

Abstract
Availability of standardized metabolite panels and genome-wide single-nucleotide polymorphism data endorse the comprehensive analysis of gene–metabolite association. Currently, many studies use genome-wide association analysis to investigate the genetic effects on single metabolites (mGWAS) separately. Such studies have identified several loci that are associated not only with one but with multiple metabolites, facilitated by the fact that metabolite panels often include metabolites of the same or related pathways. Strategies that analyse several phenotypes in a combined way were shown to be able to detect additional genetic loci. One of those methods is the phenotype set enrichment analysis (PSEA) that tests sets of metabolites for enrichment at genes. Here we applied PSEA on two different panels of serum metabolites together with genome-wide data. All analyses were performed as a two-step identification–validation approach, using data from the population-based KORA cohort and the TwinsUK study. In addition to confirming genes that were already known from mGWAS, we were able to identify and validate 12 new genes. Knowledge about gene function was supported by the enriched metabolite sets. For loci with unknown gene functions, the results suggest a function that is interrelated with the metabolites, and hint at the underlying pathways.
INTRODUCTION
Metabolites are small molecules of diverse biochemical properties, including, for example, amino acids, lipids and xenobiotics like caffeine, that can be measured in body fluids such as blood, serum or urine. They represent endpoints of biological processes and therefore enable a direct readout of related pathways (1,2). Recent studies demonstrate that although metabolites are very sensitive to environmental factors, e.g. nutrition, physical activity and medication intake (3), metabolite changes due to genetic variation of underlying biochemical processes by factors like enzymes or transporters can be identified (4–8). Insights into the so-called genetically influenced metabotypes are important preconditions to analyse pathways and processes that can improve the understanding of disease. The knowledge of the genetic determination of metabolites can guide the improvement of diagnostics and therapies. Moreover, understanding the interrelation of metabolite profiles, genes and environmental factors can be used for personalized medicine approaches (1,4).
Recent improvements and development of new bioanalytical techniques to measure metabolites promote the systematic and simultaneous analysis of hundreds of metabolites. For large cohorts, using metabolite panels, which capture a wide range of different pathways, is a feasible strategy with respect to time and cost for analyzing the metabolome of large numbers of participants. The genetic analysis of hundreds of metabolites and millions of single-nucleotide polymorphisms (SNPs) is computational challenging and demands an appropriate strategy. Several studies analyse metabolites gained from metabolite panels with genome-wide association studies (GWASs) on all metabolite traits (mGWAS) separately (1,4–12). mGWAS have recently been demonstrated to be an effective tool in identifying genes that are associated with metabolites. A biomedical and pharmaceutical impact can be described for many identified loci (4). Other studies have followed a different approach by analysing selected genes that are known from previous studies rather than the whole genome. Such studies incorporated metabolites as intermediate phenotypes with the integrative analysis of known candidate genes (13,14). Although both approaches analyse metabolites separately, they have found that many identified genes are associated not only with one metabolite but with a group of several metabolites (4,7,8,14). In many cases, these metabolites belong to the same pathway or the same biochemical group.
An approach to account for the dependency structure of metabolite panel data is to simultaneously analyse multiple metabolites together rather than separately. Various strategies have been developed that analyse multiple phenotypes at a time (e.g. 15–22). Some of these approaches were successfully applied to metabolomics data (e.g. 23). Exploiting the information shared by several metabolites makes it possible to identify additional loci, while mGWAS that are focused on single metabolites neglect such information. In this study, we have applied one of these methods, namely, the phenotype set enrichment analysis (PSEA) (22), on two large panels of serum metabolites and genome-wide SNP data. The same data were previously analysed in mGWAS (4,8). PSEA analyses genetic association of sets of phenotypes, e.g. metabolites, which can be defined in various ways using prior knowledge or the data itself. Those phenotype sets are tested genome wide for gene enrichment with a permutation test that compares the enrichment of the set under investigation with enrichment of sets of permuted phenotypes. The PSEA method was developed following ideas of gene set enrichment strategies and has been previously published (22). It was shown that PSEA could detect loci associated with blood and iron phenotypes that were known from large meta-analysis but that could not be detected in GWAS using the same sample size. Therefore, we expect that using PSEA will allow us to identify additional loci associated with metabolites when compared with mGWAS on the same data. The metabolite sets that are found to be associated with a gene might also point to the gene function. In the present study, we applied two different strategies to define the metabolite sets: (i) Gaussian graphical modelling (GGM), a data-driven method for reconstructing metabolite pathways (24,25) that is used to identify biologically meaningful metabolite sets and (ii) a method based on the association of single metabolites at genes. The advantages of PSEA are that it can test high numbers of metabolite sets that are freely defined, and that it deals with a minimal number of assumptions. Importantly, by applying PSEA as a multi-metabolite analysis strategy on two different panels of metabolites, we were able to identify 12 new loci associated with metabolites.
