] [PubMed] [Google Scholar]
- 4.Cushing MC, et al. Fibroblast growth factor represses Smad-mediated myofibroblast activation in aortic valvular interstitial cells. FASEB J. 2008;22(6):1769–77. doi: 10.1096/fj.07-087627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Clark-Greuel JN, et al. Transforming growth factor-beta1 mechanisms in aortic valve calcification: increased alkaline phosphatase and related events. Ann Thorac Surg. 2007;83(3):946–53. doi: 10.1016/j.athoracsur.2006.10.026. [DOI] [PubMed] [Google Scholar]
- 56.Kim KM, et al. Calcification in aging canine aortic valve. Scan Electron Microsc. 1986;(Pt 3):1151–6. [PubMed] [Google Scholar]
- 57.Aupperle H, Disatian S. Pathology, protein expression and signaling in myxomatous mitral valve degeneration: comparison of dogs and humans. J Vet Cardiol. 2012;14(1):59–71. doi: 10.1016/j.jvc.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 58.Hsu HH, Tawfik O, Sun F. Mechanism of dystrophic calcification in rabbit aortas: temporal and spatial distributions of calcifying vesicles and calcification-related structural proteins. Cardiovasc Pathol. 2004;13(1):3–10. doi: 10.1016/S1054-8807(03)00093-0. [DOI] [PubMed] [Google Scholar]
- 59.Rajamannan NM, et al. Experimental hypercholesterolemia induces apoptosis in the aortic valve. J Heart Valve Dis. 2001;10(3):371–4. [PubMed] [Google Scholar]
- 60.Hsu HH, et al. Induction of calcification in rabbit aortas by high cholesterol diets: roles of calcifiable vesicles in dystrophic calcification. Atherosclerosis. 2002;161(1):85–94. doi: 10.1016/s0021-9150(01)00623-2. [DOI] [PubMed] [Google Scholar]
- 61.Swiatek PJ, et al. Notch1 is essential for postimplantation development in mice. Genes Dev. 1994;8(6):707–19. doi: 10.1101/gad.8.6.707. [DOI] [PubMed] [Google Scholar]
- 62.Nus M, et al. Diet-Induced Aortic Valve Disease in Mice Haploinsufficient for the Notch Pathway Effector RBPJK/CSL. Arterioscler Thromb Vasc Biol. 2011;31(7):1580–8. doi: 10.1161/ATVBAHA.111.227561. [DOI] [PubMed] [Google Scholar]
- 63.Hjortnaes J, et al. Arterial and aortic valve calcification inversely correlates with osteoporotic bone remodelling: a role for inflammation. Eur Heart J. 2010;31(16):1975–84. doi: 10.1093/eurheartj/ehq237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nakashima Y, et al. ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler Thromb. 1994;14(1):133–40. doi: 10.1161/01.atv.14.1.133. [DOI] [PubMed] [Google Scholar]
- 65.Sanan DA, et al. Low density lipoprotein receptor-negative mice expressing human apolipoprotein B-100 develop complex atherosclerotic lesions on a chow diet: no accentuation by apolipoprotein(a) Proc Natl Acad Sci U S A. 1998;95(8):4544–9. doi: 10.1073/pnas.95.8.4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Treuting PMaSMD. Comparative Anatomy and Histology: A Mouse and Human Atlas. 1. Elsevier; 2011. [Google Scholar]
- 67.Aikawa E, et al. Human semilunar cardiac valve remodeling by activated cells from fetus to adult: implications for postnatal adaptation, pathology, and tissue engineering. Circulation. 2006;113(10):1344–52. doi: 10.1161/CIRCULATIONAHA.105.591768. [DOI] [PubMed] [Google Scholar]
- 68.Chen JH, Simmons CA. Cell-matrix interactions in the pathobiology of calcific aortic valve disease: critical roles for matricellular, matricrine, and matrix mechanics cues. Circ Res. 2011;108(12):1510–24. doi: 10.1161/CIRCRESAHA.110.234237. [DOI] [PubMed] [Google Scholar]
- 69.Yip CY, et al. Calcification by valve interstitial cells is regulated by the stiffness of the extracellular matrix. Arterioscler Thromb Vasc Biol. 2009;29(6):936–42. doi: 10.1161/ATVBAHA.108.182394. [DOI] [PubMed] [Google Scholar]
