

Abstract
Background
Genome subtyping approaches could provide useful epidemiological information regarding food pathogens. However, the full genomic diversity of strains that show similar subtyping results has not yet been completely explored. Most subtyping methods are based on the differences of only a portion of the genome. We investigated two draft genome sequences of Listeria monocytogenes strain F2-382 and NIHS-28, which have been identified as closely related strains by subtyping (identical multi-virulence-locus sequence typing and multiple-locus variable number tandem repeat analysis sequence types and very similar pulsed-field gel electrophoresis patterns), despite their different sources.
Results
Two closely related strains were compared by genome structure analysis, recombination analysis, and single nucleotide polymorphism (SNP) analysis. Both genome structure analysis and recombination analysis showed that these two strains are more closely related than other strains, from a whole-genome perspective. However, the analysis of SNPs indicated that the two strains differ at the single nucleotide level.
Conclusion
We show the relationship between the results of genome subtyping and whole-genome sequencing. It appears that the relationships among strains indicated by genome subtyping methods are in accord with the relationships indicated by whole-genome analysis. However, our results also indicate that the genetic distance between the closely related strains is greater than that between clonal strains. Our results demonstrate that subtyping methods using a part of the genome are reliable in assessing the genetic distance of the strains. Furthermore, the genetic differences in the same subtype strains may provide useful information to distinguish the bacterial strains.
Electronic supplementary material
The online version of this article (doi:10.1186/s12866-014-0309-0) contains supplementary material, which is available to authorized users.
Keywords: Listeria monocytogenes, MLVA, MVLST, Next-generation sequencing, Whole-genome shotgun sequence
Background
Listeria monocytogenes is a rod-shaped, gram-positive, and non-sporulating foodborne infectious pathogen that can cause serious diseases such as septicemia and meningitis, particularly in high-risk groups (e.g., pregnant woman, neonates, and immunocompromised individuals) with a high mortality rate of 20%–30% [1]. In particular, ready-to-eat (RTE) foods, which do not require heat cooking, are a main source of foodborne listeriosis cases [2–4]. Molecular subtyping approaches allow us to evaluate the similarity of strains isolated from geographically or temporally different sources with high accuracy and reproducibility for epidemiological studies or trace-back surveys [5]. Several molecular subtyping methods such as PFGE, ribotyping, MLST including MVLST, and MLVA have been developed for subtyping of L. monocytogenes [6–10]. These methods have categorized the microorganism into 4 lineages and several clonal complexes (CCs). The subtyping data is highly consistent with pathogenicity and other characteristics, and provides useful information for epidemiologic or phylogenetic studies [11–13]. However, the whole-genome structure of strains that show similar subtyping results is unknown, because most subtyping methods focus on only a subset of loci such as endonuclease restriction sites or alleles.
Next-generation sequencing (NGS) technology has facilitated the analysis of bacterial genomes from a whole-genome perspective by generating relatively high data output in recent years. Using NGS, some studies have already reported the relationships between molecular subtyping results and whole-genome sequences for Salmonella enterica and Escherichia coli, and presented useful information on phylogeny and virulence [14–18]. L. monocytogenes has also been investigated at the whole-genome level to reveal its pathogenicity and evolutionary history [19–21]. However, the relationship between the results of genome subtyping and whole-genome sequencing in L. monocytogenes has not yet been fully explored.
We compared two draft genome sequences of L. monocytogenes to reveal the relationship between subtyping results and whole-genome diversity. Strains F2-382 and NIHS-28 were isolated from epidemic patients in the United States and Japan, respectively. These strains were not considered to be clonal because the patients had not engaged in travel around the time of onset. However, these strains showed identical MVLST and MLVA sequence types and strikingly similar PFGE patterns [22]. Thus, they are regarded to be extremely closely related strains, regardless of their source. In the present study, their draft sequences were compared to determine the extent of the differences between their genomes. In addition, published whole-genome sequences from 22 strains were compared to evaluate the genetic distance.
Results
General properties of the draft genome sequence
Using the shotgun sequencing method, a total of 46,603,422 bp and 107,127 reads with an average read length of 435.0 bp were obtained for the F2-382 strain. A total of 64,444,465 bp with 132,436 reads and an average read length of 486.6 bp were obtained for strain NIHS-28. Next, 99.58% of the total bp of the F2-382 strain and 99.71% of the total bp of the NIHS-28 strain were aligned in the de novo assembly. In total, 56 contigs were obtained from the de novo assembly of the F2-382 strain, with a total length of 2,911,674 bp, an N50-contig of 86,218 bp, and a GC content of 37.9%. The alignment depths were distributed from 1 to 140, with a peak depth of 13. For strain NIHS-28, 35 contigs were obtained with a total length of 2,908,138 bp, an N50-contig of 120,766 bp, and a GC content of 37.9%. The alignment depths were distributed from 1 to 180 with a peak depth of 19.
Large-scale differences in genome structure
The alignment analysis constructed using MAUVE software for strains F2-382, NIHS-28, and F2365 showed that their overall genomic structures are similar (Figure 1). In addition, all of the contigs were aligned to the reference sequence, indicating that the sequenced strains did not possess plasmids. Five gap loci were identified in strains F2-382 and NIHS-28 when aligned to strain F2365 (Figure 1; gaps 1–5). However, between strains F2-382 and NIHS-28, only one locus for each sequence was identified as a gap (Figure 1; gap 6–7). Thus, strains F2-382 and NIHS-28 exhibit more similarity to each other than to F2365.
Figure 1.