RESULTS
We used PSEA to analyse 151 metabolites measured in blood serum with the BIOCRATES AbsoluteIDQTM p150 kit (Supplementary Material, Table S1) and 193 metabolites measured with a technique supplied by Metabolon (Supplementary Material, Table S2). Both technologies were applied to individuals from the population-based KORA cohorts and the TwinsUK study (26).
The basic principles of PSEA are presented in Figure 1A. The metabolite sets, either defined by GGM (GGM sets, Fig. 1B) or single association-defined metabolite sets (SAD sets, Fig. 1C), were tested for enrichment at genes to identify additional genetic loci that determine the metabolic make-up. We applied a two-step identification–validation approach: initially, promising enrichments of phenotype sets at genes were identified in KORA F4 (P < 10−4). Thereafter, those enrichments were validated in the independent TwinsUK study (see Fig. 1D and E, and Materials and Methods). PSEA is a gene-based method but uses SNP genotype data. Due to the strategy of mapping SNPs to genes (see Materials and Methods), proximate genes are often based partially on the same SNPs and are therefore not independent. A group of genes that share SNPs are named gene group in the following. In our data, 2319 gene groups were derived from the 20 801 genes. SNPs that are shared by several genes can lead to enrichment of the same metabolite set for all these genes. Of course, such enrichments are not independent, as they probably represent the effects of the same SNPs. We therefore introduced a number for independent promising enrichments, which counts multiple enrichments of the same metabolite set only once per gene group. This assured that the number of identified enrichments is not artificially increased by counting the same enrichment at one gene multiple times. It is used to correct the significance level in the validation step.
Figure 1.
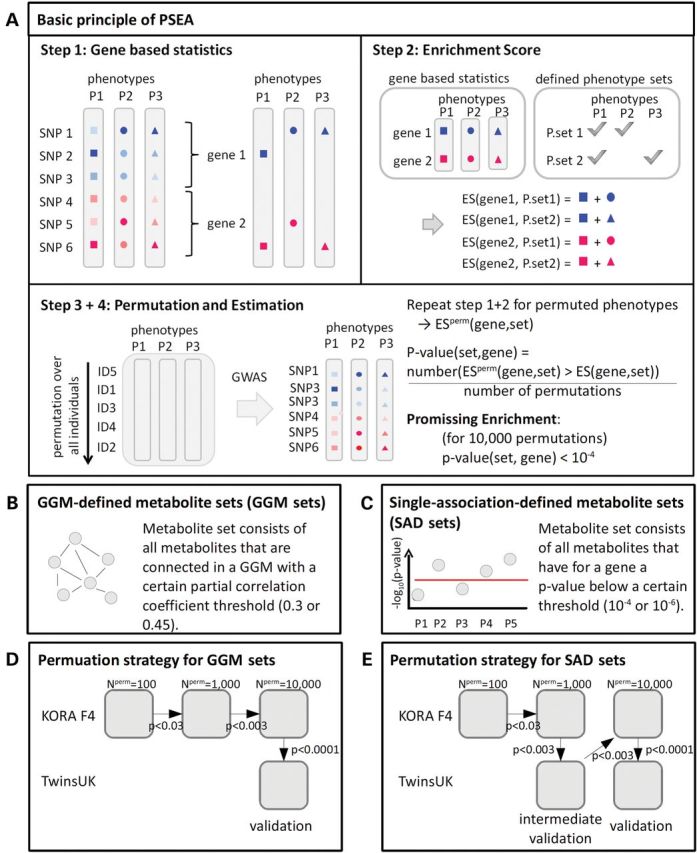
Schematic overview of the general method of PSEA (A), the definition of metabolite sets in this study (B and C) and the applied permutation schemes (D and E). In part (A), different shapes represent different phenotypes, different colours represent different genes and the intensity of the colour represents the association strength. (P, phenotype; p, P-value; P.set, phenotype set; ES, enrichment score; ESperm, enrichment score for permuted phenotypes; Nperm, number of permutations)