- 70.Gu X, Masters KS. Regulation of valvular interstitial cell calcification by adhesive peptide sequences. J Biomed Mater Res A. 2010;93(4):1620–30. doi: 10.1002/jbm.a.32660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Benton JA, Kern HB, Anseth KS. Substrate properties influence calcification in valvular interstitial cell culture. J Heart Valve Dis. 2008;17(6):689–99. [PMC free article] [PubMed] [Google Scholar]
- 72.Kunz-Schughart LA, et al. Three-dimensional tissue structure affects sensitivity of fibroblasts to TGF-beta 1. Am J Physiol Cell Physiol. 2003;284(1):C209–19. doi: 10.1152/ajpcell.00557.2001. [DOI] [PubMed] [Google Scholar]
- 73.Butcher JT, Nerem RM. Porcine aortic valve interstitial cells in three-dimensional culture: Comparison of phenotype with aortic smooth muscle cells. Journal of Heart Valve Disease. 2004;13(3):478–485. [PubMed] [Google Scholar]
- 74.Vesely I, Noseworthy R. Micromechanics of the fibrosa and the ventricularis in aortic valve leaflets. J Biomech. 1992;25(1):101–13. doi: 10.1016/0021-9290(92)90249-z. [DOI] [PubMed] [Google Scholar]
- 75.Sewell-Loftin MK, et al. A novel technique for quantifying mouse heart valve leaflet stiffness with atomic force microscopy. J Heart Valve Dis. 2012;21(4):513–20. [PMC free article] [PubMed] [Google Scholar]
- 76.Merryman WD, et al. The effects of cellular contraction on aortic valve leaflet flexural stiffness. J Biomech. 2006;39(1):88–96. doi: 10.1016/j.jbiomech.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 77.Haskett D, et al. Microstructural and biomechanical alterations of the human aorta as a function of age and location. Biomech Model Mechanobiol. 2010;9(6):725–36. doi: 10.1007/s10237-010-0209-7. [DOI] [PubMed] [Google Scholar]
- 78.Merryman WD. Mechano-potential etiologies of aortic valve disease. Journal of Biomechanics. 2010;43(1):87–92. doi: 10.1016/j.jbiomech.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Akat K, Borggrefe M, Kaden JJ. Aortic valve calcification: basic science to clinical practice. Heart. 2009;95(8):616–23. doi: 10.1136/hrt.2007.134783. [DOI] [PubMed] [Google Scholar]
- 80.Cooke JP. Flow, NO, and atherogenesis. Proc Natl Acad Sci U S A. 2003;100(3):768–70. doi: 10.1073/pnas.0430082100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ng CP, Hinz B, Swartz MA. Interstitial fluid flow induces myofibroblast differentiation and collagen alignment in vitro. J Cell Sci. 2005;118(Pt 20):4731–9. doi: 10.1242/jcs.02605. [DOI] [PubMed] [Google Scholar]
- 82.Fisher CI, Chen J, Merryman WD. Calcific nodule morphogenesis by heart valve interstitial cells is strain dependent. Biomech Model Mechanobiol. 2013;12(1):5–17. doi: 10.1007/s10237-012-0377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Balachandran K, et al. Elevated cyclic stretch induces aortic valve calcification in a bone morphogenic protein-dependent manner. Am J Pathol. 2010;177(1):49–57. doi: 10.2353/ajpath.2010.090631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Simmons CA, et al. Mechanical stimulation and mitogen-activated protein kinase signaling independently regulate osteogenic differentiation and mineralization by calcifying vascular cells. J Biomech. 2004;37(10):1531–41. doi: 10.1016/j.jbiomech.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 85.Merryman WD, et al. Synergistic effects of cyclic tension and transforming growth factor-beta1 on the aortic valve myofibroblast. Cardiovasc Pathol. 2007;16(5):268–76. doi: 10.1016/j.carpath.2007.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Walker GA, et al. Valvular myofibroblast activation by transforming growth factor-beta: implications for pathological extracellular matrix remodeling in heart valve disease. Circ Res. 2004;95(3):253–60. doi: 10.1161/01.RES.0000136520.07995.aa. [DOI] [PubMed] [Google Scholar]
