

Abstract
Glioma is a heterogeneous disease process with differential histology and treatment response. It was previously thought that the histological features of glial tumors indicated their cell of origin. However, the discovery of continuous neuro-gliogenesis in the normal adult brain and the identification of brain tumor stem cells within glioma have led to the hypothesis that these brain tumors originate from multipotent neural stem or progenitor cells, which primarily divide asymmetrically during the postnatal period. Asymmetric cell division allows these cell types to concurrently self-renew whilst also producing cells for the differentiation pathway. It has recently been shown that increased symmetrical cell division, favoring the self-renewal pathway, leads to oligodendroglioma formation from oligodendrocyte progenitor cells. In contrast, there is some evidence that asymmetric cell division maintenance in tumor stem-like cells within astrocytoma may lead to acquisition of treatment resistance. Therefore cell division mode in normal brain stem and progenitor cells may play a role in setting tumorigenic potential and the type of tumor formed. Moreover, heterogeneous tumor cell populations and their respective cell division mode may confer differential sensitivity to therapy. This review aims to shed light on the controllers of cell division mode which may be therapeutically targeted to prevent glioma formation and improve treatment response.
Keywords: Brain tumor, Astrocytoma, Oligodendroglioma, Asymmetric cell division, Symmetrical cell division, Stem cell, Progenitor cell, Cancer stem cell, Chemotherapy, Cell of origin
Introduction
The regulation of brain tumor development and the ability of brain tumors to overcome conventional treatment have been intensely studied over past decades. Improved understanding of neural stem and progenitor cell division modes and mechanisms has led to the hypothesis that disruption of the tightly controlled ratio between asymmetric and symmetric cell division of stem and progenitor cells may lead to an aberrant predominance of symmetrical cell division, producing daughter cells with increased replicative potential and thus susceptibility to tumorigenic transformation [1]. Despite substantial insights provided by these studies, some open questions still remain. This review intends to discuss the cell division mode involved in the cell of origin and maintenance of a stem or progenitor like population within astrocytoma and oligodendroglioma and how the pathogenesis of these brain tumor types differ from each other in relation to cell division mode. Specific asymmetric cell division controllers will also be discussed for their potential role in tumorigenesis and treatment resistance, and for their actionable potential, as with further research these could be translated to a clinical setting.
The adult brain is predominantly made up of neurons, astrocytes, oligodendrocytes, ependymal cells and meningeal cells and there are brain tumors that histologically resemble each of these cell types, sometimes in combination with each other. Cancers are clonal in origin and until recently it has been unclear how a single cell type in the brain, including terminally differentiated neurons, has the ability to give rise to heterogenous brain tumors. However, the capability of stem cells to generate many differential progeny provides a probable explanation for this process, especially for brain tumors that may contain a mixture of cell types. Therefore stem and progenitor cells are likely cells of origin for the development of tumors in the brain and the disruption of asymmetric cell division is one mechanism for neoplastic transformation.
The term neural stem cell may be used to refer to radial glial cells due to their capacity for continued self-renewal in conjunction with production of cells for the differentiation pathway. Within gliomas there exists a subpopulation of cancer cells which have similar properties to the normal neural stem cells. Asymmetric cell division in the brain is a process by which neural stem or progenitor cells are able to divide to both self-renew and produce daughter cells for the differentiation pathway. This involves the asymmetric distribution of cell fate determinants between the daughter cells, through the establishment of cellular polarity prior to cell division. Thus stem and progenitor cells are able to produce two different cell types during a single mitosis. In contrast, symmetrical cell division is characterized by the production of two identical daughter cells that contain equivalent concentration of fate determining proteins.
Based on studies in Drosophila, the regulation of asymmetric cell division of neural stem cells requires a concerted effort of polarity proteins, mitotic spindle regulators and cell fate determinants. Many genes involved in asymmetric cell division are conserved and they range from kinases, phosphatases, GTPases, etc. Thus, many asymmetric cell division regulators have actionable potential. Therefore, pharmacological agents which target asymmetric cell division have high translational potential and may possibly be used to prevent brain tumorigenesis or overcome treatment resistance. However, further basic and preclinical testing is required in order to clarify the regulation of cell division mode and the role of compounds targeting asymmetric cell division controllers in brain tumor treatment. Another comprehensive review of asymmetric cell division and cancer has recently been published [2], although the current review will be focused specifically on glioma and will discuss the translational implications of studies on asymmetric cell division regulators.
Cell division in mammalian brain development
As in most adult tissues, the differentiation hierarchy in the brain includes stem cells, transit amplifying cells, lineage-committed progenitor cells and mature cells of various types. During brain development in mammals, there is evidence of neural stem cells utilizing both symmetrical and asymmetric modes (Figure 1).
Figure 1.
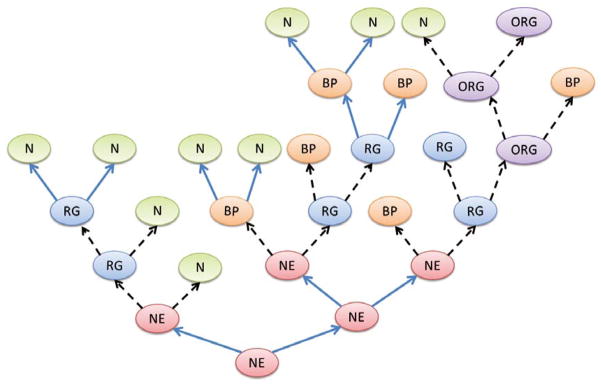
Diagram depicting asymmetric and symmetrical cell divisions and their contributions to the differentiation hierarchy in the brain during neurogenesis. Blue filled arrow denotes symmetrical cell divisions and dashed black arrows indicate asymmetric cell divisions. NE- neuroepithelial cell, RG- radial glial cell, BP- basal progenitor cell, ORG- outer radial glial cell, N- neuron.
Neuroepithelial cells and radial glia cells
The brain is developmentally derived from the neural plate, which becomes the neural tube from which neuroepithelial cells arise [3]. During a subsequent proliferative period, neuroepithelial cells undergo predominantly symmetrical proliferative divisions to increase the pool of neuroepithelial cells with self-renewal capacity. It has been shown in dissected Drosophila optic lobe neuroepithelial cells that this cell type completes several symmetrical cell divisions before transitioning to asymmetric cell division in order to produce the multiple cell types that make up the central nervous system [4, 5] (Table 1). This transition from the proliferative to the neurogenic phase occurs when Sox 1 transcription factor expression is reduced in favor of Pax 6, which drives the formation of radial glial cells over the less differentiated neuroepithelial cells [6].
Table 1.
Table of cell genesis in the mammalian brain.
| Cell type | Location | Predominate mode of cell division | Possible daughter cells |
|---|---|---|---|
| Neuroepithelial cell | Neural tube | Symmetrical during expansion and asymmetric during the neurogenic period | Neuroepithelial cell, radial glial cell, basal progenitor and neuron |
| Radial glial cell | Ventricular zone | Symmetrical during expansion and asymmetric during the neurogenic period | Radial glial cell, outer radial glial cell, basal progenitor, neuron, astrocyte, ependymal cell |
| Outer radial glial cell | Subventricular zone | Asymmetrical | Outer radial glial cell, basal progenitor and neuron |
| Basal progenitor cell | Subventricular zone | Symmetrical | Basal progenitor and neuron |
| Glial restricted progenitor cell | Throughout the brain | Unknown | Glial restricted progenitor, astrocyte and oligodendrocyte progenitor |
| Oligodendrocyte progenitor cell | Throughout the brain, predominantly white matter | Symmetrical and asymmetrical | Oligodendrocyte progenitor cell, oligodendrocyte |
| Astrocyte | Throughout the brain | Symmetrical | Astrocyte |
Neuroepithelial cells may produce one neuron or basal progenitor cell, also known as an intermediate progenitor, during asymmetric cell division [7] (Table 1). These more differentiated daughter cells lose both the apical and basal process to migrate to the subventricular zone. Basal progenitor cells divide symmetrically to produce two neurons, or rarely two basal progenitors [7–9]. In addition to being produced by neuroepithelial cells, basal progenitor cells may also be produced by asymmetric division of radial glial cells or outer radial glial cells.
Radial glial cells are located within the ventricular zone and have both apical and basal processes used to contact the lumen of the ventricle as well as the pial surface of the neural tube contacting the meninges [8]. Between radial glial cells are tight and adherens junctions at the apical end feet, maintained through the actions of Numb and Numbl and are required for the maintenance of radial glial cell polarity [10, 11]. Radial glial cells are able to divide either symmetrically or asymmetrically (Figure 1) and have been shown to undergo proliferative symmetrical cell divisions where radial glial cells or basal progenitors are produced, symmetrical neurogenic cell divisions where two neurons are produced or asymmetric cell divisions where radial glial cells, outer radial glial cells, basal progenitors or neurons are produced in combination with each other [12, 13] (Figure 1 and Table 1). Once radial glial cells become post-mitotic, they transition from Pax6 expression normally exhibited by radial glial cells to Tbr2 when they become progenitor cells and then finally Tbr1 once they reach the neuronal phenotype in the developing cortex [14, 15].
The outer radial glial cells and outer subventricular zone
More recently, a second radial glial cell type was discovered in the subventricular zone. These are derived from asymmetric division of radial glial cells, after which they migrate from the ventricular zone to the subventricular zone. These cells retain the basal fiber previously belonging to the mother radial glial cell and like their mother radial glial cells express Pax 6 [16–18]. Thus they have been termed outer radial glial cells and the area that they populate is called the outer subventricular zone. Outer radial glial cells may divide asymmetrically to both self-renew and produce either a basal progenitor or neuron [17, 19] (Figure 1 and Table 1). This is a process which relies on integrin signaling and involves the more basally located daughter cell once again inheriting the basal fiber to become another outer radial glial cell and the more apical daughter cell undergoing differentiation [17, 20]. The outer subventricular zone in humans is much larger than in rodents and due to its high proliferative activity of outer radial glial cells and their transit amplifying progeny, it is believed to be crucial for the massive increase in neuron number in the human neocortex [21].
It was previously thought that the neurogenic phase was the only time during which new neurons were produced [22]. However, it is now clear that neurogenesis is sustained at postnatal stages and possibly throughout adult life in both vertebrates and invertebrates alike, indeed in the human brain [23]. This occurs largely in the subventricular zone surrounding the lateral ventricles and in the subgranular zone of the dentate gyrus within the hippocampus in mammals, where there is a resident pool of stem cells derived from radial glial cells that persists throughout adulthood [24]. A recent study has also shown that cortical interneurons may have a non-epithelial stem cell origin within the human ganglionic eminences, which are transitory brain structures evident during the embryonic and fetal stages of development [25]. This non-epithelial stem cell had not previously been reported and is characterized by the lack of radial fibers [25].
Gliogenesis
Astrocytes
Gliogenesis becomes predominant over neurogenesis following cortical development. Mek1, Mek2, Notch, STAT3 and bone morphogenic proteins have been experimentally shown to be required for this transition in radial glial cells generally referred to as the neurogenic-gliogenic switch [26–32].
Astrocytes are glial cells in the central nervous system, which support the structure of the blood-brain barrier and help to maintain brain homeostasis. It has been demonstrated that astrocytes are derived either directly from a radial glia cells or via a glia restricted progenitor [33] (Table 1).
Another pathway of astrocyte production may be through an intermediary progenitor cell [33]. There are likely several different classes of heterogenous glial restricted progenitor cells, although the scarcity of markers to differentiate between them has led to some confusion and controversy in the field. Furthermore, there is great diversity in morphology and transcriptional regulation of gene expression between different populations of astrocytes, like the grey matter and white matter astrocytes, suggesting a differential origin [34]. However, there is also evidence of astrocyte production through an astrocyte specific progenitor, which is derived from a glial restricted progenitor cell characterized by A2B5 expression [35] (Table 1). Astrocytes may also be generated locally by symmetrical division of existing astrocytes (Table 1) [36]. Therefore it is likely that astrocytes are generated by multiple different pathways both including and excluding glial restricted progenitor cells [37].
Oligodendrocytes
Oligodendrocytes are the myelinating glial cells of the brain and are derived from oligodendrocyte progenitor cells characterized by platelet-derived growth factor receptor-alpha, NG2 proteoglycan and Olig2 expression [38, 39]. This is a separate progenitor cell lineage than that evident for neurogenesis, although this pathway may overlap with the genesis of astrocytes [40, 41]. Wnt, Olig1 and Olig2 signaling are important for the stimulation of oligodendrocyte production [40, 42], whereas Pax 6 has been shown to selectively inhibit production of oligodendrocytes [42]. Clonal analyses of O2A oligodendrocyte progenitor cells in vitro suggested that they initially undergo a finite number of symmetrical cell divisions, followed by a final asymmetric cell division to both self-renew and produce oligodendroglia. The oligodendrocyte progenitor division mode at adult stages may be more evenly distributed between asymmetric and symmetric divisions [41, 43, 44] (Table 1).
Cell division mode regulation and cancer
It has previously been shown that cells need to acquire several mutations before becoming cancerous [45], such that there is commonly intermediate transition to dysplasia before cells gain the final mutations to be classified as neoplastic. Furthermore, cancer cells often exhibit a multitude of genetic mutations, only a small subset of which may be required for tumorigenic transformation to occur - these mutations are known as driver mutations [45, 46]. Due to this stepwise accumulation of a few genes to produce the hallmarks of cancer, the number of cells with replicative potential and self-renewal capacity is tightly controlled, particularly for organisms that live a long time, as the lineage of such cells may be maintained throughout the lifespan. Therefore the control of cell division mechanisms which dictate stem cell self-renewal are of vast importance and are closely regulated in the brain.
Asymmetric cell divisions differ slightly between species and between cell types, but their general principle and many genes encoding for asymmetric cell division regulators are conserved. The induction and maintenance of asymmetric cell division is conducted through cooperation of several different factors, including intrinsic fate determinants, mitotic spindle orientation and extracellular cues. The full extent of the mechanisms controlling cell division mode in mammalian neural stem and progenitor cells are yet to be elucidated. However, this process has been extensively studied in Drosophila neural stem cells and studies showed that the mammalian homologs of fate determinants and polarity controllers play a similar role in the mammalian central nervous system. Therefore previous studies focused on Drosophila cell division mode controllers could have implications for tumorigenesis in the human brain, although further investigation is required in order to validate this hypothesis.