We analysed metabolite sets of three different batches by PSEA. The first batch of metabolite sets was defined by GGM, which is a valid tool for reconstruction of metabolite networks by using pairwise partial correlation. The GGM sets consist of the connected components in such networks at a specific partial-correlation threshold (see Fig. 1B, and Materials and Methods for details). Two other metabolite sets were defined using the single metabolite associations at each gene under two different conditions. SAD sets include all metabolites for which the minimum association P-value at this gene is below a specific threshold (see Fig. 1C and Materials and Methods for details). Our intention was to analyse all single metabolites at a specific gene that were associated with the gene at a promising low P-value as a set. With this, we aimed to find enrichments at especially those loci, for which the association of the gene and each single metabolite was not significant genome wide. Two P-value levels for the promising single association, 10−4 and 10−6, were used to define SAD sets. In contrast to the GGM sets that were analysed for all genes, each SAD set is gene dependent and was evaluated only at the gene at which it was defined.
PSEA on metabolite sets confirmed gene–metabolite associations known from mGWAS on large metabolite panels but furthermore revealed new genes that have not been previously published to be associated with metabolites. Table 1 summarizes the number of independent and validated enrichments and the number of loci with promising or validated enrichments of one or more metabolite sets. Table 2 gives the details on loci for which at least one metabolite set enrichment was validated but which had not been previously identified in an mGWAS.
Table 1.
Result counts for PSEA on Biocrates and Metabolon metabolite sets
| Number of metabolite sets | Number of promising enrichments, (independent promising enrichments) and number of gene groups (GG) | Number of validated enrichments with number of gene groups (GG) | Number of novel enrichments with number of gene groups (GG) | |
|---|---|---|---|---|
| GGM-defined metabolite sets (GGM sets) | ||||
| Biocrates | 38 | 354 (92) at 61 GG | 123 at 7 GG | 0 |
| Metabolon | 50 | 344 (78) at 58 GG | 75 at 8 GG | 0 |
| Single association-defined phenotype sets (SAD sets; threshold 10−6) | ||||
| Biocrates | 71 | 0 | 0 | 0 |
| Metabolon | 86 | 96 (20) at 13 GG | 96 at 13 GG | 0 |
| Single association-defined phenotype sets (SAD sets; threshold 10−4) | ||||
| Biocrates | 7942 | 62 (45) at 22 GG | 46 at 15 GG | 7 at 6 GG |
| Metabolon | 10 951 | 203(107) at 47 GG | 131 at 23 GG | 23 at 6 GG |
This table summarises the number of analysed metabolite sets, the number of enrichments that were found to be promising in KORA and the number of those that were validated in TwinsUK. Moreover, the number of gene groups is given at which the metabolite sets were enriched. The number of independent promising enrichments counts the enrichment of the same metabolite only once per gene group (GG). This number was used to correct P-value in the replication stage. Enrichments that could not be analysed for replication in the TwinsUK due unavailable metabolite sets are not included in these numbers.
Table 2.