- 87.Hutcheson JD, et al. 5-HT(2B) antagonism arrests non-canonical TGF-beta1-induced valvular myofibroblast differentiation. J Mol Cell Cardiol. 2012;53(5):707–14. doi: 10.1016/j.yjmcc.2012.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wipff PJ, et al. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J Cell Biol. 2007;179(6):1311–23. doi: 10.1083/jcb.200704042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hutcheson JD, et al. Cadherin-11 regulates cell-cell tension necessary for calcific nodule formation by valvular myofibroblasts. Arterioscler Thromb Vasc Biol. 2013;33(1):114–20. doi: 10.1161/ATVBAHA.112.300278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Towler DA. Molecular and cellular aspects of calcific aortic valve disease. Circ Res. 2013;113(2):198–208. doi: 10.1161/CIRCRESAHA.113.300155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Peacock JD, et al. Reduced sox9 function promotes heart valve calcification phenotypes in vivo. Circ Res. 2010;106(4):712–9. doi: 10.1161/CIRCRESAHA.109.213702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shao JS, et al. Msx2 promotes cardiovascular calcification by activating paracrine Wnt signals. J Clin Invest. 2005;115(5):1210–20. doi: 10.1172/JCI24140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yip CY, et al. Inhibition of pathological differentiation of valvular interstitial cells by C-type natriuretic peptide. Arterioscler Thromb Vasc Biol. 2011;31(8):1881–9. doi: 10.1161/ATVBAHA.111.223974. [DOI] [PubMed] [Google Scholar]
- 94.Rajamannan NM, et al. Atorvastatin inhibits hypercholesterolemia-induced calcification in the aortic valves via the Lrp5 receptor pathway. Circulation. 2005;112(9 Suppl):I229–34. doi: 10.1161/01.CIRCULATIONAHA.104.524306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Branchetti E, et al. Antioxidant enzymes reduce DNA damage and early activation of valvular interstitial cells in aortic valve sclerosis. Arterioscler Thromb Vasc Biol. 2013;33(2):e66–74. doi: 10.1161/ATVBAHA.112.300177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Miller JD, et al. Dysregulation of antioxidant mechanisms contributes to increased oxidative stress in calcific aortic valvular stenosis in humans. J Am Coll Cardiol. 2008;52(10):843–50. doi: 10.1016/j.jacc.2008.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ann EJ, et al. Inhibition of Notch1 signaling by Runx2 during osteoblast differentiation. J Bone Miner Res. 2011;26(2):317–30. doi: 10.1002/jbmr.227. [DOI] [PubMed] [Google Scholar]
- 98.Acharya A, et al. Inhibitory role of Notch1 in calcific aortic valve disease. PLoS One. 2011;6(11):e27743. doi: 10.1371/journal.pone.0027743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liberman M, et al. Oxidant generation predominates around calcifying foci and enhances progression of aortic valve calcification. Arterioscler Thromb Vasc Biol. 2008;28(3):463–70. doi: 10.1161/ATVBAHA.107.156745. [DOI] [PubMed] [Google Scholar]
- 100.Langenbach F, Handschel J. Effects of dexamethasone, ascorbic acid and beta-glycerophosphate on the osteogenic differentiation of stem cells in vitro. Stem Cell Res Ther. 2013;4(5):117. doi: 10.1186/scrt328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gronowicz G, et al. In vitro mineralization of fetal rat parietal bones in defined serum-free medium: effect of beta-glycerol phosphate. J Bone Miner Res. 1989;4(3):313–24. doi: 10.1002/jbmr.5650040305. [DOI] [PubMed] [Google Scholar]
- 102.Yu Z, et al. Tumor necrosis factor-alpha accelerates the calcification of human aortic valve interstitial cells obtained from patients with calcific aortic valve stenosis via the BMP2-Dlx5 pathway. J Pharmacol Exp Ther. 2011;337(1):16–23. doi: 10.1124/jpet.110.177915. [DOI] [PubMed] [Google Scholar]
- 103.Al-Aly Z, et al. Aortic Msx2-Wnt calcification cascade is regulated by TNF-alpha-dependent signals in diabetic Ldlr−/− mice. Arterioscler Thromb Vasc Biol. 2007;27(12):2589–96. doi: 10.1161/ATVBAHA.107.153668. [DOI] [PubMed] [Google Scholar]