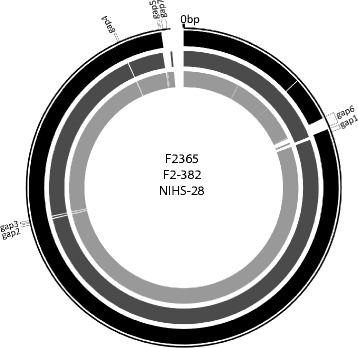
Alignment of genome sequences of strains F2-382, NIHS-28, and F2365. The seven frames on the outer ring show gap regions larger than 5 kbp. Each lane shows the sequence of one strain; F2365 (black); F2-382 (dark gray); NIHS-28 (light gray).
Each gap region between strains F2-382 and NIHS-28 contained several open reading frames (ORFs), which have been inserted by phage infection (Figure 1; gaps 6–7, Additional file 1 and Additional file 2). These gap regions were assumed to be derived from the individual phages, because they showed partial similarity in the alignment details for the region (Additional file 3).
Recombination analysis
All 24 strains were clustered into three major clades, which corresponded to previously described lineages (Figure 2, Table 1) [20,21,23–29]. Strains F2-382 and NIHS-28 were both clustered in the lineage I clade (Figure 2, Table 1). Lineage I was further divided into six clades. At the finest level of detail, strains F2-382 and NIHS-28 were clustered in the same clade as strains F2365 and SLCC2378 (Figure 2). Thus, strains F2-382 and NIHS-28 exhibit greater similarity to each other in their whole-genome sequence than to the other strains, consistent with the results of MVLST analysis and MLVA.
Figure 2.
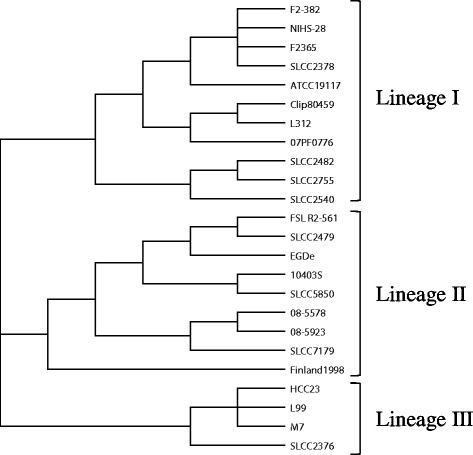
The cluster diagram constructed by recombination analysis using a wide range of genome sequences. Three independent datasets yielded identical clustering results.
Table 1.
Isolates and genome subtyping results
| Isolates | Genbank accession No. | Serotype | Lineage | MVLST a | MLVA | Reference |
|---|---|---|---|---|---|---|
| F2-382 | BAZC01000001–01000056 | 4b | N/A | 1-3-1-3-1-3 | 13-18-9 | This study |
| NIHS-28 | BAZD01000001–01000036 | 4b | N/A | 1-3-1-3-1-3 | 13-18-9 | This study |
| F2365 | NC_002973 | 4b | I | 1-3-1-3-1-3 | 14-18-9 | Nelson et al. [25] |
| EGDe | NC_003210 | 1/2a | II | 7-9-8-6-4-2 | 25-11-7 | Glaser et al. [26] |
| HCC23 | NC_011660 | 4a | III | 4-5-4-1-*-4 | 8-22-4 | Steele et al. [27] |
| Clip80459 | NC_012488 | 4b | I | 2-4-2-5-2-3 | 15-20-5 | Hain et al. [20] |
| 08-5578 | NC_013766 | 1/2a | II | 7-8-7-6-3-1 | 34-22-7 | Gilmour et al. [28] |
| 08-5923 | NC_013768 | 1/2a | II | 7-8-7-6-3-1 | 34-22-7 | Gilmour et al. [28] |
| L99 | NC_017529 | 4a | III | 4-5-4-1-*-4 | 8-22-4 | Hain et al. [20] |
| M7 | NC_017537 | 4a | N/A | 5-5-4-1-*-4 | 8-22-4 | Chen et al. [23] |
| 10403S | NC_017544 | 1/2a | N/A | 8-12-6-6-3-2 | 20-11-7 | den Bakker et al. [29] |
| FSL R2-561 | NC_017546 | 1/2c | N/A | 7-9-8-6-4-2 | 21-11-7 | den Bakker et al. [29] |
| Finland1998 | NC_017547 | 1/2a | N/A | 7-8-7-6-3-2 | 25-14-7 | den Bakker et al. [29] |
| 07PF0776 | NC_017728 | 4b | N/A | 2-4-2-5-2-3 | 14-18-9 | McMullen et al. [24] |
| ATCC19117 | NC_018584 | 4a | I | 2-1-3-3-1-3 | 17-13-6 | Kuenne et al. [21] |
| SLCC2378 | NC_018585 | 4e | I | 1-3-1-3-1-3 | 17-18-9 | Kuenne et al. [21] |
| SLCC2540 | NC_018586 | 3b | I | 3-7-2-4-2-3 | 10-11-5 | Kuenne et al. [21] |
| SLCC2755 | NC_018587 | 1/2b | I | 2-6-3-3-2-3 | 15-16-5 | Kuenne et al. [21] |
| SLCC2479 | NC_018589 | 3c | II | 7-9-8-6-4-2 | 21-11-7 | Kuenne et al. [21] |
| SLCC2376 | NC_018590 | 4c | III | 6-2-5-2-*-4 | 9-17-4 | Kuenne et al. [21] |
| SLCC2482 | NC_018591 | 7 | I | 2-6-3-3-2-3 | 15-16-6 | Kuenne et al. [21] |
| SLCC5850 | NC_018592 | 1/2a | II | *-10-6-6-3-2 | 19-11-7 | Kuenne et al. [21] |
| SLCC7179 | NC_018593 | 3a | II | 7-11-6-6-3-2 | 10-12-9 | Kuenne et al. [21] |
| L312 | NC_018642 | 4b | I | 2-4-2-5-2-3 | 15-15-5 | Kuenne et al. [21] |
aAsterisks (*) indicate that the genome region corresponding to the allele was not found.