Control of asymmetric cell division in Drosophila neuroblasts has been thoroughly reviewed in several recent papers [2, 47, 48]. In brief, neuroblast asymmetric cell divisions are characterized by the production of a self-renewing neuroblast daughter cell and concurrently a smaller, more differentiated ganglion mother cell from which mature neurons are derived. Cell polarity is established through the formation of an apical complex, which leads to correct spindle orientation and the creation of a basal complex [49].
The apical complex is made up of atypical protein kinase C (aPKC), Partition defective 3 also known as Bazooka (Par 3), Partition defective 6 (Par 6), Inscuteable (Insc), Partner of inscuteable (Pins), Gαi, Dig, Canoe and mushroom body defect (Mud) [50–54]. This apical complex is preferentially distributed into the self-renewing neuroblast daughter cell, whereas the basal complex becomes localized within the ganglion mother cell [55]. The basal complex is made up of Numb, Partner of numb (Pon), Prospero (Pros), Miranda (Mira), brain tumor (Brat) and Staufen (Stau) [56, 57]. These fate determining factors are known as the cell intrinsic controllers of asymmetric cell division. Many studies have indicated that all of the intrinsic controllers of asymmetric cell division are interrelated, such that if one of these factors is disrupted, then this may result in aberrant size ratios or cell fates of the daughter cells produced [49, 50, 58].
Asymmetric cell division regulation in mammalian neural cells
Many mammalian homologs of Drosophila cell fate determinants have been identified. These substances have been shown to act in the same manner within mammalian radial glial cells and in some cases have been able to perform the same function within Drosophila neuroblasts when used to replace their corresponding invertebrate homologs. Illustrating this, both Drosophila and mammalian Numb are able to perform normal interactions with aPKC in vitro [59]. Moreover, when applied to Numb mutant Drosophila embryos, mammalian numb is asymmetrically distributed into the cell fated to the neuronal pathway [60]. Similarly, the Drosophila fate determinant Brat is thought to be homologous to mammalian TRIM32, which during asymmetric cell division becomes preferentially located within the more differentiated daughter cell [61].
Current studies are directed at determining the degree of functional conservation in the regulators of cell fate determination and asymmetric cell division in mammalian radial glial cells. As described previously, radial glial cells have both an apical and basal process, which aids in their ability to interact with the mammalian brain microenvironment and effectively produce the heterogeneous cell types that make up the brain. During asymmetric cell division, the basal process of radial glial cells is inherited by the more basal daughter cell. Within this process, Cyclin D2 is also asymmetrically inherited by this daughter cell and is involved in the promotion of a self-renewing cell fate [62].
Despite the complex nature of mammalian asymmetric cell division and the specialized shape of radial glial cells a few of the key regulators have been identified, although it is expected that in due time more will be elucidated (Figure 2). As mentioned previously, mammalian numb has been found to perform similar functions to its Drosophila counterparts. Indeed, demonstration of asymmetric distribution of Numb between daughter cells in embryonic mice has shown that Numb was preferentially segregated into the b-tubulin III-positive daughter cell destined for the neuronal pathway (Figure 2)[63]. Interestingly, this phenomenon was more pronounced as time progressed from embryonic days 12–14 [63]. Furthermore, Numb knockout mice have been shown to undergo significantly fewer asymmetric cell divisions than their wild type counterparts [63].
Figure 2.

Mammalian asymmetric cell division. SR- self renewing cell, D- cell for the differentiation pathway.
Studies have also found evidence of asymmetric distribution of epidermal growth factor receptor (EGFR), which was mediated by Notch signaling [64, 65]. The EGFR inheritance influenced cell fate, as FACS sorting of the cellular population with high EGFR expression revealed that these cells formed twice as many neurospheres than their low EGFR expressing counterparts [66]. Another study showed that the daughter cell with high levels of EGFR also displays markers for radial glial cells and thus is the self-renewing daughter cell (Figure 2) [65]. Additionally, the mRNA binding protein Stau2 has been found to be asymmetrically segregated during cortical development in the mouse into the more differentiated daughter cell to promote a neuronal cell fate, such that its knockdown resulted in neuronal mass formation in the peri-ventricular region [67].
As has been well characterized in Drosophila neuroblasts, Par 3 is also important for the asymmetric cell division of mammalian radial glial cells [68]. Par 3 becomes asymmetrically localized and is preferentially distributed into the less differentiated daughter cell during asymmetric cell divisions in the developing mouse neocortex (Figure 2)[68]. Furthermore, either the inhibition of Par 3 or its ectopic expression resulted in a significant reduction in the proportion of asymmetric cell divisions in favor of symmetrical cell division when compared with control constructs [68].
Musashi-1 (Msi1) is integral to the asymmetric cell division of precursor cells during Drosophila sensory organ development [69]. In mammals it has been shown to enhance Notch inhibition of Numb to increase self-renewal of neural stem cells [70]. However, when the Msi1 gene was disrupted in neural stem cells, there was no significant difference in their ability to selfrenew [71]. Therefore, further investigation is required to determine if Msi1 performs a similar asymmetric cell division regulatory function in mammalian neural stem cells as in Drosophila sensory organ precursors.
In contrast to the discussed asymmetry controllers, abnormal spindle-like microcephaly-associated protein (ASPM) promotes symmetrical cell division in mammalian cells. In mouse embryonic neuroepithelial cells ASPM is downregulated when the transition from proliferative symmetric cell divisions to neurogenic asymmetric cell divisions occurs [72]. The mechanism of this switch may be through decreased ASPM microtubule interactions that lead to alterations in the cleavage plane of the cells, influencing the mode of cell division [72]. Therefore ASPM knockdown results in decreased neuroepithelial cell numbers through reduced proliferative symmetrical cell divisions [72].
In addition to discussing the potential roles of classical asymmetric cell division controllers in brain tumor development, maintenance and treatment, this review will also demonstrate the dysregulation of factors with a less well-characterized role in asymmetric cell division in central nervous system malignancies. The importance of this is that many asymmetric cell division controllers are able to be targeted pharmacologically, which in the future may have therapeutic benefits for patients suffering from glial brain tumors.
Glioma
The term glioma encompasses all brain tumors characterized by histological evidence of neoplastic glial cells, which includes astrocytoma, oligodendroglioma and ependymoma. This review is focused predominantly on astrocytoma and oligodendroglioma (Table 2). Glioma makes up a large proportion of brain tumor sufferers and may affect patients throughout any stage of life (Table 2).
Table 2.
Comparison of astrocytoma and oligodendroglioma.
| Astrocytoma | Oligodendroglioma | |
|---|---|---|
| Incidence | most common primary brain tumor [73], most common childhood brain tumor (40% of all pediatric brain tumors)[74–77] | rarer than astrocytoma |
| Grade | Grades I and II-low grade, grade III and IV - high grade, Grade IV astrocytoma - (glioblastoma multiforme, GBM) most aggressive brain tumor currently incurable | Grades I and II -low grade, grade III - high grade |
| Progression | low grade commonly progress to higher grades | generally do not progress to higher grades |
| Treatment response | chemoresistant | chemoresponsive, nosignificant difference in survival for complete versus incomplete resections [78] |
| survival | overall survival low grade astrocytoma - 4.5 years [79], median overall survival grade III anaplastic astrocytoma- 15 months [80], median survival GBM - 8 months [81, 82] | overall survival anaplastic oligodendroglioma grade III - 42 months [80] |
Recently, glioblastoma multiforme (GBM) have been classified according to four molecular subtypes based on gene expression studies. These are the proneural, neural, classical and mesenchymal molecular profiles. The classical subtype has been characterized by EGFR mutation, and has been shown to be the variant which responds most favorably to aggressive treatment [83]. The mesenchymal subtype has a high frequency of NF1 mutations and the proneural displayed evidence of alterations in PDGFRA/IDH1 along with poor response to aggressive therapy [83].
The only treatment combination that has improved overall survival of glioma patients is radiation in conjunction with the alkylating agent temozolomide [84]. Currently under investigation is Avastin, also known as bevacizumab, a monoclonal antibody against vascular endothelial growth factor and as such is an anti-angiogenic drug. However, this treatment has only been able to provide symptomatic improvement for patients and ultimately makes gliomas more aggressive upon resistance development and relapse [85, 86]. Therefore the development of alternative treatment strategies is of vast importance and this review will discuss the role that asymmetric cell division controllers may have in improvement of therapy for glioma. Furthermore, the current review will be focused predominantly on the role of cell division mode and its control in the development, maintenance, recurrence and treatment-resistance of gliomas.
Brain tumors are largely distinguished from each other based on their histological features and cell types present. In the past it was thought that these histologically evident cell types were the cells of origin for different brain tumors. For example, astrocytomas were thought to be derived from astrocytes, oligodendrogliomas from oligodendrocytes, ependymomas from ependymal cells and meningiomas from meningeal cells that had undergone tumorigenic transformation. However, given this model, it remained unclear how terminally differentiated cells could become transformed and how cells of a committed lineage could give rise to a heterogeneous tumor.
Cancer is characterized by cells that do not rely on external proliferative stimulation, are able to evade growth suppression and death signals from the tumor microenvironment including the immune system, the ability to grow new blood vessels, invasion of surrounding tissue and the ability to metastasize [87, 88]. These features are brought about by genetic alterations which generate diverse cellular features and lead to a pro-inflammatory environment conducive to tumor growth [89, 90]. Replicative ability is required for the acquisition of mutations that lead to neoplasia in the brain and throughout the body. Therefore stem cells, with their self-renewal properties and high replicative potential are likely candidates for brain tumor cell of origin. As such, it has recently been proposed and subsequently demonstrated in genetically engineered mouse models that instead of being resultant from differentiated cells acquiring tumorigenic capacity, that brain tumors originate rather from neural stem and progenitor cells, with multipotent radial glial cells being a likely candidate [91]. Moreover, stem cells with the ability to self-renew require fewer mutations in order to become cancerous, when compared with their more differentiated counterparts [92]. However, it remains unclear how temporal and regional differences in neural stem and progenitor cells may influence mechanisms of malignant transformation and the development of different brain tumor types.
Discovery of the exact cell of origin for different types of brain tumors is of vast importance as it may have implications for treatment and preventative measures [93]. However, it is important to note that the cell of mutation and cell of origin for cancer may be distinct entities, as the initial cell of mutation may not acquire enough mutations to allow for tumorigenic transformation, but rather a more lineage restricted progenitor cell derived from the cell of mutation may be the actual cell of origin through inheritance of the preliminary mutation [94, 95].
It is possible that control of asymmetric cell division is more robust in cells higher in the stem cell hierarchy, such as neural stem cells. Thus progenitor cells may be more susceptible to malignant conversion than their counterparts at an earlier developmental stage [95].
In contrast, neural stem cells have increased replicative potential over their derivative progenitor cells. Progenitor cells have been shown to complete a finite number of replications before becoming post-mitotic [96, 97], whereas stem cells have unlimited capacity for self-renewal [98]. Specific mutations may confer a predilection for malignant transformation at particular stages of differentiation and could be responsible for the heterogeneity of brain tumor histology and differential genetic profiles evident in each subtype. For example, K-ras activation is sufficient for tumorigenesis during the early stages of radial glial cell development, but must be combined with p53 inactivation to have the same effect in more differentiated cells at a later time point in development [99]. The variance in susceptibility of cells along the hierarchy of differentiation to tumorigenic transformation highlights the fact that genetic changes are context dependent and may involve complex interactions with the brain microenvironment at different stages of development [100]. This must be taken into account when formulating measures to prevent glioma development, which may be able to be achieved through manipulation of cell division mode in the pre-neoplastic brain.
In conjunction with the differences in self-renewal capacity between stem and progenitor cells in the brain, there are also regional differences in differentiation potential [101]. This has implications for brain tumor development, as demonstrated using a genetically engineered mouse models of common glioma driver mutations, where neurospheres were derived from the wall of the third ventricle and from the lateral ventricles [102]. p53 inactivation caused proliferation in both locations, whereas PTEN loss only affected the subventricular zone derived neurospheres and both Nf1 loss or KIAA1549:BRAF overexpression only affected the third ventricle derived neurospheres in this way [102]. This may explain why gliomas with specific genetic lesions are commonly found in specific brain areas. This information is important, as the brain regions venerable to malignant transformation may be those that should be therapeutically targeted to preserve the normal cell division mode.
Dysfunction of asymmetric cell division through disruption of its control mechanisms is sufficient for cancer development in the brain, as has been shown in invertebrates [103]. For example, Drosophila neuroblasts with mutations in Pins, Mira, Numb or Pros had significantly increased proliferative potential, as evidenced by a 100 fold increase in brain tissue size resulting in mortality within 2 weeks [103]. Upon transplantation into new hosts, this tissue was found to be tumorigenic with associated genomic instability [103]. Improved understanding of this phenomenon may lead to the development of preventative treatments or improved therapeutic options for brain tumor patients through the identification of novel targets that are involved in the control of asymmetric cell division in human brain tissue.
Cell division mode in glioma origin
Some researchers have proposed a common cell of origin for all glioma tumors since astrocytes, oligodendrocytes and ependymal cells are all derived from a common ancestor, the radial glial cell. However it has also been suggested that any cell along the developmental pathway that leads to astrocyte and oligodendrocyte production, including the most mature cells, may be transformed to become tumorigenic. In contrast to the radial glial theory of glioma origin, other researchers have proposed that oligodendrogliomas originate predominantly from oligodendrocyte progenitor cells, whilst astrocytomas originate from the more stem-like radial glial cells (Table 3). Furthermore, many of the genetic mutations experimentally used to recapitulate human gliomas as closely as possible have been implicated in control of cell division mode. Therefore it is possible that disruption of asymmetric cell division, in favor of symmetrical cell divisions, promotes increased proliferative potential and thus commonly results in tumor formation. However, this hypothesis has yet to be exhaustively tested.
Table 3.