Validated enrichments of metabolite sets for the 12 novel loci
| Genes | Elements of the phenotype seta |
|---|---|
| (1) GGM sets of Biocrates metabolites | |
| MFSD2A, MYCL1 | Lysophosphatidylcholines: acyl C16:0, acyl C17:0, acyl C18:1, acyl C20:4, acyl C18:0, |
| DKFZp686O1327 | Carnitine, hydroxyhexadecadienylcarnitine, octadecanoylcarnitine, arginine, threonine, phosphatidylcholines: diacyl C36:5, diacyl C36:6, diacyl C40:3, acyl–alkyl C36:5, lysophosphatidylcholines: acyl C18:1, acyl C20:4 |
| PDCD6IP | Decanoylcarnitine(C10), decanoylcarnitine (C10:1), decadienylcarnitine, tetradecenoylcarnitine (C14:1), hydroxyhexadecenoylcarnitine, octadecenoylcarnitine (C18:1), hexanoylcarnitine, pimeloylcarnitine, octanoylcarnitine, lysophosphatidylcholines: acyl C16:0, acyl C18:0, acyl C18:2 |
| IL3 | Hexadecanoylcarnitine (C16), octadecenoylcarnitine (C18:1), octadecadienylcarnitine, propionylcarnitine, valerylcarnitine, pimeloylcarnitine, phosphatidylcholine acyl–alkyl C40:5, hydroxysphingomyeline C14:1 |
| C12orf75 | Decadienylcarnitine, hydroxytetradecadienylcarnitine, hydroxybutyrylcarnitine, hexanoylcarnitine, valerylcarnitine, phosphatidylcholines: diacyl C36:2, acyl–alkyl C40:2, hydroxysphingomyeline: C14:1, C16:1, C22:1, C22:2, C24:1 |
| INTS8 | Decadienylcarnitine, phosphatidylcholines: diacyl C36:1, diacyl C42:2 |
| (2) GGM sets of Metabolon metabolites | |
| CYP4B1 | Tyrosine, haeme, carnitine C3:0, glutaroyl carnitine, fatty acid C11:1(10Z), gamma-glutamyltyrosine |
| KIAA0494 | Tyrosine, haeme, carnitine 3:0, fatty acid C11:1(10Z), gamma-glutamyltyrosine |
| CYP4A11, CYP4X1 | Fatty acid C11:1(10Z), phosphatidylcholines: diacyl C16:1(9Z)/C0:0, diacyl C14:0/C0:0 |
| CYP4Z2P | Fatty acid C11:1(10Z), phosphatidylcholine: diacyl C16:1(9Z)/C0:0, glutaroyl carnitine |
| DIRAS3 | 3-Methyl-2-oxopentanoate, glycerate, glycerol, phosphatidylcholine: acyl–alkyl C18:1(9Z) |
| MIR138-1 | Creatinine, phosphatidylcholines: diacyl C20:3(8Z,11Z,14Z)/C0:0, diacyl C18:2(9Z,12Z)/C0:0, diacyl C0:0/C18:1(9Z), gamma-glutamyltyrosine |
| LINGO2 | Aspartate, betaine, creatine, S-glutathionyl-l-cysteine, glutamate, methionine, pyroglutamine, 2-tetradecenoyl carnitine, isovalerylcarnitine, glycerol (C18:2(9Z, 12Z)/C0:0/C0:0), glycerol (C18:1(9Z)/C0:0/C0:0), phosphatidylcholine: acyl–alkyl C18:2(9Z,12Z)/C0:0, fatty acid C11:1(10Z), fatty acid C20:4(5Z, 8Z, 11Z, 14Z), fatty acid C20:3(n-3/n-6), xanthine, DSGEGDFXAEGGGVR, phenylsulfate |
| UBL3 | Indolepropionate, N-acetylornithine, p-cresol, lactate, cortisone, dehydroepiandrosterone sulphate, 3-dehydrocarnitine, hydroxy fatty acid C16:0, hydroxy fatty acid C18:0, fatty acid C20:4(5Z, 8Z, 11Z, 14Z) |
| LOC440131 | Indolepropionate, N-acetylornithine, p-cresol, lactate, cortisone, dehydroepiandrosterone sulphate, 3-dehydrocarnitine, hydroxy fatty acid C16:0, hydroxy fatty acid C18:0, fatty acid C20:4(5Z, 8Z, 11Z, 14Z), phosphatidylcholine: diacyl C20:4(5Z, 8Z, 11Z, 14Z)/C0:0, theophylline |
| CALR, DNASE2, GCDH, MAST1, PRDX2, RTBDN | Glutaroyl carnitine, glycerophosphorylcholine, erythritol |
| DAND5, FARSA, KLF1, RAD23A, SYCE2 | Arabitol, glutaroyl carnitine, glycerophosphorylcholine, oleamide C18:2(9Z), erythritol |
| GADD45GIP1 | Arabitol, fructose, glutaroyl carnitine, oleamide C18:2(9Z), erythritol |
| NFIX | Arabitol, fructose, glutaroyl carnitine, glycerophosphorylcholine, oleamide C18:2(9Z), erythritol |
This table specifies the composition of all validated metabolite set enrichments at genes that were not previously found in mGWAS on large metabolite panels. Part 1 of the table shows all novel genes found with Biocrates metabolite sets. Analogously, part 2 refers to all corresponding genes on Metabolon metabolites. Results for gene groups are grey shaded. Further details on metabolites are given in the Supplementary Material, Tables S1 and S2.