A comparison of the clustering results between methods
Recombination analysis of a set of large-genome sequences showed some differences in the relationships between lineages, compared to other subtyping methods (Table 1; Figures 2, 3 and 4). In the recombination analysis, the three lineages clustered independently. In contrast, MVLST subtyping showed that lineages I and II are more closely related to each other than to lineage III (Figure 3). MLVA subtyping showed that lineage I could be separated into two major clusters, and that lineage II and III comprised a large cluster that did not include lineage I. However, the relationships among the strains within the lineage cluster were similar in all methods.
Figure 3.

The cluster diagram constructed by MVLST analysis using six virulence gene alleles.
Figure 4.
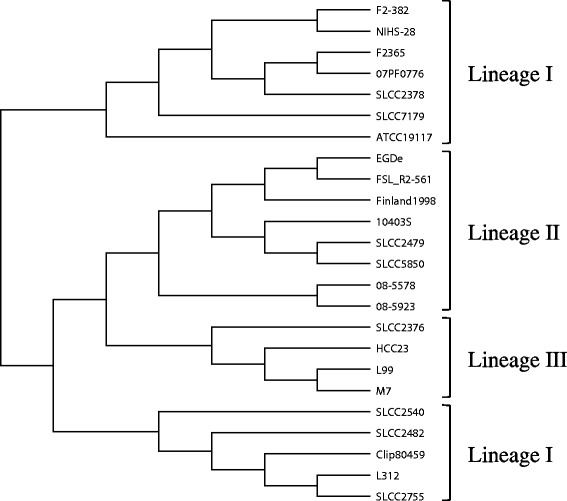
The cluster diagram constructed by MLVA using three tandem repeat regions.
The results of MVLST analysis and MLVA of strains F2-382 and NIHS-28 were similar to those of strains F2365 and SLCC2378, which were identical to the results of recombination analysis. These four strains showed the same sequence type in MVLST analysis. MLVA results showed that strains F2-382 and NIHS-28 had the same MLVA type, and were distinct from other strains. In addition, strains F2365, SLCC2378, and 07PF0776 showed MLVA types similar to those exhibited by strains F2-382 and NIHS-28, and comprised a parent cluster.
Single nucleotide polymorphism analysis of the four most similar strains
Strains F2-382, NIHS-28, F2365, and SLCC2378, which were clustered into the smallest clade in recombination analysis, were evaluated by single nucleotide polymorphism (SNP) analysis. Strains F2-382 and NIHS-28 were barely affected by the subculture, because they had been stored at the freezing temperature. Using strain F2365 as a reference, 424 SNPs were identified in strain F2-382, 317 SNPs in NIHS-28, and 106 SNPs in SLCC2378 (Figure 5). However, when comparing strains F2-382 and NIHS-28, only 48 SNPs were found at the same loci with the same polymorphism. Thus, most of the SNPs were unique to one of the two strains. The numbers of unique SNPs in strains F2-382 and NIHS-28 were 366 and 263, respectively, in contrast to 58 in the SLCC2378 strain. Thus, strains F2-382 and NIHS-28 have unique sequences, whereas strains F2365 and SLCC2378 have very similar sequences.
Figure 5.
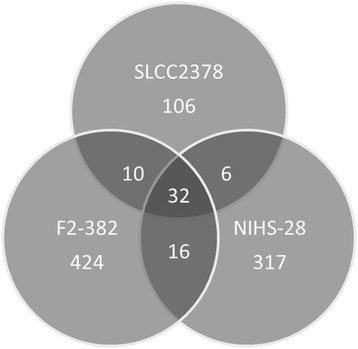
The number of SNPs in each strain compared to strain F2365. The numbers under each strain name indicate the number of SNPs in the strain. The numbers in the overlap region indicate the number of SNPs with the same polymorphism at the same locus between strains.
Discussion
Differences in genome structure
We examined the differences between the draft genome sequences of strains showing similar genome subtyping results by using NGS technology. The backbone of the genome (e.g., GC content, number of rRNAs or tRNAs) is known to be similar among the different strains of L. monocytogenes regardless of the serotype and lineage, with the exception of a partial mutation by gene deletion and/or transfer [19,29]. Strains F2-382 and NIHS-28 also showed more similar genomic structure in the set of large genome sequences based on alignment with strain F2365, compared to the other strains. Furthermore, fewer gaps were observed between strains F2-382 and NIHS-28 compared to strain F2365. Thus, it appears that these two strains, which were shown to be closely related by genome subtyping analysis, are more similar to each other than they are to other strains in the genomic structure that is formed by insertion and deletion, not just in the genomic backbone of GC content.