Cell type specific mutations which result in glioma formation.
| Mutation | Cell type which forms tumors | Cell type unable to form tumors | Tumor histology | Reference |
|---|---|---|---|---|
| p53 deletion, active c-Myc, Akt | primary astrocytes | astrocytoma | [104] | |
| p53 deletion, Nf1 | neural stem cells, astrocytes, neurons | astrocytoma | [105] | |
| Nf1, Pten, inactivation of p53 | stem and progenitor cells | mature astrocytes | astrocytoma | [106] |
| p53 and Pten mutation | oligodendrocyte progenitor cells | proneural GBM | [107] | |
| p53 mutation, Hras amplified | oligodendrocyte progenitor cells, neural stem cells | GBM | [108] | |
| Ink4a/Arf deletion, EGFR activated | neural stem cells, astrocytes | high grade glioma | [109] | |
| Pten deletion | GFAP-positive cells | no tumor | [110] | |
| Pten deletion, p53 deletion | GFAP-positive cells | high grade astrocytoma | [110] | |
| EGFR | GFAP-positive cells | increased astrocyte number, no tumor | [111] | |
| EGFRvIII | GFAP-positive cells | increased astrocyte number, no tumor | [111] | |
| verb B | oligodendrocyte progenitor cells | oligodendroglioma | [44, 112, 113] | |
| PDGF-B | oligodendrocyte progenitor cells | oligodendroglioma | [114] | |
| PDGF-B | nestin expressing cells | oligodendroglioma | [115] | |
| PDGF-B | GFAP-positive cells | oligodendroglioma, oligoastrocytoma | [115] | |
| EGFR | oligodendrocyte progenitor cells | hyperplasia | [116] | |
| V12Ha-ras, EGFRvIII | GFAP-positive cells | oligodendroglioma, oligoastrocytoma | [111] |
In an attempt to determine the glioma cell of origin, a p53/Nf2 mutation driven mouse model of glioma was utilized to show that even when the initial cell of mutation is a neural stem cell, the cell of tumor origin is in fact an oligodendrocyte progenitor cell [95]. The loss of p53 in cancer favors symmetrical cell division. This has been shown in mammospheres, where restoration of p53 was correlated with resumed asymmetric cell division [117]. Therefore it is also possible that the p53 mutation in neural stem cells may contribute to an aberrant increase in symmetrical cell divisions in their derivative oligodendrocyte progenitor cells, but not in the neural stem cells themselves. Although the effect of p53 mutation on asymmetric cell division of neural stem and progenitor cells has yet to be established, it would be of interest for future studies to determine if there is a lineage and maturity-specific effect of p53 mutation on the mode of cell division in the brain.
The p53 gene is mutated in approximately 48% of anaplastic astrocytoma and 31% of GBM [118]. Mutation in p53 alone is not sufficient for astrocytoma formation [119]. However it does confer proliferative advantage to the slow dividing neural stem cells and fast proliferating progenitor cells of the subventricular zone in adult mice carrying p53 deletion [119]. However, in mice which harbor p53 and Nf1 deletion, astrocytomas develop, the majority of which display characteristics of GBM [120]. Nf1 is deleted in approximately 18% of GBM and 53% of the mesenchymal molecular subtype of GBM [83]. When inactivated, Nf1 results in pro-mitotic Ras signaling. Furthermore, these tumors first appear in the regions of the brain in which stem cell populations reside, like the subventricular zone [120].
Based on mathematical modeling, it has been proposed that mutations which promote symmetric cell division, like overexpression of PDGF or p53 deletion, result in a progenitor cell of origin for astrocytoma, whereas other mutations which do not increase symmetrical cell division result in a neural stem cell of origin [121]. Recent studies from within our lab have shown that neural stem and progenitor cells have distinct sensitivities to MAPK pathway activation with regards to their cell division mode, such that asymmetric cell division is more sensitive to disruption in progenitor cells when compared to their neural stem cell counterparts (Lerner R., unpublished data).
Alternatively, it is also possible that several central nervous system cells in the differentiation hierarchy have the potential to become neoplastic, but that different mutation combinations may have a predilection for cell division mode disruption in specific cell types (Table 3)[105, 122]. In addition, there is evidence to suggest that neural stem cells at the same stage of differentiation, but from different germinal zones, may display differential reactions to the same genetic stimulus [102].
Interestingly when a construct carrying two shRNAs targeting p53 and Nf1 was used to transduce neural stem cells, astrocytes and mature neurons the tumors derived from mature neurons and astrocytes displayed high levels of progenitor markers, like Nestin and Sox2, suggesting de-differentiation is involved in tumor formation [105]. Evidence for the possibility of an astrocyte origin for astrocytic tumors was also provided when murine primary astrocytes on a p53−/− background were transduced with c-Myc and Akt [104]. Upon in vitro cultivation, these cells showed stem cell marker expression, indicative of de-differentiation, and in vivo were tumorigenic when implanted into C57BL/6 mice [104].
It has been proposed that c-Myc is integral to this de-differentiation step of tumor formation in mature cell types. For instance, neural stem cells deficient for Pten and p53 showed increased self-renewal, proliferation and failed to respond to differentiation signals in vitro [123, 124]. In contrast, this same differentiation stimulus caused the cells with a wild type genetic profile or only one of these mutations to flatten and lose Nestin staining [123, 124]. The pro-differentiation environment also resulted in increased c-Myc in the Pten and p53 double null neural stem cells. Furthermore, the resistance to differentiation stimuli was abolished when the activity of c-Myc was inhibited [123, 124].
Some studies have shown that only self-renewing cell types and not terminally differentiated cells are able to be genetically transformed to result in tumor formation particularly with Pten deletion [106, 110] (Table 3). The tumor suppressor gene Pten has been proposed to play a role in asymmetric cell division maintenance, although further studies are required to validate this theory [125]. This proposition is based on mouse studies where Pten knockdown was localized in central nervous system tissues at mid-gestation, resulting in increased cell proliferation, cell size and decreased apoptosis, which led to increased brain size and histological abnormalities [126]. In addition, when Pten is knocked down in patient derived glioblastoma cells, the tumor inhibitor lethal giant larvae (Lgl) is phosphorylated and thus inactivated by aPKC that leads to the maintenance of the self-renewing phenotype in these cells [127]. However, differentiation of this self-renewing cancer cell population was initiated when Pten levels were restored to normal levels, when Lgl was constitutively activated and when aPKC was knocked down [127]. Therefore each of these factors are in all likelihood involved in the maintenance of asymmetric cell division in astrocytoma during tumor development and should be tested as possible targets to disrupt this cell division mode and possibly sensitize GBM cells to standard chemotherapeutic agents.
Based on the discussed studies, it is likely that there is no single cell of origin for astrocytoma, but rather that they may arise from any of the cells along the neural stem cell, gliogenic and neurogenic differentiation hierarchy. In this scenario, there are multiple pathways by which astrocytic tumors can be formed, which may account for their high incidence. However, these studies also suggest that neural stem cells, progenitor cells and more differentiated cell types have distinct propensity for tumorigenic transformation, with the least differentiated cells requiring fewer mutations in key pathways driving tumor growth and therefore most likely to be transformed. A possible reason for this is increased replicative potential of cells at less differentiated stages. Nevertheless, it is also conceivable that de-differentiation is required before mature astrocytes or neurons may undergo malignant transformation, thus providing an extra barrier to tumor formation in this cell type. This could explain why experimental studies have shown that some mutations are unable to elicit tumor formation from these cell types.
Moreover, the literature suggests that the predominant mode of cell division that leads to tumor formation may be cell of origin specific, with differentiation state dictating whether asymmetric cell division is disrupted. Therefore, further studies are required to elucidate the exact mechanisms involved in astrocytoma development and if changes in cell division mode are causally involved in this process.
In contrast to astrocytic tumors, the cell of origin for oligodendroglioma is much more consistent within the literature. By far, the most common cell of origin for oligodendroglial tumors has been found to be oligodendrocyte progenitor cells [112]. This predominantly different cell of origin to those evident for astrocytomas may be responsible for the improved therapeutic response evident in oligodendroglioma patients when compared to patients with astrocytomas of the same grade.
As previously discussed in this document, oligodendrocyte progenitor cells may divide either symmetrically or asymmetrically. Our group has shown that increased symmetrical cell division in lieu of asymmetrical cell division in oligodendrocyte progenitor cells leads to oligodendroglioma formation [44]. This study was performed using a genetically modified mouse model of glioma expressing verbB with a S100β promoter, to overexpress EGFR in neural stem cell progeny that have lost GFAP expression, like oligodendrocyte progenitor cells [112, 113]. The described mouse model produces oligodendroglioma tumors from oligodendrocyte progenitor cells in close association with white matter tracts, as identified by NG2 and olig2 expression [112]. Furthermore, gene expression profiling of the resultant murine tumors showed an expression profile that closely resembled oligodendrocyte progenitor cells rather than that of neural stem cells [112].
To evaluate asymmetric cell division, NG2 expression was characterized in daughter cells that had recently been produced by mitosis of an oligodendrocyte progenitor cell. NG2 proteoglycan was asymmetrically localized in 41% of oligodendrocyte progenitor cell pairs derived from wild type mice, whereas this was decreased to 22% in S100β-verbB p53+/− mice, as evaluated by an in vitro pair cell assay [44]. In conjunction with this decrease in asymmetric cell division, there was also evidence of aberrant self-renewal, impaired oligodendrocyte differentiation and increased tumor initiating potential [44]. This was mirrored by the human condition, as NG2+ cells isolated from oligodendroglial tissue showed reduced asymmetric cell divisions when compared to control tissue from epileptic patients with no evidence of neoplasia [44]. This study has far reaching implications, as cell division controllers may be able to be targeted to avoid disruption of asymmetric cell division in oligodendrocyte progenitor cells and perhaps prevent oligodendroglioma tumor formation. Therefore further studies should aim to identify pharmacological targets with the ability to preserve asymmetric cell division and thereby restore differentiation and prevent aberrant self-renewal in non-neoplastic oligodendrocyte progenitor cells.
Further supporting the oligodendrocyte progenitor origin theory of oligodendroglioma, a Ctv-a mouse model where PDGF-B driven tumor induction is restricted to oligodendrocyte progenitor cells, showed tumor formation in 33% of animals [114]. Likewise, retroviral infection of cells with the marker profile of oligodendrocyte progenitor cells to express a EGFR-GFP fusion protein on postnatal day 3 in rats, demonstrated proliferation resulting in hyperplasia of this cell type [116].
However, not all experimental models show that oligodendrocyte progenitor cells are the origins for oligodendroglioma. Oligodendroglial tumors and mixed oligoastrocytomas have also been produced from genetic manipulation of GFAP expressing cells, thus excluding oligodendrocyte progenitor cells. For example, both oligodendroglial tumors and oligoastrocytomas developed from GFAP-positive cells, like neural stem cells or astrocytes, in GFAP-V12Ha-ras; GFAP-EGFRvIII transgenic mice [111]. Additionally, transfer of PDGF-B to nestin expressing cells, like neural stem and progenitor cells, led to formation of oligodendroglioma in 60% of mice as evaluated at 12 weeks of age [115]. In conjunction, transfer of PDGF-B to GFAP expressing cells resulted in a mixture of oligodendrogliomas and oligoastrocytoma formation in 40% of mice at the same time point [115].
Therefore, it is likely that the transformation of different cell types into tumors with histological features of oligodendroglioma may be mutation specific. However, the vast majority of experimentally induced oligodendroglioma originate from an oligodendrocyte progenitor origin. This may have implications for treatment, as it has been proposed that the mechanism which controls asymmetric cell division in these cells is more susceptible to disruption than that evident in more stem like cells, although as of yet this has not been experimentally proven.
Effect of cell of origin and cell division mode on treatment
Tumors with the same genetic mutations from different cells of origin have been shown to respond differently to preventative treatment. Combination treatment with a Ptgs2 inhibitor and an EGFR inhibitor prevented tumorigenic growth of oligodendrocyte progenitor cells, but not neural stem cells transformed with amplified HRas and p53 mutation [108]. It is unclear if this phenomenon was due to the hierarchal differentiation stage of these two cell populations. It is possible that the machinery that determined the cell division mode of these cells were affected differently, both by the initial genetic mutation and by the subsequent treatment. Thus, asymmetric cell divisions may have been maintained in neural stem cells to provide resistance to treatment, but disrupted to become more symmetrical in oligodendrocyte progenitor cells, rendering them more susceptible to therapeutic intervention.
Additionally, there is some evidence to suggest that tumors originating in stem and progenitor cells are inherently more difficult to treat and show increased aggressive behavior. Clinical and magnetic resonance imaging data from 91 patients with GBM were analyzed to determine if proximity to the germinal niche affected outcome [128]. Those patients with tumors in contact with the subventricular zone showed poorer outcome and more rapid disease progression when compared with the patients who had cortical GBM [128]. It is possible that derivation from a stem or progenitor cell population enriches astrocytic tumors with cancer cells that resemble these normal brain cell types.
Tumor propagating cells
Heterogeneous cancer cell populations have been identified within brain tumors [129–131]. In this context, cancer cell heterogeneity refers to cancer cell populations with distinct genetic mutations, differentiation status and responses to external stimuli. These include tumor populations characterized by greater tumorigenic potential as evidenced by transplantation assays into immune compromised mice, when compared to the remaining tumor cell subpopulations [132, 133]. These populations have been called cancer stem cells or stem-like tumor propagating cells (TPC) for their intrinsic similarities to neural stem cells, which include expression of stem cell markers such as CD133, drug resistance, self-renewal and the generation of multi-lineage progeny. TPC have repeatedly been shown to be associated with poorer outcome due to their resistance to chemotherapeutic agents and radiotherapy [92, 134, 135]. More recently it has been demonstrated that tumor cells with expression of lineage restricted progenitor markers OLIG2 and NG2 can be highly malignant as well [136] pointing to heterogeneity in the tumor propagating population.
In vitro sphere forming assays have also been used to identify TPC. It has recently become clear that these spheres are heterogeneous and contain both stem and progenitor-like TPC [137]. Therefore, populations referred to as cancer stem cells in some studies, may in fact contain multiple populations of cancer cells at various stages of differentiation.
Stem-like TPC are slow growing with self-renewal properties [132, 138, 139], but progenitor-like TPC are faster dividing cells which make up a large bulk of the tumor cells [133, 137]. The distinction between these tumor cell subpopulations is imperative, as they may respond differently to therapeutic agents and thus if not correctly identified, may lead to the generation of misleading results for drug screening assays. Further investigation is also required to determine how stem and progenitor-like TPC relate to each other, whether progenitor-like TPC are hierarchically derived from stem-like TPC or if these cells are able plastically transition in both directions via differentiation and de-differentiation.
It is also important to note the distinction between neural stem cells and stem-like TPC. TPC are not necessarily derived from neural stem cells, although there are similarities between them in terms of marker expression and behavior in vitro. For instance, both neural stem cells and stem-like TPC commonly exhibit Y-box binding protein 1 (YB-1) expression, Sox-2, nestin, and musashi-1 (Msi1) expression and with increased differentiation these markers are lost [140]. It is possible that TPC are direct descendants neural stem cells as suggested in genetically engineered mouse models of glioma. For example, using Nestin-ΔTK-IRES-GFP transgenic mice that were used to mark the quiescent subventricular zone neural cell population, it was shown that when these mice were crossed with Nf1, p53 and Pten deleted mice, tumor growth ensued and that a subpopulation of the tumor cells were also marked by green fluorescent protein, likely representing the TPC population [141]. However, it is also possible that TPC originate from dedifferentiation of other more differentiated tumor cell subpopulations. Furthermore, there is increased heterogeneity of marker expression and molecular profiles for TPC over neural stem cells [142].