aCa:b indicates a chain of ‘a’ carbon atoms, including ‘b’ double bounds. For phosphatidylcholines measured by Biocrates, the accumulated number of carbon atoms and double bounds of both ligated fatty acid chains are given. For Metabolon phosphatidylcholines and glycerol, the number of carbon atoms in each ligated fatty acid is given and separated by a ‘/’, and the position of double bounds is given in brackets.
The analysis of 38 Biocrates- and 50 Metabolon-based GGM sets (Supplementary Material, Tables S3 and S4) revealed seven and eight independent gene groups, respectively, with validated enrichments of at least one metabolite set (Table 1; Supplementary Material, Tables S5 and S6). These findings confirm gene metabolite associations found in mGWAS on the same data (4,8). The elements of the metabolite sets fit well to the known metabolite associations (1,7,8). For example, at ACADM, the enrichment of one Biocrates and two Metabolon GGM sets were validated. The Biocrates set consisted of two carnitines with a carbon atom chain length of 8 and 10, and the Metabolon GGM sets included both carnitines with carbon 6, 8 and 10 carbon atoms and one of those additional 2-tetradecenoyl carnitine. In mGWAS, SNPs in ACADM were found to be associated with acylcarnitines with a medium chain length (4,8). This association reflects the gene function of ACADM, which is a key enzyme in the β-oxidation with its strongest substrate affinity to acyl-CoAs with chains of 4–12 carbon atoms.
The analysis of SAD sets with the threshold of 10−6 validated the enrichment of Metabolon metabolites sets at 13 gene groups. Genes of all gene groups are known from mGWAS (Supplementary Material, Fig. S1 and Table S7). For Biocrates, no metabolite set reached a sufficient P-value in the intermediate validation step (see Material and Methods). In total, 7942 different Biocrates and 10 951 Metabolon metabolite sets were identified as SAD sets for at least one gene with the higher P-value threshold of 10−4. Testing these sets led to 15 and 23 independent gene groups with validated enrichments of at least one set of Biocrates or Metabolon metabolites (Fig. 2 and Fig. 3; Supplementary Material, Tables S8 and S9). Eight and 16 of those gene groups with enrichment of Biocrates- and Metabolon-based SAD sets, respectively, were already known from previous mGWAS using the same data (4,8). One special case is SLC22A1, which was known from mGWAS on Metabolon metabolites (associated with isobutyrylcarnitine) (4) but not from mGWAS on Biocrates metabolites; however, it was identified in this study with a promisingly enriched phenotype set of Biocrates metabolites (six carnitines including butyrylcarnitine, five phosphatidylcholines and one amino acid). In addition to comparing mGWAS on the same data, we used other published mGWAS (1,4–6,8–12) on large metabolite panels to investigate our findings. This revealed that one additional gene (SLC1A4), identified by both Biocrates- and Metabolon-based SAD sets, was found to be previously reported with metabolite levels (5). For the remaining 12 loci identified with PSEA using SAD sets (threshold 10−4) on Biocrates metabolites (six loci) and on Metabolon metabolites (six loci), no association with a metabolite had been previously reported in mGWAS on a large metabolite panel (Table 2). Therefore, these 12 loci were newly identified for association with metabolites. For 8 of these 12 novel loci, the corresponding SAD set of one gene was promisingly enriched and validated (DKFZp686O1327, PDCD6IP, IL3, C12orf75, INTS8, DIRAS3, MIR138–1 and LINGO2). At the other four loci, SAD-defined phenotype sets showed validated enrichment for several genes of a gene group (MFSD2A and MYCL1, UBL3 and LOC440131, several genes of the Cytochrome P450 family 4, GCDH and 12 other genes at chromosome 19). The promisingly enriched and validated SAD-defined phenotype sets were identical or similar within the gene group. For example, the overlapping genes UBL3 and LOC440131 showed enrichment of two different SAD sets. The SAD set of LOC440131 includes the same metabolites as the SAD set of UBL3 as well as two additional ones.