Both F2-382 and NIHS-28 had only a single gap in their genomes (Figure 1; gap 6–7) when we set the minimum gap size as 5 kbp. The smaller gaps could be unreadable regions in the shotgun sequencing or could be generated by misassembly. Annotation of the region containing the gaps indicated that both gaps were derived from a phage infection (Additional file 1 and Additional file 2). Phage infection is a primary type of genome rearrangement that can adapt to a wide range of environments [20,21,25,30]. Interestingly, both prophage regions encode a gene related to sugar phosphorylation (Additional file 1 and Additional file 2; F2382_00310, LmNIHS28_02116). Several paralogs of this type of gene are known to be present in the genome, and play a role in energy production in host cells in the absence of sugar [20,31]. We can hypothesize that the genes encoded in the prophage region are a result of adaptation, as both F2-382 and NIHS-28 were isolated from epidemic patients.
Genetic distance between strains F2-382 and NIHS-28
To compare genetic distances in the whole genome, Deloger et al. established a maximal unique matches index (MUMi) [32]. However, we were not able to calculate this index because the two draft whole-genome sequences obtained in this study were not of sufficient length for an accurate calculation. Therefore, we compared the strains in the aligned region by establishing three large datasets. Large dataset recombination analysis showed that the strains formed clusters corresponding to lineages, as described in a previous study (Figure 2, Table 1) [20,21,23–29]. It appears that the lineage perspective that is reflected by partial genome analysis is consistent with the relationships indicated by comparison of whole-genome sequences. This hypothesis has been supported by Kuenne et al., who reported that phylogenetic analysis using 2,018 gene sequences showed high correspondence with the lineages [21]. Furthermore, the present study shows that the set of large genome sequences, including non-functional regions, are associated with lineages.
Recombination analysis indicated that both strains F2-382 and NIHS-28 belong to the lineage I cluster. Lineage I can be classified into at least five CCs [12]. In the present study, the strains that clustered in lineage I formed the six smallest clades (Figure 2). Strains F2-382 and NIHS-28 were both identified as belonging to the smallest clade, indicating that they are more closely related than the other strains.
Strains F2365 and SLCC2378 were included in the same clade with strains F2-382 and NIHS-28 (Figure 2). Kuenne et al. reported that these two strains have extremely similar genome sequences [21]. We used SNP analysis to compare the four strains to determine genomic similarity at the single nucleotide level (Figure 5). One limitation of analysis using NGS technology is false-positive SNPs caused by sequencing errors. To avoid this problem, we excluded SNPs with quality scores of <40, ensuring an accuracy of >99.99%. Thus, since <1000 SNPs were detected, the number of false-positives should be <1. Furthermore, to avoid false-positive SNPs caused by homopolymers, which is particularly problematic in 454 sequencing, we detected only SNPs with depths of >10 in the assembly result. We also ensured that all sequence reads showed identical nucleotides at a given position. For SNP detection, strain F2365 was used as a reference for comparison with the other three strains. In strains F2-382 and NIHS-28, the majority of the SNPs detected were unique to each strain (F2-382, 366; NIHS-28, 263). However, only 10%–15% of the SNPs (F2-382, 48/424; NIHS-28, 48/317) involved identical mutations at the same locus. In contrast, only 58 SNPs were unique to strain SLCC2378. Gilmour et al. compared complete genome sequences of strains isolated from the same outbreak, and reported only 36 SNPs [28]. Based on the above results, it appears that the genome sequences of strain F2-382 and NIHS-28 are very similar, but less similar than those of clonal strains.
Relationship between genome subtyping results and whole-genome sequencing
The recombination analysis result in the present study generated from an accumulation of point mutations. Accordingly, this result can be considered to indicate the vertical relationship among the strains. When the recombination analysis result was compared to MVLST analysis and MLVA, different relationships were observed for each subtyping method between clusters corresponding to lineages. The three lineages were shown to be independent by large genome sequence recombination analysis. In contrast, MVLST indicated that lineages I and II were more closely related to each other than to lineage III. We presume that this is because MVLST is based only on virulence genes [33]. Thus, the lineage III strains, which were rarely associated with human listeriosis cases, were located distantly from lineage I and II strains. The relationships between lineages indicated by MLVA also differed from the results of other analyses. It is known that the number of tandem repeats can be altered several times in a short period of time in E. coli O157:H7 and Vibrio parahaemolyticus [34,35]. Based on this knowledge, we assumed that the characteristics of the tandem repeat region biased the results. While the relationships among the lineages are different, as previously mentioned, the relationships among the strains under the lineage cluster were almost identical in all analyses. Thus, genome subtyping methods are valid for the identification of strains because the alleles require adequate time for mutation. In addition, we concluded that the results of genome subtyping are correlated with the phylogenetic relationships between strains when considering a limited group such as a lineage.
Accuracy of genome subtyping methods
Our results suggest that strains that show similar subtyping results have similar whole-genome sequences, according to the analysis of DNA sequence, which is the origin of the genome subtyping methods. In a limited group of strains, the relationships indicated by the genome subtyping methods were correlated with the results of whole-genome sequencing. These results support the reliability of the phylogenetic relationships that were inferred using genome subtyping methods in previous studies. However, our observations also demonstrate that a more accurate classification is possible by single nucleotide analysis within the whole-genome perspective.
Strains F2-382 and NIHS-28, which showed similar subtyping results, were isolated from the United States and Japan, respectively. Listeriosis is rare in Japan, and only one foodborne case has been reported [36,37]. In contrast, cases have been sporadically reported in the United States and in European nations [38,39]. However, foodborne illnesses are no longer geographically limited since the globalization of food, including RTE foods, has been promoted worldwide [40–42]. Therefore, it is necessary to evaluate the virulence or to trace the origin of L. monocytogenes strains in foods [39,41]. The present study demonstrates that the genome typing results can be considered as genetic distances regardless of their geographically or temporally independent sources. This observation may improve the reliability of subtyping results in surveys.