TPC have been identified in many human gliomas [143, 144]. Populations of TPC are able to not only recapitulate the pathophysiology of the original tumor from which they were isolated, but are also able to produce the corresponding diversity of cell populations within the tumor mass in an experimental setting. Thus, a single glioma TPC is able to produce multiple heterogeneous cell types [139, 145]. It has been proposed that a combination of asymmetric and symmetric cellular divisions allows TPC to both expand the tumor mass and produce differential progeny.
Evidence of this is presented in an in vitro study using single cell lineage tracing analysis of glioma TPC isolated from human surgical glioma specimens, in order to determine the mode of TSC division. This study revealed that glioma TPC predominantly perform symmetric cell divisions under expansion conditions, functioning to build up the bulk of the tumor, whilst completing principally asymmetric cell divisions under growth factor deficient conditions [146]. In this case CD133 was asymmetrically segregated between the two daughter cells, as determined by mitotic paired cell analysis [146].
This abnormally low proportion of asymmetric cell divisions, which has been identified in gliomas, may contribute to impaired patient survival through influence on treatment response. Following treatment with gamma knife surgery, radiation or chemotherapy, gliomas are enriched for TPC positively labeled with CD133 [147–149]. Whether stem-like TPC are being selectively spared from treatment induced cell death, or alternatively if the treatment stimulates expansion of this cancer cell population is difficult to address in human patients.
Two studies suggest that the first scenario is more likely. First, studies in genetically engineered mouse models suggest that the nestin-positive TPC survive treatment with temozolamide and grow the recurring tumor [141]. Secondly, clonal analyses of 118 human GBM samples obtained from The Cancer Genome Atlas has shown that the recurrent tumor is frequently genetically distinct from the primary tumor with a small proportion of overlap [131]. Thus, in some patients the recurrent tumor contains differential proportions of specific cancer cell subpopulations when compared to the primary tumor and in other patients it may contain additional subpopulations which were not present at all in the primary tumor [131].
The homeobox gene PROX1 is the mammalian homolog of Drosophila Pros, which, is part of the basal complex that maintains asymmetric cell division of neuroblasts. PROX1 has been associated with poor outcome in human glioma and was also more prevalent in high grade astrocytic gliomas, as determined by immunohistochemistry in a series of 56 human gliomas of differing grade [150, 151]. This poorer outcome could be due to increased asymmetric cell divisions in the PROX1 labeled cell populations within these tumors [152]. Asymmetric cell division has been proposed to contribute to therapy resistance in the slow replicating stem-like TPC populations of other cancer types including gastric cancer and acute lymphoblastic leukemia [153, 154]. Therefore, a similar mechanism may confer poorer outcome to brain tumor patients with malignancies which contain a proportion of asymmetrically dividing stem-like TPC.
As previously discussed, Msi1 is a RNA-binding protein and functions to enhance Notch signaling and thus inhibit Numb, promoting neural stem cell self-renewal [70]. In a study of primary human central nervous system tumors, Msi1 expression was increased, particularly in malignant gliomas, when compared to non-neoplastic tissue and other brain tumor types [155]. This expression was related to both tumor malignancy and proliferation rate [155, 156]. In GBM cells with shRNA knockdown of Msi1, an increased number of cells were evident in G2 or M phase of the cell cycle, meaning that this knockdown increased the time these cells spent in mitosis [157]. Further, when these cells were orthotopically xenografted into NOD/SCID mice, the resultant tumors were 96.6% smaller than their non-Msi1 knockdown counterparts [157]. Therefore, further studies should be performed to determine if targeting Msi1 could be used to improve treatment of gliomas by disrupting asymmetric cell division of TPC and possibly improving tumor response to standard glioma treatment strategies.
Numb is a classical controller of asymmetric cell division and has been found to be asymmetrically segregated between daughter cells of dividing GBM stem-like TPC [158]. In this study, Numb was localized into the CD133-positive daughter cell and excluded from its CD133-negative counterpart [158]. In contrast, a study using Lgl1 knockout mice showed that brain progenitor cells are only able to symmetrically distribute Numb between daughter cells, resulting in a failure of asymmetric cell division [159]. This led to a hyperproliferative phenotype where cells lack features of differentiation [159]. These data suggest that normal Numb function and distribution is required for asymmetric cell division of TPC, which may inhibit excessive proliferation of this population, but may also contribute to treatment resistance and tumor propagation in astrocytomas.
Supporting this, studies have shown that Numb is expressed in all grades of astrocytomas [160]. Numb deletions or very low levels of Numb have been found within the proneural GBM subtype, whereas much higher expression is evident in the classical and mesenchymal subtypes, which are associated with comparatively poorer outcomes for patients [158]. Thus, increased Numb in astrocytoma, specifically in classical and mesenchymal GBM may contribute to an increased proportion of asymmetrically dividing stem-like TPC within the tumor mass, leading to impaired treatment response. However this theory has not yet been validated experimentally.
Notch is important for the maintenance of stem cells and inhibition of the differentiation pathway. In astrocytoma and GBM tissue samples, Notch is elevated when compared with non-neoplastic tissue [161, 162]. Although Numb acts to inhibit Notch signaling and Numb has been associated with poor treatment response in GBM, Notch is also a hallmark of poorer prognosis for glioma patients [163]. Notch signaling has been shown to induce increased colony forming capacity, increased self-renewal and decreased differentiation in GBM neurosphere cultures [164]. Similarly, in neural stem cells, NOTCH2 activation results in hyperplasia of the neurogenic niche and impaired neuronal differentiation. In this same study, Notch 2 signaling in GBM TPC resulted in enhanced proliferation and decreased apoptosis [165].
NOTCH activation can be promoted by the neural stem cell niche endothelial cells [166, 167]. This may in part explain why tumors located in close proximity to the ventricles produce poorer patient outcomes [128]. In conjunction, using a three dimensional culture system for surgical GBM specimens to simulate the normal tumor and tumor stroma cellular arrangement, when Notch signaling is pharmacologically inhibited with the g-secretase inhibitor DAPT (N-[N-(3,5-Difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester), not only is there inhibition of tumor cell proliferation and self-renewal, but also a decrease in endothelial cells [168]. Therefore, Notch signaling likely plays an important role in tumor associated angiogenesis and its inhibition may be beneficial in treating astrocytoma. When Notch 1 is inhibited in stem-like TPC, their tumorigenicity is impaired when injected into Balb/c nude mice [169]. Furthermore, siRNA knockdown of NOTCH1 in established tumors significantly slowed the cancerous growth when compared to control tumors [169]. Studies have also shown that with Notch pathway inhibition, with either a g-secretase inhibitor or arsenic trioxide, depletes the stem-like TPC population within GBM [170–173]. Furthermore, CD133-positive and CD133-negative cells, thought to represent TPC and non-TPC respectively, were isolated from human glioma surgical specimens, and treated with g-secretase inhibitors [174]. This resulted in increased sensitivity to radiation therapy in the TPC population but not the non-TPC population, which is already considered to be radiosensitive [174]. Thus, Notch pathway blockade by a g-secretase inhibitor has been suggested for therapeutic intervention following GBM diagnosis and in conjunction with standard treatment options. These standard treatment options have long been thought to selectively spare or possibly promote the expansion of stem-like TPC, meaning that this intervention aimed at increasing TPC sensitivity to treatment could have a substantial clinical impact.
Mutation or amplification of EGFR is very common among adult astrocytoma, particularly GBM. The asymmetric cell division regulators Numb and Notch have both been shown to regulate EGFR expression. Knockdown of Numb in U87 glioblastoma cells increased EGFR expression [158] and NOTCH1 knockdown in U251 malignant glioma cells by shRNA downregulated EGFR expression [161]. EGFR has consistently been shown to be instrumental in the formation and propagation of many astrocytoma tumors. In cells derived from patient GBM samples, it has been demonstrated that EGFR-positive subpopulations sorted by FACS were more tumorigenic than their EGFR-negative counterparts as evaluated by intracranial implantation into nude mice [129]. As such, EGFR inhibitors have gone into clinical trials for the treatment of high grade glioma and have shown significantly improved patient survival, although treatment resistance is a significant issue with this treatment [175, 176].
Studies have shown that EGFR is unevenly distributed between daughter cells during asymmetric cell division of neural stem and oligodendrocyte progenitor cells [44, 66]. The role of EGFR is to promote proliferation and self-renewal in the daughter cell into which it has been distributed during asymmetric cell division [44]. However, if asymmetric cell division is disrupted, then EGFR may become active in both daughter cells, thus increasing the replicative potential of the cellular population. It is currently unclear if EGFR is an asymmetric cell division controller itself, or if it is simply maneuvered asymmetrically by other factors. Similarly, at present the effect of EGFR inhibition on cell division mode in astrocytoma TPC and the possible implications for patient outcome has not yet been determined.
The Wnt pathway is frequently mutated in astrocytoma and is associated with hereditary syndromes that predispose patients to central nervous system tumor formation [177, 178]. Wnt, through the actions of beta catenin, is important for the maintenance of asymmetric cell divisions in the brain [179, 180]. GBM cells treated with Wnt ligands exhibit decreased proliferation and promotion of the differentiation pathway [181]. Another study has shown that the Wnt pathway antagonist, recombinant sFRP1, is effective at inhibiting patient derived glioma TPC self-renewal and proliferation [182].
Therefore further investigation is warranted to understand the role of the Wnt pathway in regulation of TPC division mode and the possible implications for treatment response. Towards this goal, an intracranial xenograft model of U373MG and GBM578 GBM was used to determine that the Wnt pathway is activated following radiation treatment with concurrent enrichment for a stem cell population within the remaining tumor [183]. This result suggests that Wnt and its role in maintenance of asymmetric cell division, may contribute to astrocytoma treatment resistance. Furthermore, siRNA inhibition of the Wnt pathway resulted in reduced survival of GBM cells following radiation treatment when compared to the wild type cell line [183].
Polo-like kinase 1 (Plk1) is an important component of the machinery that governs spindle orientation and mitotic progression of mammalian neural progenitor cells [184] and has been shown to be important for asymmetric cell division in non-mammalian experimental models [185, 186]. Plk1 expression is increased in a manner correlated with degree of anaplasia in human glioma cell lines [187]. Similarly, astrocytoma TPC show an increase in the expression of Plk1 more than 100 fold above that evident for non-neoplastic astrocytes [188]. Furthermore, inhibition of Plk1 in these TPC decreased proliferation and caused G2/M arrest, ultimately leading to apoptosis [188]. Concurrently, there was a decrease in SOX2 marker expression with Plk1 inhibition, indicating that the TPC lost some of their stem cell properties [188]. Therefore it is possible that Plk1 inhibition could be used in conjunction with standard therapeutic agents, to deplete the normally treatment resistant stem-like cancer cell population within astrocytoma. Further studies should be performed to determine if Plk1 is involved in the maintenance of asymmetric cell division in mammalian TPC and if its inhibition leads to a predominance of symmetrical cell divisions over asymmetric cell divisions.
Oligodendroglioma are far more susceptible to standard therapeutic approaches than astrocytoma. Both oligodendroglioma cells and oligodendrocyte progenitor cells extracted from genetically modified mouse models are more susceptible to temozolomide when compared with astrocytoma cells or neural stem cells [112]. The fact that both the normal cells and their derived tumors exhibit this sensitivity suggests that the therapeutic resistance seen in astrocytoma and susceptibility evident in oligodendroglioma stems from their proposed differential cell of origin.
Another possible reason for this difference is that oligodendrogliomas are less enriched for a stem-like TPC population, but rather harbor more progenitor-like TPC when compared to astrocytoma [112, 189]. Furthermore, NG2-positive progenitor-like TPC derived from human oligodendroglioma have a far greater tumor forming capacity when injected into mice, compared to their NG2-negative counterparts [112]. As previously discussed, it has been proposed that asymmetric cell division promotes resistance to therapeutic agents targeted towards actively cycling cells due to their slow proliferation rate. However, symmetrically faster dividing cells are suggested to be more susceptible to these same agents. Thus, the increased rate of symmetrical cell divisions evident in oligodendroglioma NG2-positive progenitor-like TPC may explain why oligodendroglioma are more chemosensitive than their stem-like TPC enriched astrocytoma counterparts [44].
Tumor microenvironment and cell division mode
As previously discussed for glioma, CD133 has been shown to be asymmetrically distributed between daughter cells suggesting that glioma TPC undergo asymmetric cell divisions. In GBM cell lines established from surgical tissue samples, the CD133-positive cells showed increased rate of proliferation in neurosphere culture conditions and increased potential for neuronal differentiation [190]. Whether these asymmetric cell divisions are part of a tumor cell hierarchy similar to that generated by radial glial cells in the developing brain remains to be determined.
In brain tissue placed under hypoxic conditions stem and progenitor cells populations expand [191]. Astrocytomas, in particularly GBM, commonly contain areas within the center of the tumor that are hypoxic due to the inadequate expansion of blood vessels for the nutritional needs of the neoplastic mass [192]. It is thought that these hypoxic regions enrich the tumor for TPC, in a similar manner to the normal brain, which under hypoxic conditions is enriched for neural stem cells [191]. This is supported by the fact that TPC have been found to be most prevalent within the center or inner core of GBM tumor masses, where hypoxic environments are commonly found [193].
CD133 expression increases when GBM cells are cultured in 7% oxygen, compared with 20% oxygen [194]. Low oxygen also decreased the proliferation and enhanced the differentiation capacity of these cells [194]. Similarly, when GBM cell lines were cultured in 21% oxygen and hypoxic conditions of 3% oxygen, the CD133-positive content of the cell lines were altered from 69% to 92% respectively [195]. Another effect of hypoxic conditions is an increase in the expression of Notch pathway ligands in GBM [196]. Further, in patient derived GBM TPC, culture in hypoxia not only increased CD133 expression, but also increased the ability of the cells to form neurospheres [197]. These data suggest that culture in low oxygen levels selects for CD133-labeled TPC cells or preferentially promote their expansion, which may be a predominantly asymmetrically dividing population. Supporting this, asymmetric cell division has previously been shown to be increased in lung cancer cells upon exposure to a hypoxic environment [198]. Thus extrinsic environmental factors like oxygen content may be important for regulation of cell division mode in astrocytoma TPC, in turn controlling treatment resistance.