- 104.Rusanescu G, Weissleder R, Aikawa E. Notch signaling in cardiovascular disease and calcification. Curr Cardiol Rev. 2008;4(3):148–56. doi: 10.2174/157340308785160552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zeng Q, et al. Cross-talk between the Toll-like receptor 4 and Notch1 pathways augments the inflammatory response in the interstitial cells of stenotic human aortic valves. Circulation. 2012;126(11 Suppl 1):S222–30. doi: 10.1161/CIRCULATIONAHA.111.083675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yang X, et al. Pro-osteogenic phenotype of human aortic valve interstitial cells is associated with higher levels of Toll-like receptors 2 and 4 and enhanced expression of bone morphogenetic protein 2. J Am Coll Cardiol. 2009;53(6):491–500. doi: 10.1016/j.jacc.2008.09.052. [DOI] [PubMed] [Google Scholar]
- 107.O’Brien KD. Pathogenesis of calcific aortic valve disease: a disease process comes of age (and a good deal more) Arterioscler Thromb Vasc Biol. 2006;26(8):1721–8. doi: 10.1161/01.ATV.0000227513.13697.ac. [DOI] [PubMed] [Google Scholar]
- 108.Kaden JJ, et al. Receptor activator of nuclear factor kappaB ligand and osteoprotegerin regulate aortic valve calcification. J Mol Cell Cardiol. 2004;36(1):57–66. doi: 10.1016/j.yjmcc.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 109.Kawaguchi H, et al. Distinct effects of PPARgamma insufficiency on bone marrow cells, osteoblasts, and osteoclastic cells. J Bone Miner Metab. 2005;23(4):275–9. doi: 10.1007/s00774-005-0599-2. [DOI] [PubMed] [Google Scholar]
- 110.Kennedy JA, et al. Inhibition of calcifying nodule formation in cultured porcine aortic valve cells by nitric oxide donors. Eur J Pharmacol. 2009;602(1):28–35. doi: 10.1016/j.ejphar.2008.11.029. [DOI] [PubMed] [Google Scholar]
- 111.Rajamannan NM, et al. Atorvastatin inhibits calcification and enhances nitric oxide synthase production in the hypercholesterolaemic aortic valve. Heart. 2005;91(6):806–10. doi: 10.1136/hrt.2003.029785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Byon CH, et al. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. J Biol Chem. 2008;283(22):15319–27. doi: 10.1074/jbc.M800021200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Galeone A, et al. Aortic valvular interstitial cells apoptosis and calcification are mediated by TNF-related apoptosis-inducing ligand. Int J Cardiol. 2013;169(4):296–304. doi: 10.1016/j.ijcard.2013.09.012. [DOI] [PubMed] [Google Scholar]
- 114.Benton JA, et al. Statins block calcific nodule formation of valvular interstitial cells by inhibiting alpha-smooth muscle actin expression. Arterioscler Thromb Vasc Biol. 2009;29(11):1950–7. doi: 10.1161/ATVBAHA.109.195271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cai ZJ, et al. Endoplasmic Reticulum Stress Participates in Aortic Valve Calcification in Hypercholesterolemic Animals. Arteriosclerosis Thrombosis and Vascular Biology. 2013;33(10):2345–2354. doi: 10.1161/ATVBAHA.112.300226. [DOI] [PubMed] [Google Scholar]
- 116.Caira FC, et al. Human degenerative valve disease is associated with up-regulation of low-density lipoprotein receptor-related protein 5 receptor-mediated bone formation. J Am Coll Cardiol. 2006;47(8):1707–12. doi: 10.1016/j.jacc.2006.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Connolly JM, et al. Triglycidylamine Crosslinking of Porcine Aortic Valve Cusps or Bovine Pericardium Results in Improved Biocompatibility, Biomechanics, and Calcification Resistance: Chemical and Biological Mechanisms. Am J Pathol. 2005;166(1):1–13. doi: 10.1016/S0002-9440(10)62227-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bertazzo S, et al. Nano-analytical electron microscopy reveals fundamental insights into human cardiovascular tissue calcification. Nature Materials. 2013;12(6):576–83. doi: 10.1038/nmat3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Otero EU, et al. Raman spectroscopy for diagnosis of calcification in human heart valves. Spectroscopy-an International Journal. 2004;18(1):75–84. [Google Scholar]