The identity of individual strains from a whole-genome perspective is suggested by the present results. Genome-wide SNP analysis demonstrated that strains F2-382 and NIHS-28, which have been shown to be very similar by a previous subtyping method [22], are not clonal strains. The development of novel DNA sequencing technologies, including NGS, allows the analysis of microorganisms, including L. monocytogenes, from a whole-genome perspective [14–21]. We anticipate that the observations reported here will be useful as a reference for future studies that require validation of strain identity.
Conclusions
We revealed the genome differences between two similar subtype strains, F2-382 and NIHS-28, from a whole-genome perspective, and estimated the concordance of the whole-genome sequence and genome subtyping results. Strains that show similar subtypes were shown to also have similar sequences at the whole-genome scale. We observed that the relationships among strains indicated by genome subtyping methods are consistent with the relationships indicated by whole-genome analysis. However, we also revealed that the genetic distance between closely related strains is greater than that between clonal strains, from a single nucleotide perspective. These findings could facilitate improvement of the reliability of genome subtyping results, in that they may be valuable references for estimating genetic distances by using subtyping methods. We anticipate that our findings will be useful for evaluating the identity of the strains.
Methods
Bacterial isolates
L. monocytogenes strain F2-382 was isolated from a patient in the United States and was kindly provided by Dr. Martin Wiedmann (Cornell University, Ithaca, NY). Strain NIHS-28 was isolated from a patient in Japan. Both strains were of serotype 4b. Ethical approval was not required, as the clinical isolates were collected as part of standard patient care. These strains showed identical MVLST and MLVA sequence types and only one difference in the PFGE band pattern [22]. However, they were considered as non-clonal strains, because the patients had not engaged in travel near the time of onset of listeriosis. After isolation, these strains had been stored at freezing temperature until pre-culture to extract DNA.
Whole-genome sequences were obtained for 22 additional strains from the GenBank/EMBL/DDBJ databases (http://www.insdc.org/) for comparison (Table 1) [20,21,23–29]. The strains contained in the reference showed nine serotypes (1/2a, 4a, 1/2b, 3b, 4b, 1/2c, 3c, 4c, and 7).
Whole-genome shotgun sequence
Strains F2-382 and NIHS-28 were cultured overnight in brain-heart infusion broth (Eiken Chemical, Tochigi, Japan) at 37°C. Bacterial DNA was extracted using the phenol-chloroform extraction and ethanol precipitation methods [43,44]. For whole-genome shotgun sequencing, the GS Junior platform (Roche, Basel, Switzerland) was employed using a GS Junior Rapid Library Preparation Kit and a GS Junior emPCR Kit (Lib-L; Roche), according to the manufacturer’s protocol. The read sequences were used to construct a contig without a reference sequence by de novo assembly, using the GS De Novo Assembler (Roche). For this assembly, the program parameters were set to: seed step, 12; seed length, 16; seed count, 1; minimum overlap, 10; and minimum identity, 90.
Genome structural analysis
To construct the scaffolds from the contigs, the software MAUVE v2.3.1 was employed, with the move contig script using the default settings [45]. Strain F2365 was chosen as a reference because it was aligned with the largest number of contigs in the 22 reference strains. The aligned contigs were constructed into a scaffold by joining with an N of 100 bp between each contig.
Annotation
ORFs were extracted from each contig by using the Glimmer v3.02 software [46]. The ORFs were imported into the Genome Traveler software (In Silico Biology, Kanagawa, Japan) for annotation. The microbial database included in the software was used in combination with the BLAST algorithm for annotation. ORFs that showed less than 90% identity during the annotation process were excluded from subsequent analyses; this threshold indicated that the gene coding region had low reliability.
Recombination analysis
Recombination analysis was conducted according to Cao et al.’s method [16]. To identify the core local collinear blocks (LCBs), which are the sequence segments common to all strains, the two scaffold sequences from F2-382 and NIHS-28 were aligned with the complete whole-genome sequences of the 22 reference strains with the MAUVE software, using the progressiveMauve script [47]. Using this alignment, 258 core LCBs were identified. Three independent data sets were constructed by randomly extracting approximately 500-kbp sequences from core LCBs, and evaluated by recombination analysis using ClonalFrame software v1.1 [48]. For this analysis, the parameters were set to 25,000 rounds of burn within the 50,000 generations and 100 thinning intervals. To rule out any bias among the three independent data sets, the results were evaluated using Gelman-Rubin statistics.
In silico MVLST and MLVA
The genome regions for the targets of MVLST and MLVA were extracted for cluster analysis from the 24 L. monocytogenes sequences. The target loci for MVLST analysis were chosen according to Zhang et al.’s method [33]. The loci for MLVA were chosen according to Miya et al.’s method [22]. Clustering analysis was conducted using BioNumerics software v. 4.0 (Applied Maths, Sint-Martens-Latem, Belgium) by using the categorical coefficient and UPGMA method.
Single nucleotide polymorphisms
SNPs were identified for four strains (F2-382, NIHS-28, F2365, and SLCC2378) that clustered in a single group in recombination analysis. MUMmer software v3.23 was employed to identify SNPs by alignment with the reference sequence, F2365 [49]. To avoid false-positives caused by sequencing errors and the problematic 454 sequencing-related homopolymers for strains F2-382 and NIHS-28, the polymorphisms that showed a quality score of <40 or a depth of <10 in the de novo assembly results were excluded from the SNP results.