Conclusion
Gliomas with different histological features are likely derived from a differential cell of origin with specific modes of cell division involved in this process. For instance, the disruption of asymmetric cell division in favor of faster and more expansive symmetric cell divisions in oligodendrocyte progenitor cells leads to oligodendroglioma formation, a process that has been well characterized. In contrast, astrocytomas seem to maintain asymmetric cell division of radial glial cells or dedifferentiated cells further along the gliogenic or neurogenic pathway. Thus, astrocytoma development and the cell of origin of astrocytoma are less well characterized than for oligodendroglioma in the context of cell division mode. There may also be some overlap between oligodendroglioma and astrocytoma development cell of origin. These tumors are likely on a spectrum, where the most oligodendroglial-like tumors are derived from oligodendrocyte progenitor cells and become enriched for progenitor-like TPC, whilst the most astrocytic tumors may be derived from multiple different cell types and become more enriched for stem-like TPC.
The cell types present within a glioma neoplastic mass and their cell division mode should inform tumor treatment, as faster symmetrically dividing progenitor-like TPC are thought to be more susceptible to standard therapeutic agents, whereas slower asymmetrically dividing stem-like TPC are resistant to therapeutic agents targeted at cycling proliferative cancer cells. Future studies should be aimed at conclusively determining the stem and progenitor-like TPC population ratios in oligodendroglioma and astrocytoma with the intention of confirming the cell division modes prevalent within these subgroups in each tumor type.
There are multiple factors which control cell division mode and future studies ought to focus on identifying actionable proteins which can be manipulated to prevent the disruption of asymmetric cell division, which has been shown to result in oligodendroglioma development. Conversely, targets aimed at disrupting the asymmetric cell division of TPC in astrocytoma thought to be responsible for treatment resistance would also be of vast importance. A major challenge may be to alleviate the proposed negative aspects of asymmetric cell division maintenance in astrocytoma without inadvertently causing the expansion of tumor growth by symmetrically dividing oligodendrocyte progenitor cells. Therefore it is likely that therapeutic agents targeting cell division mode need to be cell type specific to avoid unwanted tumorigenic side effects. Furthermore, research into cell division mode controllers and their role in glioma formation and treatment response could have implications for many other tumor types, as stem and progenitor cells have been proposed as the cell of origin for cancers that develop in many organs other than the brain.
Acknowledgments
Funding: Research in the Petritsch lab is supported by the National Cancer Institute and the National Institute of Neurological Disorders and Stroke of the National Institute of Health under award numbers R01CA 164746 and R01NS080619 (C.P.).
Abbreviations
- aPKC
atypical protein kinase C
- ASPM
abnormal spindle-like microcephaly-associated protein
- Brat
brain tumor
- EGFR
epidermal growth factor receptor
- Insc
inscuteable
- Lgl
lethal giant larvae
- Mira
Miranda
- Msi1
musashi-1
- Mud
Mushroom body defect
- Nf1
neurofibromatosis type 1
- Par 3
Partition defective 3
- Par 6
partition defective 6
- Pins
partner of inscuteable
- Plk1
polo like kinase 1
- Pon
partner of numb
- Pros
prospero
- Stau
staufen
- TPC
tumor propagating cells
- YB-1
Y-box binding protein 1
Footnotes
The authors state that they have no conflict of interest in regards to this manuscript. The authors thank Dr. Julie Harness for help editing this manuscript.
Author contributions: Writing the draft of the manuscript: Kate Marie Lewis. Critical revision of the manuscript for important intellectual content: Claudia Petritsch and Kate Marie Lewis.
The content of this publication is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- 1.Ashkenazi R, Gentry SN, Jackson TL. Pathways to tumorigenesis - modeling mutation acquisition in stem cells and their progeny. Neoplasia. 2008;10:1170–1182. doi: 10.1593/neo.08572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gomez-Lopez S, Lerner RG, Petritsch C. Asymmetric cell division of stem and progenitor cells during homeostasis and cancer. Cell Mol Life Sci. 2013 doi: 10.1007/s00018-013-1386-1. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gotz M, Huttner WB. The cell biology of neurogenesis. Nat Rev Mol Cell Biol. 2005;6:777–788. doi: 10.1038/nrm1739. [DOI] [PubMed] [Google Scholar]
- 4.Egger B, Gold KS, Brand AH. Notch regulates the switch from symmetric to asymmetric neural stem cell division in the Drosophila optic lobe. Development. 2010;137:2981–2987. doi: 10.1242/dev.051250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gotz M, Barde YA. Radial glial cells defined and major intermediates between embryonic stem cells and CNS neurons. Neuron. 2005;46:369–372. doi: 10.1016/j.neuron.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 6.Suter DM, Tirefort D, Julien S, Krause KH. A Sox1 to Pax6 switch drives neuroectoderm to radial glia progression during differentiation of mouse embryonic stem cells. Stem Cells. 2009;27:49–58. doi: 10.1634/stemcells.2008-0319. [DOI] [PubMed] [Google Scholar]
- 7.Haubensak W, Attardo A, Denk W, Huttner WB. Neurons arise in the basal neuroepithelium of the early mammalian telencephalon: a major site of neurogenesis. Proc Natl Acad Sci USA. 2004;101:3196–3201. doi: 10.1073/pnas.0308600100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Noctor SC, Martinez-Cerdeno V, Ivic L, Kriegstein AR. Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci. 2004;7:136–144. doi: 10.1038/nn1172. [DOI] [PubMed] [Google Scholar]
- 9.Attardo A, Calegari F, Haubensak W, Wilsch-Bräuninger M, Huttner WB. Live imaging at the onset of cortical neurogenesis reveals differential appearance of the neuronal phenotype in apical versus basal progenitor progeny. PLoS One. 2008;3:e2388. doi: 10.1371/journal.pone.0002388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stelzer S, Worlitzer MM, Bahnassawy L, Hemmer K, Rugani K, Werthschulte I, et al. JAM-C is an apical surface marker for neural stem cells. Stem Cells Dev. 2012;21:757–766. doi: 10.1089/scd.2011.0274. [DOI] [PubMed] [Google Scholar]
- 11.Rasin MR, Gazula VR, Breunig JJ, Kwan KY, Johnson MB, Liu-Chen S, et al. Numb and Numbl are required for maintenance of cadherin-based adhesion and polarity of neural progenitors. Nat Neurosci. 2007;10:819–827. doi: 10.1038/nn1924. [DOI] [PubMed] [Google Scholar]
- 12.Dave RK, Ellis T, Toumpas MC, Robson JP, Julian E, Adolphe C, et al. Sonic hedgehog and notch signaling can cooperate to regulate neurogenic divisions of neocortical progenitors. PLoS One. 2011;6:e14680. doi: 10.1371/journal.pone.0014680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miyata T, Kawaguchi D, Kawaguchi A, Gotoh Y. Mechanisms that regulate the number of neurons during mouse neocortical development. Curr Opin Neurobiol. 2010;20:22–28. doi: 10.1016/j.conb.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 14.Glickstein SB, Monaghan JA, Koeller HB, Jones TK, Ross ME. Cyclin D2 is critical for intermediate progenitor cell proliferation in the embryonic cortex. J Neurosci. 2009;29:9614–9624. doi: 10.1523/JNEUROSCI.2284-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Englund C, Fink A, Lau C, Pham D, Daza RA, Bulfone A, et al. Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J Neurosci. 2005;25:247–251. doi: 10.1523/JNEUROSCI.2899-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.LaMonica BE, Lui JH, Hansen DV, Kriegstein AR. Mitotic spindle orientation predicts outer radial glial cell generation in human neocortex. Nat Commun. 2013;4:1665. doi: 10.1038/ncomms2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang X, Tsai JW, LaMonica B, Kriegstein AR. A new subtype of progenitor cell in the mouse embryonic neocortex. Nat Neurosci. 2011;14:555–561. doi: 10.1038/nn.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pilz GA, Shitamukai A, Reillo I, Pacary E, Schwausch J, Stahl R, et al. Amplification of progenitors in the mammalian telencephalon includes a new radial glial cell type. Nat Commun. 2013;4:2125. doi: 10.1038/ncomms3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hansen DV, Lui JH, Parker PR, Kriegstein AR. Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature. 2010;464:554–561. doi: 10.1038/nature08845. [DOI] [PubMed] [Google Scholar]
- 20.Fietz SA, Kelava I, Vogt J, Wilsch-Bräuninger M, Stenzel D, Fish JL, et al. OSVZ progenitors of human and ferret neocortex are epithelial-like and expand by integrin signaling. Nature Neurosci. 2010;13:690–699. doi: 10.1038/nn.2553. [DOI] [PubMed] [Google Scholar]
- 21.Lui JH, Hansen DV, Kriegstein AR. Development and evolution of the human neocortex. Cell. 2011;146:18–36. doi: 10.1016/j.cell.2011.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alvarez-Buylla A, Garcia-Verdugo JM. Neurogenesis in adult subventricular zone. J Neurosci. 2002;22:629–634. doi: 10.1523/JNEUROSCI.22-03-00629.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alvarez-Buylla A. Mechanism of neurogenesis in adult avian brain. Experientia. 1990;46:948–955. doi: 10.1007/BF01939388. [DOI] [PubMed] [Google Scholar]
- 24.Merkle FT, Tramontin AD, Garcia-Verdugo JM, Alvarez-Buylla A. Radial glia give rise to adult neural stem cells in the subventricular zone. Proc Natl Acad Sci USA. 2004;101:17528–17532. doi: 10.1073/pnas.0407893101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hansen DV, Lui JH, Flandin P, Yoshikawa K, Rubenstein JL, Alvarez-Buylla A, et al. Non-epithelial stem cells and cortical interneuron production in the human ganglionic eminences. Nat Neurosci. 2013;16:1576–1587. doi: 10.1038/nn.3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qian X, Shen Q, Goderie SK, He W, Capela A, Davis AA, et al. Timing of CNS cell generation: a programmed sequence of neuron and glial cell production from isolated murine cortical stem cells. Neuron. 2000;28:69–80. doi: 10.1016/s0896-6273(00)00086-6. [DOI] [PubMed] [Google Scholar]
- 27.Li X, Newbern JM, Wu Y, Morgan-Smith M, Zhong J, Charron J, et al. MEK is a key regulator of gliogenesis in the developing brain. Neuron. 2012;75:1035–1050. doi: 10.1016/j.neuron.2012.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu X, Jin L, Feng L. Erk1/2 but not PI3K pathway is required for neurotrophin 3-induced oligodendrocyte differentiation of post-natal neural stem cells. J Neurochem. 2004;90:1339–1347. doi: 10.1111/j.1471-4159.2004.02594.x. [DOI] [PubMed] [Google Scholar]
- 29.Rodriguez-Martinez G, Molina-Hernandez A, Velasco I. Activin A promotes neuronal differentiation of cerebrocortical neural progenitor cells. PLoS One. 2012;7:e43797. doi: 10.1371/journal.pone.0043797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boku S, Nakagawa S, Takamura N, Kato A, Takebayashi M, Hisaoka-Nakashima K, et al. GDNF facilitates differentiation of the adult dentate gyrus-derived neural precursor cells into astrocytes via STAT3. Biochem Biophys Res Comm. 2013;434:779–784. doi: 10.1016/j.bbrc.2013.04.011. [DOI] [PubMed] [Google Scholar]
- 31.Zhou ZD, Kumari U, Xiao ZC, Tan EK. Notch as a molecular switch in neural stem cells. IUBMB Life. 2010;62:618–623. doi: 10.1002/iub.362. [DOI] [PubMed] [Google Scholar]
- 32.Namihira M, Kohyama J, Semi K, Sanosaka T, Deneen B, Taga T, et al. Committed neuronal precursors confer astrocytic potential on residual neural precursor cells. Dev Cell. 2009;16:245–255. doi: 10.1016/j.devcel.2008.12.014. [DOI] [PubMed] [Google Scholar]
- 33.Ravin R, Hoeppner DJ, Munno DM, Carmel L, Sullivan J, Levitt DL, et al. Potency and fate specification in CNS stem cell populations in vitro. Cell Stem Cell. 2008;3:670–680. doi: 10.1016/j.stem.2008.09.012. [DOI] [PubMed] [Google Scholar]
- 34.Wang DD, Bordey A. The astrocyte odyssey. Prog Neurobiol. 2008;86:342–367. doi: 10.1016/j.pneurobio.2008.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Y, Han SS, Wu Y, Tuohy TM, Xue H, Cai J, et al. CD44 expression identifies astrocyte-restricted precursor cells. Dev Biol. 2004;276:31–46. doi: 10.1016/j.ydbio.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 36.Ge WP, Miyawaki A, Gage FH, Jan YN, Jan LY. Local generation of glia is a major astrocyte source in postnatal cortex. Nature. 2012;484:376–380. doi: 10.1038/nature10959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Strathmann FG, Wang X, Mayer-Proschel M. Identification of two novel glial-restricted cell populations in the embryonic telencephalon arising from unique origins. BMC Dev Biol. 2007;7:33. doi: 10.1186/1471-213X-7-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dawson MR, Polito A, Levine JM, Reynolds R. NG2-expressing glial progenitor cells: an abundant and widespread population of cycling cells in the adult rat CNS. Mol Cell Neurosci. 2003;24:476–488. doi: 10.1016/s1044-7431(03)00210-0. [DOI] [PubMed] [Google Scholar]