Figure 2.
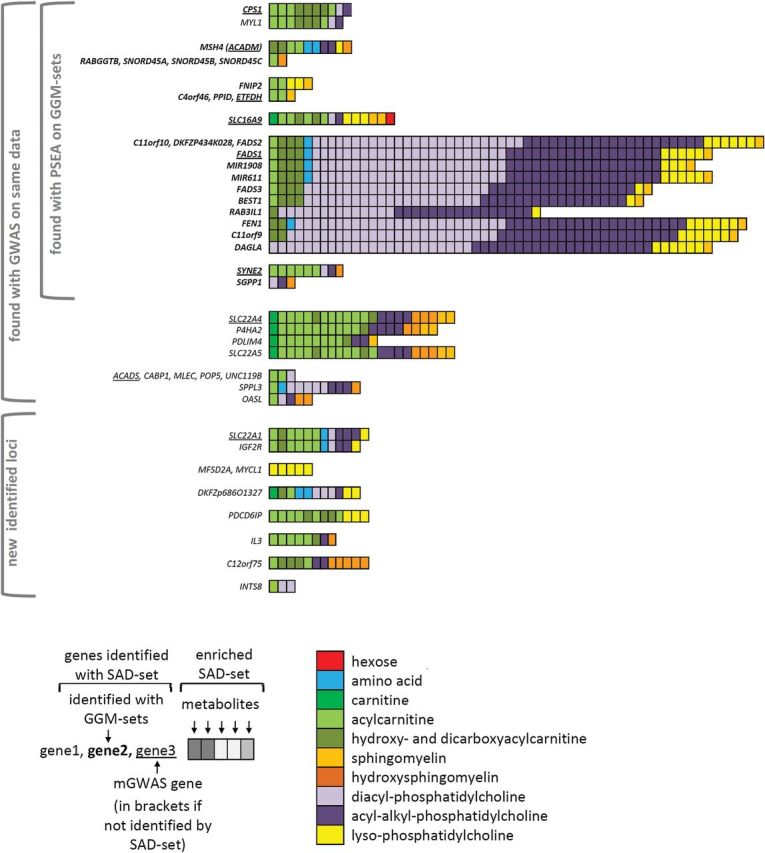
PSEA results on single association-defined metabolite sets (SAD set; threshold: 10−4) on Biocrates metabolites. All metabolite sets that were promisingly enriched in KORA F4 and validated in TwinsUK are presented along with the genes at which they were identified. Gene groups are separated by horizontal space. Details of the presentation are explained in below the figure.
Figure 3.
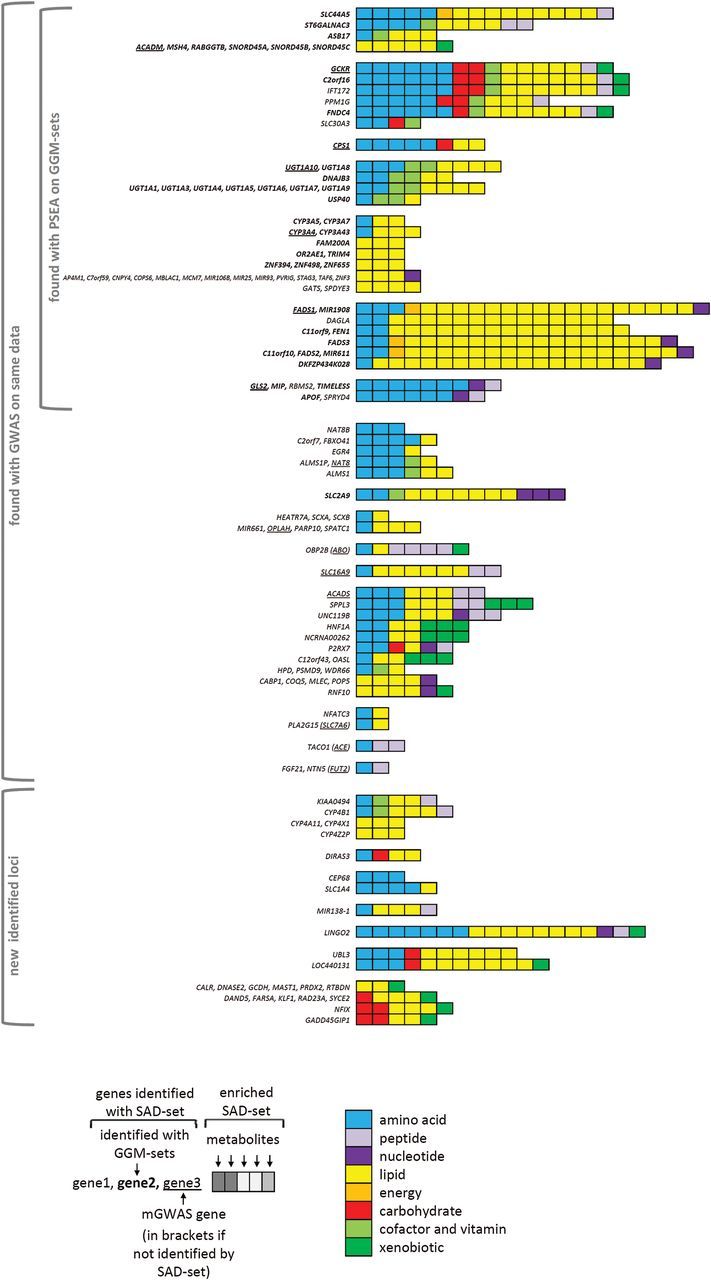
PSEA results on single association-defined metabolite sets (SAD set; threshold: 10−4) on Metabolon metabolites. All metabolite sets that were promisingly enriched in KORA F4 and validated in TwinsUK are presented along with their respective genes. Gene groups are separated by a horizontal space. Details of the presentation are explained below the figure.
DISCUSSION
By applying the multiple phenotype approach PSEA to metabolites, 12 novel associations of genes and metabolites were identified that have not been published before in any mGWAS of a large metabolite panel. This method additionally confirmed several loci with known metabolite associations. For both known and unknown loci, the enriched phenotype sets carried information about networks and pathways.
The gene function of the genes that were newly found to be associated with metabolites is discussed in the Supplementary Material, Text S1. Three genes are exemplarily discussed here:
IL3