Availability of supporting data
The draft genome sequences of strains F2-382 and NIHS-28 were deposited in the Genbank/EMBL/DDBJ database with the accession numbers BAZC01000001–BAZC01000056 and BAZD01000001–BAZD01000035, respectively.
Acknowledgments
This work was supported by a Grant-in-Aid for Scientific Research (B 24380115) from the Ministry of Education, Culture, Sports, Science and Technology, Japan. We thank Dr. Martin Wiedmann, Cornell University, USA, for kindly providing strain F2-382.
Abbreviations
- PFGE
Pulsed-field gel electrophoresis
- MLST
Multi locus sequence typing
- MVLST
Multi-virulence-locus sequencing typing
- MLVA
Multi-locus variable number of tandem repeat analysis
- ORF
Open reading frame
- SNP
Single nucleotide polymorphism
- NGS
Next-generation sequencing
- LCB
Local collinear block
Additional files
F2-382 CDSs in the gap region.
NIHS-28 CDSs in the gap region.
The alignment result of gap 6 of strain F2-382 and gap 7 of strain NIHS-28.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Concept and design of this study: HT, BK. Laboratory work and data analysis: DK, HT, SI. Manuscript writing, review and revision: DK, HT, SM, TK. All authors have read and approved the final manuscript.
Contributor Information
Daisuke Kyoui, Email: d131006@kaiyodai.ac.jp.
Hajime Takahashi, Email: hajime@kaiyodai.ac.jp.
Satoko Miya, Email: s-miya@kaiyodai.ac.jp.
Takashi Kuda, Email: kuda@kaiyodai.ac.jp.
Shizunobu Igimi, Email: igimi@nihs.go.jp.
Bon Kimura, Email: kimubo@kaiyodai.ac.jp.
References
- 1.Swaminathan B, Gerner-Smidt P. The epidemiology of human listeriosis. Microbes Infect. 2007;9:1236–1243. doi: 10.1016/j.micinf.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 2.U.S. Department of Health and Human Service, U.S. Department of Agriculture: Quantitative Assessment of the Relative Risk to Public Health from Foodborne Listeria monocytogenes Among Selected Categories of Ready-to-Eat Foods. Washington, D.C; 2003.
- 3.Rocourt J, BenEmbarek P, Toyofuku H, Schlundt J. Quantitative risk assessment of Listeria monocytogenes in ready-to-eat foods: the FAO/WHO approach. FEMS Immunol Med Microbiol. 2003;35:263–267. doi: 10.1016/S0928-8244(02)00468-6. [DOI] [PubMed] [Google Scholar]
- 4.World Health Organization, Food and Agriculture Organization . Risk Assessment of Listeria monocytogenes in Ready-to-Eat Foods. Geneva: Microbiological risk assessment series 5; 2004. [Google Scholar]
- 5.Jadhave S, Bhave M, Palombo EA. Methods used for the detection and subtyping of Listeria monocytogenes. J Microbiol Methods. 2012;88:327–341. doi: 10.1016/j.mimet.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 6.Fugett EB, Schoonmaker-Bopp D, Dumas NB, Corby J, Wiedmann M. Pulsed-Field Gel Electrophoresis (PFGE) analysis of temporally matched Listeria monocytogenes isolates from human clinical cases, foods, ruminant farms, and urban and natural environments reveals source-associated as well as widely distributed PFGE types. J Clin Microbiol. 2007;45:865–873. doi: 10.1128/JCM.01285-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen Y, Zhang W, Knebel SJ. Multi-virulence-locus sequence typing identifies single nucleotide polymorphisms which differentiate epidemic clones and outbreak strains of Listeria monocytogenes. J Clin Microbiol. 2007;45:835–846. doi: 10.1128/JCM.01575-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miya S, Kimura B, Sato M, Takahashi H, Ishikawa T, Suda T, Takakura C, Fujii T, Wiedmann M. Development of a multilocus variable-number of tandem repeat typing method for Listeria monocytogenes serotype 4b strains. Int J Food Microbiol. 2008;124:239–249. doi: 10.1016/j.ijfoodmicro.2008.03.023. [DOI] [PubMed] [Google Scholar]
- 9.Sperry KEV, Kathariou S, Edwards JS, Wolf LA. Multiple-locus variable-number tandem-repeat analysis as a tool for subtyping Listeria monocytogenes strains. J Clin Microbiol. 2008;46:1435–1450. doi: 10.1128/JCM.02207-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rasmussen OF, Skouboe P, Dons L, Rossen L, Olsen JE. Listeria monocytogenes exists in at least three evolutionary lines: evidence from flagellin, invasive associated protein and listeriolysin O genes. Microbiology. 1995;141:2053–2061. doi: 10.1099/13500872-141-9-2053. [DOI] [PubMed] [Google Scholar]
- 11.Cantinelli T, Chenal-Francisque V, Diancourt L, Frezal L, Leclercq A, Wirth T, Lecuit M, Brisse S. “Epidemic clones” of Listeria monocytogenes are widespread and ancient clonal groups. J Clin Microbiol. 2013;51:3770–3779. doi: 10.1128/JCM.01874-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ragon M, Wirth T, Hollandt F, Lavenir R, Lecuit M, Monnier AL, Brisse S. A new perspective on Listeria monocytogenes evolution. PLoS Pathog. 2008;4:e1000146. doi: 10.1371/journal.ppat.1000146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Orsi RH, den Bakker HC, Wiedmann M. Listeria monocytogenes lineages: genomics, evolution, ecology, and phenotypic characteristics. Int J Med Microbiol. 2011;301:79–96. doi: 10.1016/j.ijmm.