- 39.Gregori N, Proschel C, Noble M, Mayer-Proschel M. The tripotential glial-restricted precursor (GRP) cell and glial development in the spinal cord: generation of bipotential oligodendrocyte-type-2 astrocyte progenitor cells and dorsal-ventral differences in GRP cell function. J Neurosci. 2002;22:248–256. doi: 10.1523/JNEUROSCI.22-01-00248.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ortega F, Gascon S, Masserdotti G, Deshpande A, Simon C, Fischer J, et al. Oligodendrogliogenic and neurogenic adult subependymal zone neural stem cells constitute distinct lineages and exhibit differential responsiveness to Wnt signalling. Nat Cell Biol. 2013;15:602–613. doi: 10.1038/ncb2736. [DOI] [PubMed] [Google Scholar]
- 41.Yakovlev AY, Boucher K, Mayer-Proschel M, Noble M. Quantitative insight into proliferation and differentiation of oligodendrocyte type 2 astrocyte progenitor cells in vitro. Proc Natl Acad Sci USA. 1998;95:14164–14167. doi: 10.1073/pnas.95.24.14164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Klempin F, Marr RA, Peterson DA. Modification of pax6 and olig2 expression in adult hippocampal neurogenesis selectively induces stem cell fate and alters both neuronal and glial populations. Stem Cells. 2012;30:500–509. doi: 10.1002/stem.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhu X, Hill RA, Dietrich D, Komitova M, Suzuki R, Nishiyama A. Age-dependent fate and lineage restriction of single NG2 cells. Development. 2011;138:745–753. doi: 10.1242/dev.047951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sugiarto S, Persson AI, Munoz EG, Waldhuber M, Lamagna C, Andor N, et al. Asymmetry-defective oligodendrocyte progenitors are glioma precursors. Cancer Cell. 2011;20:328–340. doi: 10.1016/j.ccr.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 46.Knudson AG. Two genetic hits (more or less) to cancer. Nat Rev Cancer. 2001;1:157–162. doi: 10.1038/35101031. [DOI] [PubMed] [Google Scholar]
- 47.Chang KC, Wang C, Wang H. Balancing self-renewal and differentiation by asymmetric division: insights from brain tumor suppressors in Drosophila neural stem cells. BioEssays. 2012;34:301–310. doi: 10.1002/bies.201100090. [DOI] [PubMed] [Google Scholar]
- 48.Saini N, Reichert H. Neural stem cells in Drosophila: molecular genetic mechanisms underlying normal neural proliferation and abnormal brain tumor formation. Stem Cells Int. 2012:486169. doi: 10.1155/2012/486169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Albertson R, Doe CQDlg. Scrib and Lgl regulate neuroblast cell size and mitotic spindle asymmetry. Nat Cell Biol. 2003;5:166–170. doi: 10.1038/ncb922. [DOI] [PubMed] [Google Scholar]
- 50.Wang C, Li S, Januschke J, Rossi F, Izumi Y, Garcia-Alvarez G, et al. An ana2/ctp/mud complex regulates spindle orientation in Drosophila neuroblasts. Dev Cell. 2011;21:520–533. doi: 10.1016/j.devcel.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 51.Speicher S, Fischer A, Knoblich J, Carmena A. The PDZ protein Canoe regulates the asymmetric division of Drosophila neuroblasts and muscle progenitors. Curr Biol. 2008;18:831–837. doi: 10.1016/j.cub.2008.04.072. [DOI] [PubMed] [Google Scholar]
- 52.Atwood SX, Chabu C, Penkert RR, Doe CQ, Prehoda KE. Cdc42 acts downstream of Bazooka to regulate neuroblast polarity through Par-6 aPKC. J Cell Sci. 2007;120:3200–3206. doi: 10.1242/jcs.014902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bowman SK, Neumuller RA, Novatchkova M, Du Q, Knoblich JA. The Drosophila NuMA Homolog Mud regulates spindle orientation in asymmetric cell division. Dev Cell. 2006;10:731–742. doi: 10.1016/j.devcel.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 54.Schaefer M, Petronczki M, Dorner D, Forte M, Knoblich JA. Heterotrimeric G proteins direct two modes of asymmetric cell division in the Drosophila nervous system. Cell. 2001;107:183–194. doi: 10.1016/s0092-8674(01)00521-9. [DOI] [PubMed] [Google Scholar]
- 55.Lee CY, Robinson KJ, Doe CQ. Lgl, Pins and aPKC regulate neuroblast self-renewal versus differentiation. Nature. 2006;439:594–598. doi: 10.1038/nature04299. [DOI] [PubMed] [Google Scholar]
- 56.Wang C, Chang KC, Somers G, Virshup D, Ang BT, Tang C, et al. Protein phosphatase 2A regulates self-renewal of Drosophila neural stem cells. Development. 2009;136:2287–2296. doi: 10.1242/dev.035758. [DOI] [PubMed] [Google Scholar]
- 57.Petritsch C, Tavosanis G, Turck CW, Jan LY, Jan YN. The Drosophila myosin VI Jaguar is required for basal protein targeting and correct spindle orientation in mitotic neuroblasts. Dev Cell. 2003;4:273–281. doi: 10.1016/s1534-5807(03)00020-0. [DOI] [PubMed] [Google Scholar]
- 58.Shen CP, Jan LY, Jan YN. Miranda is required for the asymmetric localization of Prospero during mitosis in Drosophila. Cell. 1997;90:449–458. doi: 10.1016/s0092-8674(00)80505-x. [DOI] [PubMed] [Google Scholar]
- 59.Smith CA, Lau KM, Rahmani Z, Dho SE, Brothers G, She YM, et al. aPKC-mediated phosphorylation regulates asymmetric membrane localization of the cell fate determinant Numb. EMBO J. 2007;26:468–480. doi: 10.1038/sj.emboj.7601495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhong W, Feder JN, Jiang MM, Jan LY, Jan YN. Asymmetric localization of a mammalian numb homolog during mouse cortical neurogenesis. Neuron. 1996;17:43–53. doi: 10.1016/s0896-6273(00)80279-2. [DOI] [PubMed] [Google Scholar]
- 61.Schwamborn JC, Berezikov E, Knoblich JA. The TRIM-NHL protein TRIM32 activates microRNAs and prevents self-renewal in mouse neural progenitors. Cell. 2009;136:913–925. doi: 10.1016/j.cell.2008.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tsunekawa Y, Britto JM, Takahashi M, Polleux F, Tan SS, Osumi N. Cyclin D2 in the basal process of neural progenitors is linked to non-equivalent cell fates. EMBO J. 2012;31:1879–1892. doi: 10.1038/emboj.2012.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shen Q, Zhong W, Jan YN, Temple S. Asymmetric Numb distribution is critical for asymmetric cell division of mouse cerebral cortical stem cells and neuroblasts. Development. 2002;129:4843–4853. doi: 10.1242/dev.129.20.4843. [DOI] [PubMed] [Google Scholar]
- 64.Andreu-Agullo C, Morante-Redolat JM, Delgado AC, Farinas I. Vascular niche factor PEDF modulates Notch-dependent stemness in the adult subependymal zone. Nat Neuroscience. 2009;12:1514–1523. doi: 10.1038/nn.2437. [DOI] [PubMed] [Google Scholar]
- 65.Sun Y, Goderie SK, Temple S. Asymmetric distribution of EGFR receptor during mitosis generates diverse CNS progenitor cells. Neuron. 2005;45:873–886. doi: 10.1016/j.neuron.2005.01.045. [DOI] [PubMed] [Google Scholar]
- 66.Ferron SR, Pozo N, Laguna A, Aranda S, Porlan E, Moreno M, et al. Regulated segregation of kinase Dyrk1A during asymmetric neural stem cell division is critical for EGFR-mediated biased signaling. Cell Stem Cell. 2010;7:367–379. doi: 10.1016/j.stem.2010.06.021. [DOI] [PubMed] [Google Scholar]
- 67.Kusek G, Campbell M, Doyle F, Tenenbaum SA, Kiebler M, Temple S. Asymmetric segregation of the double-stranded RNA binding protein Staufen2 during mammalian neural stem cell divisions promotes lineage progression. Cell Stem Cell. 2012;11:505–516. doi: 10.1016/j.stem.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bultje RS, Castaneda-Castellanos DR, Jan LY, Jan YN, Kriegstein AR, Shi SH. Mammalian Par3 regulates progenitor cell asymmetric division via notch signaling in the developing neocortex. Neuron. 2009;63:189–202. doi: 10.1016/j.neuron.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nakamura M, Okano H, Blendy JA, Montell C. Musashi, a neural RNA-binding protein required for Drosophila adult external sensory organ development. Neuron. 1994;13:67–81. doi: 10.1016/0896-6273(94)90460-x. [DOI] [PubMed] [Google Scholar]
- 70.Okano H, Kawahara H, Toriya M, Nakao K, Shibata S, Imai T. Function of RNA-binding protein Musashi-1 in stem cells. Exp Cell Res. 2005;306:349–356. doi: 10.1016/j.yexcr.2005.02.021. [DOI] [PubMed] [Google Scholar]
- 71.Sakakibara S, Nakamura Y, Yoshida T, Shibata S, Koike M, Takano H, et al. RNA-binding protein Musashi family: roles for CNS stem cells and a subpopulation of ependymal cells revealed by targeted disruption and antisense ablation. Proc Natl Acad Sci USA. 2002;99:15194–15199. doi: 10.1073/pnas.232087499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fish JL, Kosodo Y, Enard W, Paabo S, Huttner WB. Aspm specifically maintains symmetric proliferative divisions of neuroepithelial cells. Proc Natl Acad Sci USA. 2006;103:10438–10443. doi: 10.1073/pnas.0604066103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Khan MA, Godil SS, Tabani H, Panju SA, Enam SA. Clinical review of pediatric pilocytic astrocytomas treated at a tertiary care hospital in Pakistan. Surg Neurol Int. 2012;3:90. doi: 10.4103/2152-7806.99936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Katchy KC, Alexander S, Al-Nashmi NM, Al-Ramadan A. Epidemiology of primary brain tumors in childhood and adolescence in Kuwait. Springerplus. 2013;2:58. doi: 10.1186/2193-1801-2-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rosychuk RJ, Witol A, Wilson B, Stobart K. Central nervous system (CNS) tumor trends in children in a western Canadian province: a population-based 22-year retrospective study. J Neurol. 2012;259:1131–1136. doi: 10.1007/s00415-011-6314-4. [DOI] [PubMed] [Google Scholar]
- 76.Ramanan M, Chaseling R. Paediatric brain tumours treated at a single, tertiary paediatric neurosurgical referral centre from 1999 to 2010 in Australia. J Clin Neurosci. 2012;19:1387–1391. doi: 10.1016/j.jocn.2012.01.028. [DOI] [PubMed] [Google Scholar]
- 77.Pinho RS, Andreoni S, Silva NS, Cappellano AM, Masruha MR, Cavalheiro S, et al. Pediatric central nervous system tumors: a single-center experience from 1989 to 2009. J Pediatr Hematol Oncol. 2011;33:605–609. doi: 10.1097/MPH.0b013e31822031d9. [DOI] [PubMed] [Google Scholar]
- 78.Yamaguchi S, Kobayashi H, Terasaka S, Ishii N, Ikeda J, Kanno H, et al. The impact of extent of resection and histological subtype on the outcome of adult patients with high-grade gliomas. Jpn J Clin Oncol. 2012;42:270–277. doi: 10.1093/jjco/hys016. [DOI] [PubMed] [Google Scholar]
- 79.de Bianco AM, Miura FK, Clara C, Almeida JR, Silva CC, Teixeira MJ, et al. Low-grade astrocytoma: surgical outcomes in eloquent versus non-eloquent brain areas. Arq Neuro-psiquiatr. 2013;71:31–34. doi: 10.1590/s0004-282x2012005000017. [DOI] [PubMed] [Google Scholar]
- 80.Nuno M, Birch K, Mukherjee D, Sarmiento JM, Black KL, Patil CG. Survival and prognostic factors of anaplastic gliomas. Neurosurgery. 2013;73:458–465. doi: 10.1227/01.neu.0000431477.02408.5e. [DOI] [PubMed] [Google Scholar]
- 81.Johnson DR, Leeper HE, Uhm JH. Glioblastoma survival in the United States improved after Food and Drug Administration approval of bevacizumab: a population-based analysis. Cancer. 2013;119:3489–3495. doi: 10.1002/cncr.28259. [DOI] [PubMed] [Google Scholar]
- 82.Jeswani S, Nuño M, Folkerts V, Mukherjee D, Black KL, Patil CG. Comparison of survival between cerebellar and supratentorial glioblastoma patients: surveillance, epidemiology, and end results (SEER) analysis. Neurosurgery. 2013;73:240–246. doi: 10.1227/01.neu.0000430288.85680.37. [DOI] [PubMed] [Google Scholar]
- 83.Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 85.Galldiks N, Rapp M, Stoffels G, Fink GR, Shah NJ, Coenen HH, et al. Response assessment of bevacizumab in patients with recurrent malignant glioma using [18F]Fluoroethyl-L-tyrosine PET in comparison to MRI. Eur J Nucl Med Mol Imaging. 2013;40:22–33. doi: 10.1007/s00259-012-2251-4. [DOI] [PubMed] [Google Scholar]
- 86.Piao Y, Liang J, Holmes L, Henry V, Sulman E, de Groot JF. Acquired resistance to anti-VEGF therapy in glioblastoma is associated with a mesenchymal transition. Clin Cancer Res. 2013;19:4392–4403. doi: 10.1158/1078-0432.CCR-12-1557. [DOI] [PubMed] [Google Scholar]
- 87.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 88.Lewis KM, Harford-Wright E, Vink R, Nimmo AJ, Ghabriel MN. Walker 256 tumour cells increase substance P immunoreactivity locally and modify the properties of the blood-brain barrier during extravasation and brain invasion. Clin Exp Metastasis. 2013;30:1–12. doi: 10.1007/s10585-012-9487-z. [DOI] [PubMed] [Google Scholar]
- 89.Harford-Wright E, Lewis KM, Vink R. The potential for substance P antagonists as anti-cancer agents in brain tumours. Recent Pat CNS Drug Discov. 2013;8:13–23. doi: 10.2174/1574889811308010003. [DOI] [PubMed] [Google Scholar]
- 90.Lewis KM, Harford-Wright E, Vink R, Ghabriel MN. Targeting classical but not neurogenic inflammation reduces peritumoral oedema in secondary brain tumours. J Neuroimmunol. 2012;250:59–65. doi: 10.1016/j.jneuroim.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 91.Castillo M. Stem cells, radial glial cells, and a unified origin of brain tumors. AJNR Am J Neuroradiol. 2010;31:389–390. doi: 10.3174/ajnr.A1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Scopelliti A, Cammareri P, Catalano V, Saladino V, Todaro M, Stassi G. Therapeutic implications of cancer initiating cells. Expert Opin Biol Ther. 2009;9:1005–1016. doi: 10.1517/14712590903066687. [DOI] [PubMed] [Google Scholar]