- 120.Tzang O, et al. Detection of microcalcification in tissue with Raman spectroscopy. Cardiovascular Engineering and Technology. 2011;2(3):228–33. [Google Scholar]
- 121.Latif N, et al. Expression of smooth muscle cell markers and co-activators in calcified aortic valves. Eur Heart J. 2014 doi: 10.1093/eurheartj/eht547. [DOI] [PubMed] [Google Scholar]
- 122.Brockbank KG, Song YC. Mechanisms of bioprosthetic heart valve calcification. Transplantation. 2003;75(8):1133–5. doi: 10.1097/01.TP.0000062864.54455.E5. [DOI] [PubMed] [Google Scholar]
- 123.Chemistry, R.S.o. Atomic absorption spectroscopy. [Google Scholar]
- 124.Gregory CA, et al. An Alizarin red-based assay of mineralization by adherent cells in culture: comparison with cetylpyridinium chloride extraction. Anal Biochem. 2004;329(1):77–84. doi: 10.1016/j.ab.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 125.Utah, U.o von Kossa’s Method - Calcium
- 126.Bonewald LF, et al. von Kossa staining alone is not sufficient to confirm that mineralization in vitro represents bone formation. Calcif Tissue Int. 2003;72(5):537–47. doi: 10.1007/s00223-002-1057-y. [DOI] [PubMed] [Google Scholar]
- 127.Rungby J, et al. The von Kossa reaction for calcium deposits: silver lactate staining increases sensitivity and reduces background. Histochem J. 1993;25(6):446–51. doi: 10.1007/BF00157809. [DOI] [PubMed] [Google Scholar]
- 128.ThermoScientific. Calcium Reagent Arsenazo III. [Google Scholar]
- 129.Janssen JW, Helbing AR. Arsenazo III: an improvement of the routine calcium determination in serum. Eur J Clin Chem Clin Biochem. 1991;29(3):197–201. [PubMed] [Google Scholar]
- 130.Coulter B. Calcium oCPC. 2009. [Google Scholar]
- 131.Delogne C, et al. Characterization of the calcification of cardiac valve bioprostheses by environmental scanning electron microscopy and vibrational spectroscopy. J Microsc. 2007;228(Pt 1):62–77. doi: 10.1111/j.1365-2818.2007.01824.x. [DOI] [PubMed] [Google Scholar]
- 132.Erni R, et al. Atomic-resolution imaging with a sub-50-pm electron probe. Phys Rev Lett. 2009;102(9):096101. doi: 10.1103/PhysRevLett.102.096101. [DOI] [PubMed] [Google Scholar]
- 133.Hafner B. U.o. Minnesota, editor . Energy Dispersive Spectroscopy on the SEM: A Primer. [Google Scholar]
- 134.abcam. Alkaline Phosphatase Assay Kit (Colorimetric) ab83369. [Google Scholar]
- 135.Kaden JJ, et al. Influence of receptor activator of nuclear factor kappa B on human aortic valve myofibroblasts. Exp Mol Pathol. 2005;78(1):36–40. doi: 10.1016/j.yexmp.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 136.Grases F, Sohnel O, Zelenkova M. Ultrafine structure of human aortic valve calcific deposits. J Cytol Histol. 2014;5(2) [Google Scholar]
- 137.Jastrzebska M, et al. Supramolecular structure of human aortic valve and pericardial xenograft material: atomic force microscopy study. J Mater Sci Mater Med. 2008;19(1):249–56. doi: 10.1007/s10856-006-0049-2. [DOI] [PubMed] [Google Scholar]
- 138.Lommi JI, et al. High-density lipoproteins (HDL) are present in stenotic aortic valves and may interfere with the mechanisms of valvular calcification. Atherosclerosis. 2011;219(2):538–44. doi: 10.1016/j.atherosclerosis.2011.08.027. [DOI] [PubMed] [Google Scholar]
- 139.Merryman WD, et al. Viscoelastic properties of the aortic valve interstitial cell. J Biomech Eng. 2009;131(4):041005. doi: 10.1115/1.3049821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Merryman WD, et al. Correlation between heart valve interstitial cell stiffness and transvalvular pressure: implications for collagen biosynthesis. Am J Physiol Heart Circ Physiol. 2006;290(1):H224–31. doi: 10.1152/ajpheart.00521.2005. [DOI] [PubMed] [Google Scholar]