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 14.Allard MW, Luo Y, Strain E, Li C, Keys CE, Son I, Stones R, Musser SM, Brown EW. High resolution clustering of Salmonella enterica serovar Montevideo strains using a next-generation sequencing approach. BMC Genomics. 2012;13:32. doi: 10.1186/1471-2164-13-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Allard MW, Luo Y, Strain E, Pettengill J, Timme R, Wang C, Li C, Keys CE, Zheng J, Stones R, Wilson MR, Musser SM, Brown EW. On the evolutionary history, population genetics and diversity among isolates of Salmonella Enteritidis PFGE pattern JEGX01.0004. PLoS ONE. 2013;8:e55254. doi: 10.1371/journal.pone.0055254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cao G, Meng J, Strain E, Stones R, Pettengill J, Zhao S, McDermott P, Brown E, Allard M. Phylogenetics and differentiation of Salmonella Newport lineages by whole genome sequencing. PLoS One. 2013;8:e55687. doi: 10.1371/journal.pone.0055687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eppinger M, Mammel MK, Leclerc JE, Ravel J, Cebula TA. Genomic anatomy of Escherichia coli O157:H7 outbreaks. Proc Natl Acad Sci U S A. 2011;108:20142–20147. doi: 10.1073/pnas.1107176108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ju W, Cao G, Rump L, Strain E, Luo Y, Timme R, Allard M, Zhao S, Brown E, Meng J. Phylogenetic analysis of non-O157 shiga toxin-producing Escherichia coli strains by whole-genome sequencing. J Clin Microbiol. 2012;50:4123–4127. doi: 10.1128/JCM.02262-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.den Bakker HC, Cummings CA, Ferreira V, Vatta P, Orsi RH, Degoricija L, Barker M, Petrauskene O, Furtado MR, Wiedmann M. Comparative genomics of the bacterial genus Listeria: genome evolution is characterized by limited gene acquisition and limited gene loss. BMC Genomics. 2010;11:688. doi: 10.1186/1471-2164-11-688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hain T, Ghai R, Billion A, Kuenne CT, Steinweg C, Izar B, Mohamed W, Mraheil MA, Domann E, Schaffrath S, Kärst U, Goesmann A, Oehm S, Pühler A, Merkl R, Vorwerk S, Glaser P, Garrido P, Rusniok C, Buchrieser C, Goebel W, Chakraborty T. Comparative genomics and transcriptomics of lineage I, II, and III strains of Listeria monocytogenes. BMC Genomics. 2012;13:144. doi: 10.1186/1471-2164-13-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuenne C, Billion A, Mraheil MA, Strittmatter A, Daniel R, Goesmann A, Barbuddhe S, Hain T, Chakraborty T. Reassessment of the Listeria monocytogenes pan-genome reveals dynamic integration hotspots and mobile genetic elements as major components of the accessory genome. BMC Genomics. 2013;14:47. doi: 10.1186/1471-2164-14-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miya S, Takahashi H, Kamimura C, Nakagawa M, Kuda T, Kimura B. Highly discriminatory typing method for Listeria monocytogenes using polymorphic tandem repeat regions. J Microbiol Methods. 2012;90:285–291. doi: 10.1016/j.mimet.2012.05.023. [DOI] [PubMed] [Google Scholar]
- 23.Chen J, Xia Y, Cheng C, Fang C, Shan Y, Jin G, Fang W. Genome sequence of the nonpathogenic Listeria monocytogenes serovar 4a strain M7. J Microbiol. 2011;193:5019–5020. doi: 10.1128/JB.05501-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McMullen PD, Gillaspy AF, Gipson J, Bobo LD, Skiest DJ, Freitag NE. Genome sequence of Listeria monocytogenes 07PF0776, a cardiotropic serovar 4b strain. J Bacteriol. 2012;194:3552. doi: 10.1128/JB.00616-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nelson KE, Fouts DE, Mongodin EF, Ravel J, DeBoy RT, Kolonay JF, Rasko DA, Angiuoli SV, Gill SR, Beanan MJ, Brinkac LM, Daugherty SC, Dodson RJ, Durkin AS, Madupu R, Haft DH, Selengut J, Aken SV, Khouri H, Fedorova N, Forberger H, Tran B, Kathariou S, Wonderling LD, Uhlich GA, Bayles DO, Luchansky JB, Fraser CM. Whole genome comparisons of serotype 4b and 1/2a strains of the food-borne pathogen Listeria monocytogenes reveal new insights into the core genome components of this species. Nucleic Acids Res. 2004;32:2386–2395. doi: 10.1093/nar/gkh562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Glaser P, Frangeul L, Buchrieser C, Rusniok C, Amend A, Baquero F, Berche P, Bloecker H, Brandt P, Chakraborty T, Charbit A, Chetouani F, Couvé E, de Daruvar A, Dehoux P, Domann E, Domínguez-Bernal G, Duchaud E, Durant L, Dussurget O, Entian K-D, Fishi H, Garcia-Del Porillo F, Garrido P, Gautier L, Goebel W, Gómez-López N, Hain T, Hauf J, Jackson D, et al. Comparative genomics of Listeria species. Science. 2001;294:849–852. doi: 10.1126/science.1063447. [DOI] [PubMed] [Google Scholar]