- 93.Pei Y, Wechsler-Reya RJ. A malignant oligarchy: progenitors govern the behavior of oligodendrogliomas. Cancer Cell. 2010;18:546–547. doi: 10.1016/j.ccr.2010.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Liu C, Zong H. Developmental origins of brain tumors. Curr Opin Neurobiol. 2012;22:844–849. doi: 10.1016/j.conb.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Liu C, Sage JC, Miller MR, Verhaak RG, Hippenmeyer S, Vogel H, et al. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell. 2011;146:209–221. doi: 10.1016/j.cell.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Durand B, Gao FB, Raff M. Accumulation of the cyclin-dependent kinase inhibitor p27/Kip1 and the timing of oligodendrocyte differentiation. EMBO J. 1997;16:306–317. doi: 10.1093/emboj/16.2.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ponti G, Obernier K, Guinto C, Jose L, Bonfanti L, Alvarez-Buylla A. Cell cycle and lineage progression of neural progenitors in the ventricular-subventricular zones of adult mice. Proc Natl Acad Sci USA. 2013;110:E1045–E1054. doi: 10.1073/pnas.1219563110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Seaberg RM, van der Kooy D. Stem and progenitor cells: the premature desertion of rigorous definitions. Trends Neurosci. 2003;26:125–131. doi: 10.1016/S0166-2236(03)00031-6. [DOI] [PubMed] [Google Scholar]
- 99.Muñoz DM, Singh S, Tung T, Agnihotri S, Nagy A, Guha A, et al. Differential transformation capacity of neuro-glial progenitors during development. Proc Natl Acad Sci USA. 2013;110:14378–14383. doi: 10.1073/pnas.1303504110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Poppleton H, Gilbertson RJ. Stem cells of ependymoma. Brit J Cancer. 2007;96:6–10. doi: 10.1038/sj.bjc.6603519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Seaberg RM, Smukler SR, van der Kooy D. Intrinsic differences distinguish transiently neurogenic progenitors from neural stem cells in the early postnatal brain. Dev Biol. 2005;278:71–85. doi: 10.1016/j.ydbio.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 102.Lee da Y, Gianino SM, Gutmann DH. Innate neural stem cell heterogeneity determines the patterning of glioma formation in children. Cancer Cell. 2012;22:131–138. doi: 10.1016/j.ccr.2012.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Caussinus E, Gonzalez C. Induction of tumor growth by altered stem-cell asymmetric division in Drosophila melanogaster. Nat Genet. 2005;37:1125–1129. doi: 10.1038/ng1632. [DOI] [PubMed] [Google Scholar]
- 104.Radke J, Bortolussi G, Pagenstecher A. Akt and c-Myc induce stem-cell markers in mature primary p53(−)/(−) astrocytes and render these cells gliomagenic in the brain of immunocompetent mice. PLoS One. 2013;8:e56691. doi: 10.1371/journal.pone.0056691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Friedmann-Morvinski D, Bushong EA, Ke E, Soda Y, Marumoto T, Singer O, et al. Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science. 2012;338:1080–1084. doi: 10.1126/science.1226929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Alcantara Llaguno S, Chen J, Kwon CH, Jackson EL, Li Y, Burns DK, et al. Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer Cell. 2009;15:45–56. doi: 10.1016/j.ccr.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lei L, Sonabend AM, Guarnieri P, Soderquist C, Ludwig T, Rosenfeld S, et al. Glioblastoma models reveal the connection between adult glial progenitors and the proneural phenotype. PLoS One. 2011;6:e20041. doi: 10.1371/journal.pone.0020041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hide T, Takezaki T, Nakatani Y, Nakamura H, Kuratsu J, Kondo T. Combination of a ptgs2 inhibitor and an epidermal growth factor receptor-signaling inhibitor prevents tumorigenesis of oligodendrocyte lineage-derived glioma-initiating cells. Stem Cells. 2011;29:590–599. doi: 10.1002/stem.618. [DOI] [PubMed] [Google Scholar]
- 109.Bachoo RM, Maher EA, Ligon KL, Sharpless NE, Chan SS, You MJ, et al. Epidermal growth factor receptor and Ink4a/Arf: convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell. 2002;1:269–277. doi: 10.1016/s1535-6108(02)00046-6. [DOI] [PubMed] [Google Scholar]
- 110.Chow LM, Endersby R, Zhu X, Rankin S, Qu C, Zhang J, et al. Cooperativity within and among Pten, p53, and Rb pathways induces high-grade astrocytoma in adult brain. Cancer Cell. 2011;19:305–316. doi: 10.1016/j.ccr.2011.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ding H, Shannon P, Lau N, Wu X, Roncari L, Baldwin RL, et al. Oligodendrogliomas result from the expression of an activated mutant epidermal growth factor receptor in a RAS transgenic mouse astrocytoma model. Cancer Res. 2003;63:1106–1113. [PubMed] [Google Scholar]
- 112.Persson AI, Petritsch C, Swartling FJ, Itsara M, Sim FJ, Auvergne R, et al. Non-stem cell origin for oligodendroglioma. Cancer Cell. 2010;18:669–682. doi: 10.1016/j.ccr.2010.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Weiss WA, Burns MJ, Hackett C, Aldape K, Hill JR, Kuriyama H, et al. Genetic determinants of malignancy in a mouse model for oligodendroglioma. Cancer Res. 2003;63:1589–1595. [PubMed] [Google Scholar]
- 114.Lindberg N, Kastemar M, Olofsson T, Smits A, Uhrbom L. Oligodendrocyte progenitor cells can act as cell of origin for experimental glioma. Oncogene. 2009;28:2266–2275. doi: 10.1038/onc.2009.76. [DOI] [PubMed] [Google Scholar]
- 115.Dai C, Celestino JC, Okada Y, Louis DN, Fuller GN, Holland EC. PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes Dev. 2001;15:1913–1925. doi: 10.1101/gad.903001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ivkovic S, Canoll P, Goldman JE. Constitutive EGFR signaling in oligodendrocyte progenitors leads to diffuse hyperplasia in postnatal white matter. J Neurosci. 2008;28:914–922. doi: 10.1523/JNEUROSCI.4327-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Cicalese A, Bonizzi G, Pasi CE, Faretta M, Ronzoni S, Giulini B, et al. The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell. 2009;138:1083–1095. doi: 10.1016/j.cell.2009.06.048. [DOI] [PubMed] [Google Scholar]
- 118.Shiraishi S, Tada K, Nakamura H, Makino K, Kochi M, Saya H, et al. Influence of p53 mutations on prognosis of patients with glioblastoma. Cancer. 2002;95:249–257. doi: 10.1002/cncr.10677. [DOI] [PubMed] [Google Scholar]
- 119.Gil-Perotin S, Marin-Husstege M, Li J, Soriano-Navarro M, Zindy F, Roussel MF, et al. Loss of p53 induces changes in the behavior of subventricular zone cells: implication for the genesis of glial tumors. J Neurosci. 2006;26:1107–1116. doi: 10.1523/JNEUROSCI.3970-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhu Y, Guignard F, Zhao D, Liu L, Burns DK, Mason RP, et al. Early inactivation of p53 tumor suppressor gene cooperating with NF1 loss induces malignant astrocytoma. Cancer Cell. 2005;8:119–130. doi: 10.1016/j.ccr.2005.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hambardzumyan D, Cheng YK, Haeno H, Holland EC, Michor F. The probable cell of origin of NF1- and PDGF-driven glioblastomas. PLoS One. 2011;6:e24454. doi: 10.1371/journal.pone.0024454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Muñoz DM, Guha A. Mouse models to interrogate the implications of the differentiation status in the ontogeny of gliomas. Oncotarget. 2011;2:590–598. doi: 10.18632/oncotarget.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zheng H, Ying H, Yan H, Kimmelman AC, Hiller DJ, Chen AJ, et al. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature. 2008;455:1129–1133. doi: 10.1038/nature07443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zheng H, Ying H, Yan H, Kimmelman AC, Hiller DJ, Chen AJ, et al. Pten and p53 converge on c-Myc to control differentiation, self-renewal, and transformation of normal and neoplastic stem cells in glioblastoma. Cold Spring Harb Symp Quant Biol. 2008;73:427–437. doi: 10.1101/sqb.2008.73.047. [DOI] [PubMed] [Google Scholar]
- 125.Sherley JL. Asymmetric cell kinetics genes: the key to expansion of adult stem cells in culture. Stem Cells. 2002;20:561–572. doi: 10.1634/stemcells.20-6-561. [DOI] [PubMed] [Google Scholar]
- 126.Groszer M, Erickson R, Scripture-Adams DD, Lesche R, Trumpp A, Zack JA, et al. Negative regulation of neural stem/progenitor cell proliferation by the Pten tumor suppressor gene in vivo. Science. 2001;294:2186–2189. doi: 10.1126/science.1065518. [DOI] [PubMed] [Google Scholar]
- 127.Gont A, Hanson JE, Lavictoire SJ, Parolin DA, Daneshmand M, Restall IJ, et al. PTEN loss represses glioblastoma tumor initiating cell differentiation via inactivation of Lgl1. Oncotarget. 2013;4:1266–1279. doi: 10.18632/oncotarget.1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Jafri NF, Clarke JL, Weinberg V, Barani IJ, Cha S. Relationship of glioblastoma multiforme to the subventricular zone is associated with survival. Neuro Oncol. 2013;15:91–96. doi: 10.1093/neuonc/nos268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Mazzoleni S, Politi LS, Pala M, Cominelli M, Franzin A, Sergi Sergi L, et al. Epidermal growth factor receptor expression identifies functionally and molecularly distinct tumor-initiating cells in human glioblastoma multiforme and is required for gliomagenesis. Cancer Res. 2010;70:7500–7513. doi: 10.1158/0008-5472.CAN-10-2353. [DOI] [PubMed] [Google Scholar]
- 130.Morrison LC, McClelland R, Aiken C, Bridges M, Liang L, Wang X, et al. Deconstruction of medulloblastoma cellular heterogeneity reveals differences between the most highly invasive and self-renewing phenotypes. Neoplasia. 2013;15:384–398. doi: 10.1593/neo.13148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Andor N, Harness J, Mewes HW, Petritsch C. EXPANDS: expanding ploidy and allele frequency on nested subpopulations. Bioinformatics. 2013 doi: 10.1093/bioinformatics/btt622. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Deleyrolle LP, Harding A, Cato K, Siebzehnrubl FA, Rahman M, Azari H, et al. Evidence for label-retaining tumour-initiating cells in human glioblastoma. Brain. 2011;134:1331–1343. doi: 10.1093/brain/awr081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Foong CS, Sandanaraj E, Brooks HB, Campbell RM, Ang BT, Chong YK, et al. Glioma-propagating cells as an in vitro screening platform: PLK1 as a case study. J Biomol Screen. 2012;17:1136–1150. doi: 10.1177/1087057112457820. [DOI] [PubMed] [Google Scholar]
- 134.Hambardzumyan D, Squatrito M, Holland EC. Radiation resistance and stem-like cells in brain tumors. Cancer Cell. 2006;10:454–456. doi: 10.1016/j.ccr.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 135.Gao X, McDonald JT, Hlatky L, Enderling H. Acute and fractionated irradiation differentially modulate glioma stem cell division kinetics. Cancer Res. 2013;73:1481–1490. doi: 10.1158/0008-5472.CAN-12-3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ligon KL, Huillard E, Mehta S, Kesari S, Liu H, Alberta JA, et al. Olig2-regulated lineage-restricted pathway controls replication competence in neural stem cells and malignant glioma. Neuron. 2007;53:503–517. doi: 10.1016/j.neuron.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Richichi C, Brescia P, Alberizzi V, Fornasari L, Pelicci G. Marker-independent method for isolating slow-dividing cancer stem cells in human glioblastoma. Neoplasia. 2013;15:840–847. doi: 10.1593/neo.13662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Deleyrolle LP, Rohaus MR, Fortin JM, Reynolds BA, Azari H. Identification and isolation of slow-dividing cells in human glioblastoma using carboxy fluorescein succinimidyl ester (CFSE) J Vis Exp. 2012:3918. doi: 10.3791/3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Liu Q, Nguyen DH, Dong Q, Shitaku P, Chung K, Liu OY, et al. Molecular properties of CD133+ glioblastoma stem cells derived from treatment-refractory recurrent brain tumors. J Neurooncol. 2009;94:1–19. doi: 10.1007/s11060-009-9919-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Fotovati A, Abu-Ali S, Wang PS, Deleyrolle LP, Lee C, Triscott J, et al. YB-1 bridges neural stem cells and brain tumor-initiating cells via its roles in differentiation and cell growth. Cancer Res. 2011;71:5569–5578. doi: 10.1158/0008-5472.CAN-10-2805. [DOI] [PubMed] [Google Scholar]
- 141.Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488:522–526. doi: 10.1038/nature11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Liu W, Shen G, Shi Z, Shen F, Zheng X, Wen L, et al. Brain tumour stem cells and neural stem cells: still explored by the same approach? J Int Med Res. 2008;36:890–895. doi: 10.1177/147323000803600504. [DOI] [PubMed] [Google Scholar]
- 143.Qiu B, Zhang D, Wang Y, Ou S, Wang J, Tao J, et al. Interleukin-6 is overexpressed and augments invasiveness of human glioma stem cells in vitro. Clin Exp Metastasis. 2013;30:1009–1018. doi: 10.1007/s10585-013-9599-0. [DOI] [PubMed] [Google Scholar]
- 144.Nakada M, Nambu E, Furuyama N, Yoshida Y, Takino T, Hayashi Y, et al. Integrin alpha3 is overexpressed in glioma stem-like cells and promotes invasion. Brit J Cancer. 2013;108:2516–2524. doi: 10.1038/bjc.2013.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kushibiki T, Sakai M, Awazu K. Differential effects of photodynamic therapy on morphologically distinct tumor cells derived from a single precursor cell. Cancer Lett. 2008;268:244–251. doi: 10.1016/j.canlet.2008.03.054. [DOI] [PubMed] [Google Scholar]
- 146.Lathia JD, Hitomi M, Gallagher J, Gadani SP, Adkins J, Vasanji A, et al. Distribution of CD133 reveals glioma stem cells self-renew through symmetric and asymmetric cell divisions. Cell Death Dis. 2011;2:e200. doi: 10.1038/cddis.2011.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Tamura K, Aoyagi M, Wakimoto H, Ando N, Nariai T, Yamamoto M, et al. Accumulation of CD133-positive glioma cells after high-dose irradiation by Gamma Knife surgery plus external beam radiation. J Neurosurg. 2010;113:310–318. doi: 10.3171/2010.2.JNS091607. [DOI] [PubMed] [Google Scholar]