- 27.Steele CL, Donaldson JR, Paul D, Banes MM, Arick T, Bridges SM, Lawrence ML. Genome sequence of lineage III Listeria monocytogenes strain HCC23. J Bacteriol. 2011;193:3679–3680. doi: 10.1128/JB.05236-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gilmour MW, Graham M, Domselaar GV, Tyler S, Kent H, Trout-Yakel KM, Larios O, Allen V, Lee B, Nadon C. High-throughput genome sequencing of two Listeria monocytogenes clinical isolates during a large foodborne outbreak. BMC Genomics. 2010;11:120. doi: 10.1186/1471-2164-11-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.den Bakker HC, Desjardins CA, Griggs AD, Peters JE, Zeng Q, Young SK, Kodira CD, Yandava C, Hepburn TA, Haas BJ, Birren BW, Wiedmann M. Evolutionary dynamics of the accessory genome of Listeria monocytogenes. PLoS One. 2013;8:e67511. doi: 10.1371/journal.pone.0067511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Verghese B, Lok M, Wen J, Alessandria V, Chen Y, Kathriou S, Knabel S. comK prophage junction fragments as markers for Listeria monocytogenes genotypes unique to individual meat and poultry processing plants and a model for rapid niche-specific adaptation, biofilm formation, and persistence. Appl Environ Microbiol. 2011;77:3279–3292. doi: 10.1128/AEM.00546-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Joseph B, Przybilla K, Stühler C, Schauer K, Slaghuis J, Fuchs TM, Goebel W. Identification of Listeria monocytogenes genes contributing to intracellular replication by expression profiling and mutant screening. J Bacteriol. 2006;188:556–568. doi: 10.1128/JB.188.2.556-568.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deloger M, Karouri ME, Petit M-A. A genomic distance based on MUM indicates discontinuity between most bacterial species and genera. J Bacteriol. 2009;191:91–99. doi: 10.1128/JB.01202-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang W, Jayarao BM, Knabel SJ. Multi-virulence-locus sequence typing of Listeria monocytogenes. Appl Environ Microbiol. 2004;70:913–920. doi: 10.1128/AEM.70.2.913-920.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cooley MB, Carychao D, Nguyen K, Whitehand L, Mandrell R. Effects of environmental stress on stability of tandem repeats in Escherichia coli O157:H7. Appl Eniviron Microbiol. 2010;76:3398–3400. doi: 10.1128/AEM.02373-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.García K, Gavilán RG, Höfle MG, Martínez-Urtaza J, Espejo RT. Microevolution of pandemic Vibrio parahaemolyticus assessed by the number of repeat units in short sequence tandem repeat regions. PLoS ONE. 2012;7:e30823. doi: 10.1371/journal.pone.0030823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Makino SI, Kawamoto K, Takeshi K, Okada Y, Yamasaki M, Yamamoto S, Igimi S. An outbreak of food-borne listeriosis due to cheese in Japan, during 2001. Int J Food Microbiol. 2005;104:189–196. doi: 10.1016/j.ijfoodmicro.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 37.Okutani A, Okada Y, Yamamoto S, Igimi S. Nationwide survey of human Listeria monocytogenes infection in Japan. Epidemiol Infect. 2004;132:769–772. doi: 10.1017/S0950268804001967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cartwright EJ, Jackson KA, Johnson SD, Graves LM, Silk BJ, Mahon BE. Listeriosis outbreaks and associated food vehicles, United States, 1998–2008. Emerging Infect Dis. 2013;19:1–9. doi: 10.3201/eid1901.120393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eurosurveillance Editorial Team: The European Union summary report on trends and sources of zoonoses, zoonotic agents and food-borne outbreaks in 2010.Euro Surveill, 17. Available online: [http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=20113] [PubMed]
- 40.Lambertz ST, Nilsson C, Brådenmark A, Sylvén S, Johansson A, Jansson LM, Lindblad M. Prevalence and level of Listeria monocytogenes in ready-to-eat foods in Sweden 2010. Int J Food Microbiol. 2012;160:24–33. doi: 10.1016/j.ijfoodmicro.2012.09.010. [DOI] [PubMed] [Google Scholar]
- 41.Okutani A, Okada Y, Yamamoto S, Igimi S. Overview of Listeria monocytogenes contamination in Japan. Int J Food Microbiol. 2004;93:131–140. doi: 10.1016/j.ijfoodmicro.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 42.Wang P, Yang H, Hu Y, Yuan F, Zhao G, Zhao Y, Chen Y. Characterization of Listeria monocytogenes isolates in import food products of China from 8 provinces between 2005 and 2007. J Food Sci. 2012;77:M212–M216. doi: 10.1111/j.1750-3841.2011.02596.x. [DOI] [PubMed] [Google Scholar]
- 43.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 44.Takahashi H, Handa-Miya S, Kimura B, Sato M, Yokoi A, Goto S, Watanabe I, Koda T, Hisa K, Fujii T. Development of multilocus single strand conformation polymorphism (MLSSCP) analysis of virulence genes of Listeria monocytogenes and comparison with existing DNA typing methods. Int J Food Microbiol. 2007;118:274–284. doi: 10.1016/j.ijfoodmicro.2007.07.047. [DOI] [PubMed] [Google Scholar]
- 45.Darling ACE, Mau B, Blatter FR, Perna NT. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14:1394–1403. doi: 10.1101/gr.2289704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Delcher AL, Bratke KA, Powers EC, Salzberg SL. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics. 2007;23:673–679. doi: 10.1093/bioinformatics/btm009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Darling AE, Mau B, Perna NT. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 2010;5:e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Didelot X, Falush D. Inference of bacterial microevolution using multilocus sequence data. Genetics. 2007;175:1251–1266. doi: 10.1534/genetics.106.063305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]