- 148.Kang MK, Hur BI, Ko MH, Kim CH, Cha SH, Kang SK. Potential identity of multi-potential cancer stem-like subpopulation after radiation of cultured brain glioma. BMC Neurosci. 2008;9:15. doi: 10.1186/1471-2202-9-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Bao S, Wu Q, Li Z, Sathornsumetee S, Wang H, McLendon RE, et al. Targeting cancer stem cells through L1CAM suppresses glioma growth. Cancer Res. 2008;68:6043–6048. doi: 10.1158/0008-5472.CAN-08-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Elsir T, Qu M, Berntsson SG, Orrego A, Olofsson T, Lindstrom MS, et al. PROX1 is a predictor of survival for gliomas WHO grade II. Brit J Cancer. 2011;104:1747–1754. doi: 10.1038/bjc.2011.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Elsir T, Eriksson A, Orrego A, Lindstrom MS, Nister M. Expression of PROX1 Is a common feature of high-grade malignant astrocytic gliomas. J Neuropathol Exp Neurol. 2010;69:129–138. doi: 10.1097/NEN.0b013e3181ca4767. [DOI] [PubMed] [Google Scholar]
- 152.Elsir T, Smits A, Lindstrom MS, Nister M. Transcription factor PROX1: its role in development and cancer. Cancer Metastasis Rev. 2012;31:793–805. doi: 10.1007/s10555-012-9390-8. [DOI] [PubMed] [Google Scholar]
- 153.Yamazaki H, Xu CW, Naito M, Nishida H, Okamoto T, Ghani FI, et al. Regulation of cancer stem cell properties by CD9 in human B-acute lymphoblastic leukemia. Biochem Biophys Res Comm. 2011;409:14–21. doi: 10.1016/j.bbrc.2011.04.098. [DOI] [PubMed] [Google Scholar]
- 154.Tian T, Zhang Y, Wang S, Zhou J, Xu S. Sox2 enhances the tumorigenicity and chemoresistance of cancer stem-like cells derived from gastric cancer. J Biomed Res. 2012;26:336–345. doi: 10.7555/JBR.26.20120045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Toda M, Iizuka Y, Yu W, Imai T, Ikeda E, Yoshida K, et al. Expression of the neural RNA-binding protein Musashi1 in human gliomas. Glia. 2001;34:1–7. doi: 10.1002/glia.1034. [DOI] [PubMed] [Google Scholar]
- 156.Kanemura Y, Mori K, Sakakibara S, Fujikawa H, Hayashi H, Nakano A, et al. Musashi1, an evolutionarily conserved neural RNA-binding protein, is a versatile marker of human glioma cells in determining their cellular origin, malignancy, and proliferative activity. Differentiation. 2001;68:141–152. doi: 10.1046/j.1432-0436.2001.680208.x. [DOI] [PubMed] [Google Scholar]
- 157.Muto J, Imai T, Ogawa D, Nishimoto Y, Okada Y, Mabuchi Y, et al. RNA-binding protein Musashi1 modulates glioma cell growth through the post-transcriptional regulation of Notch and PI3 kinase/Akt signaling pathways. PLoS One. 2012;7:e33431. doi: 10.1371/journal.pone.0033431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Jiang X, Xing H, Kim TM, Jung Y, Huang W, Yang HW, et al. Numb regulates glioma stem cell fate and growth by altering epidermal growth factor receptor and Skp1-Cullin-F-box ubiquitin ligase activity. Stem Cells. 2012;30:1313–1326. doi: 10.1002/stem.1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Klezovitch O, Fernandez TE, Tapscott SJ, Vasioukhin V. Loss of cell polarity causes severe brain dysplasia in Lgl1 knockout mice. Genes Dev. 2004;18:559–571. doi: 10.1101/gad.1178004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Yan B, Omar FM, Das K, Ng WH, Lim C, Shiuan K, et al. Characterization of Numb expression in astrocytomas. Neuropathology. 2008;28:479–484. doi: 10.1111/j.1440-1789.2008.00907.x. [DOI] [PubMed] [Google Scholar]
- 161.Xu P, Qiu M, Zhang Z, Kang C, Jiang R, Jia Z, et al. The oncogenic roles of Notch1 in astrocytic gliomas in vitro and in vivo. J Neurooncol. 2010;97:41–51. doi: 10.1007/s11060-009-0007-1. [DOI] [PubMed] [Google Scholar]
- 162.El Hindy N, Keyvani K, Pagenstecher A, Dammann P, Sandalcioglu IE, Sure U, et al. Implications of Dll4-Notch signaling activation in primary glioblastoma multiforme. Neuro Oncol. 2013;15:1366–1378. doi: 10.1093/neuonc/not071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9:157–173. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 164.Kristoffersen K, Villingshoj M, Poulsen HS, Stockhausen MT. Level of Notch activation determines the effect on growth and stem cell-like features in glioblastoma multiforme neurosphere cultures. Cancer Biol Ther. 2013;14:625–637. doi: 10.4161/cbt.24595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Tchorz JS, Tome M, Cloetta D, Sivasankaran B, Grzmil M, Huber RM, et al. Constitutive Notch2 signaling in neural stem cells promotes tumorigenic features and astroglial lineage entry. Cell Death Dis. 2012;3:e325. doi: 10.1038/cddis.2012.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Gursel DB, Berry N, Boockvar JA. The contribution of Notch signaling to glioblastoma via activation of cancer stem cell self-renewal: the role of the endothelial network. Neurosurgery. 2012;70:N19–N21. doi: 10.1227/01.neu.0000410937.38828.6f. [DOI] [PubMed] [Google Scholar]
- 167.Zhu TS, Costello MA, Talsma CE, Flack CG, Crowley JG, Hamm LL, et al. Endothelial cells create a stem cell niche in glioblastoma by providing NOTCH ligands that nurture self-renewal of cancer stem-like cells. Cancer Res. 2011;71:6061–6072. doi: 10.1158/0008-5472.CAN-10-4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Hovinga KE, Shimizu F, Wang R, Panagiotakos G, Van Der Heijden M, Moayedpardazi H, et al. Inhibition of notch signaling in glioblastoma targets cancer stem cells via an endothelial cell intermediate. Stem Cells. 2010;28:1019–1029. doi: 10.1002/stem.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Wang J, Wang C, Meng Q, Li S, Sun X, Bo Y, et al. siRNA targeting Notch-1 decreases glioma stem cell proliferation and tumor growth. Mol Biol Rep. 2012;39:2497–2503. doi: 10.1007/s11033-011-1001-1. [DOI] [PubMed] [Google Scholar]
- 170.Zhen Y, Zhao S, Li Q, Li Y, Kawamoto K. Arsenic trioxide-mediated Notch pathway inhibition depletes the cancer stem-like cell population in gliomas. Cancer Lett. 2010;292:64–72. doi: 10.1016/j.canlet.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 171.Dai L, He J, Liu Y, Byun J, Vivekanandan A, Pennathur S, et al. Dose-dependent proteomic analysis of glioblastoma cancer stem cells upon treatment with gamma-secretase inhibitor. Proteomics. 2011;11:4529–4540. doi: 10.1002/pmic.201000730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Fan X, Khaki L, Zhu TS, Soules ME, Talsma CE, Gul N, et al. NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells. 2010;28:5–16. doi: 10.1002/stem.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Chen J, Kesari S, Rooney C, Strack PR, Shen H, Wu L, et al. Inhibition of notch signaling blocks growth of glioblastoma cell lines and tumor neurospheres. Genes Cancer. 2010;1:822–835. doi: 10.1177/1947601910383564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Wang J, Wakeman TP, Lathia JD, Hjelmeland AB, Wang XF, White RR, et al. Notch promotes radioresistance of glioma stem cells. Stem Cells. 2010;28:17–28. doi: 10.1002/stem.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Lo HW. EGFR-targeted therapy in malignant glioma: novel aspects and mechanisms of drug resistance. Curr Mol Pharmacol. 2010;3:37–52. doi: 10.2174/1874467211003010037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Solomon MT, Selva JC, Figueredo J, Vaquer J, Toledo C, Quintanal N, et al. Radiotherapy plus nimotuzumab or placebo in the treatment of high grade glioma patients: results from a randomized, double blind trial. BMC Cancer. 2013;13:299. doi: 10.1186/1471-2407-13-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Howng SL, Wu CH, Cheng TS, Sy WD, Lin PC, Wang C, et al. Differential expression of Wnt genes, beta-catenin and E-cadherin in human brain tumors. Cancer Lett. 2002;183:95–101. doi: 10.1016/s0304-3835(02)00085-x. [DOI] [PubMed] [Google Scholar]
- 178.Ohgaki H, Kim YH, Steinbach JP. Nervous system tumors associated with familial tumor syndromes. Curr Opin Neurol. 2010;23:583–591. doi: 10.1097/WCO.0b013e3283405b5f. [DOI] [PubMed] [Google Scholar]
- 179.Noles SR, Chenn A. Cadherin inhibition of beta-catenin signaling regulates the proliferation and differentiation of neural precursor cells. Mol Cell Neurosci. 2007;35:549–558. doi: 10.1016/j.mcn.2007.04.012. [DOI] [PubMed] [Google Scholar]
- 180.Habib SJ, Chen BC, Tsai FC, Anastassiadis K, Meyer T, Betzig E, et al. A localized Wnt signal orients asymmetric stem cell division in vitro. Science. 2013;339:1445–1448. doi: 10.1126/science.1231077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Rampazzo E, Persano L, Pistollato F, Moro E, Frasson C, Porazzi P, et al. Wnt activation promotes neuronal differentiation of glioblastoma. Cell Death Dis. 2013;4:e500. doi: 10.1038/cddis.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Sandberg CJ, Altschuler G, Jeong J, Stromme KK, Stangeland B, Murrell W, et al. Comparison of glioma stem cells to neural stem cells from the adult human brain identifies dysregulated Wnt- signaling and a fingerprint associated with clinical outcome. Exp Cell Res. 2013;319:2230–2243. doi: 10.1016/j.yexcr.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 183.Kim Y, Kim KH, Lee J, Lee YA, Kim M, Lee SJ, et al. Wnt activation is implicated in glioblastoma radioresistance. Lab Invest. 2012;92:466–473. doi: 10.1038/labinvest.2011.161. [DOI] [PubMed] [Google Scholar]
- 184.Sakai D, Dixon J, Dixon MJ, Trainor PA. Mammalian neurogenesis requires Treacle-Plk1 for precise control of spindle orientation, mitotic progression, and maintenance of neural progenitor cells. PLoS Genet. 2012;8:e1002566. doi: 10.1371/journal.pgen.1002566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Takaki T, Trenz K, Costanzo V, Petronczki M. Polo-like kinase 1 reaches beyond mitosis--cytokinesis, DNA damage response, and development. Curr Opin Cell Biol. 2008;20:650–660. doi: 10.1016/j.ceb.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 186.Budirahardja Y, Gonczy C. PPLK-1 asymmetry contributes to asynchronous cell division of elegans embryos. Development. 2008;135:1303–1313. doi: 10.1242/dev.019075. [DOI] [PubMed] [Google Scholar]
- 187.Dietzmann K, Kirches E, von B, Jachau K, Mawrin C. Increased human polo-like kinase-1 expression in gliomas. J Neurooncol. 2001;53:1–11. doi: 10.1023/a:1011808200978. [DOI] [PubMed] [Google Scholar]
- 188.Lee C, Fotovati A, Triscott J, Chen J, Venugopal C, Singhal A, et al. Polo-like kinase 1 inhibition kills glioblastoma multiforme brain tumor cells in part through loss of SOX2 and delays tumor progression in mice. Stem Cells. 2012;30:1064–1075. doi: 10.1002/stem.1081. [DOI] [PubMed] [Google Scholar]
- 189.Ducray F, Idbaih A, de Reynies A, Bieche I, Thillet J, Mokhtari K, et al. Anaplastic oligodendrogliomas with 1p19q codeletion have a proneural gene expression profile. Mol Cancer. 2008;7:41. doi: 10.1186/1476-4598-7-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Denysenko T, Gennero L, Juenemann C, Morra I, Masperi P, Ceroni V, et al. Heterogeneous phenotype of human glioblastoma. In vitro study. Cell Biochem Funct. 2013 doi: 10.1002/cbf.2988. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 191.Felling RJ, Snyder MJ, Romanko MJ, Rothstein RP, Ziegler AN, Yang Z, et al. Neural stem/progenitor cells participate in the regenerative response to perinatal hypoxia/ischemia. J Neurosci. 2006;26:4359–4369. doi: 10.1523/JNEUROSCI.1898-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Linnik IV, Scott ML, Holliday KF, Woodhouse N, Waterton JC, O’Connor B, et al. Noninvasive tumor hypoxia measurement using magnetic resonance imaging in murine U87 glioma xenografts and in patients with glioblastoma. Magn Reson Med. 2013 doi: 10.1002/mrm.24826. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 193.Pistollato F, Abbadi S, Rampazzo E, Persano L, Della Puppa A, Frasson C, et al. Intratumoral hypoxic gradient drives stem cells distribution and MGMT expression in glioblastoma. Stem Cells. 2010;28:851–862. doi: 10.1002/stem.415. [DOI] [PubMed] [Google Scholar]
- 194.McCord AM, Jamal M, Shankavaram UT, Lang FF, Camphausen K, Tofilon PJ. Physiologic oxygen concentration enhances the stem-like properties of CD133+ human glioblastoma cells in vitro. Mol Cancer Res. 2009;7:489–497. doi: 10.1158/1541-7786.MCR-08-0360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Lehnus KS, Donovan LK, Huang X, Zhao N, Warr TJ, Pilkington GJ, et al. CD133 glycosylation is enhanced by hypoxia in cultured glioma stem cells. Int J Oncol. 2013;42:1011–1017. doi: 10.3892/ijo.2013.1787. [DOI] [PubMed] [Google Scholar]
- 196.Bar EE, Lin A, Mahairaki V, Matsui W, Eberhart CG. Hypoxia increases the expression of stem-cell markers and promotes clonogenicity in glioblastoma neurospheres. Am J Pathol. 2010;177:1491–1502. doi: 10.2353/ajpath.2010.091021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Sgubin D, Wakimoto H, Kanai R, Rabkin SD, Martuza RL. Oncolytic herpes simplex virus counteracts the hypoxia-induced modulation of glioblastoma stem-like cells. Stem Cells Transl Med. 2012;1:322–332. doi: 10.5966/sctm.2011-0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Pine SR, Ryan BM, Varticovski L, Robles AI, Harris CC. Microenvironmental modulation of asymmetric cell division in human lung cancer cells. Proc Natl Acad Sci USA. 2010;107:2195–2200. doi: 10.1073/pnas.0909390107. [DOI] [PMC free article] [PubMed] [Google Scholar]