A set of six acylcarnitines, one dicarboxyacylcarnitine, one acyl–alkyl-phosphatidylcholine and one hydroxysphingomyelin was enriched for interleukin 3 (IL3) on Biocrates metabolites. In other words, the enriched metabolite set consists of seven acylcarnitines and two phospholipids. IL3 is known to be a hematopoietic growth factor that stimulates survival, multiplication and differentiation of hematopoietic cells (27). Other studies found that IL3 stimulates phospholipid synthesis (28) and suppresses lipid degradation and β-oxidation of fatty acids (29). Acylcarnitines are known to play an important role in β-oxidation and are needed for transport of activated fatty acids into the mitochondria. Fatty acids, which are part of phosphatidylcholines and sphingomyelins, are substrates of β-oxidation. This shows how the elements of the enriched phenotype set are involved in the β-oxidation. The two phospholipids also stand for the involvement of IL3 in phospholipid synthesis. Therefore, it can be stated that the elements of the enriched metabolite set underscore the previously reported role of the gene product of IL3 in the β-oxidation.
Cytochrome P450 family 4
Four genes of the cytochrome P450 family 4 (CYP4B1, CYP4A11, CYP4X1 and CYP4Z2P) and one additional gene KIAA0494, which maps to a region that overlaps with CYP4B1, were found on Metabolon metabolites with an enrichment in four slightly different SAD sets. The sets included three to six metabolites. Two glycerolipids as well as one fatty acid and two carnitines were part of several sets. The amino acid l-tyrosine and the peptide γ-glutamyltyrosine were part of two enriched phenotype sets. The cofactor haeme was identified for two genes. The cytochrome P450 monooxygenase system is a multigene superfamily of enzymes that are involved in various reactions, e.g. drug metabolism and lipid synthesis. Haeme is a cofactor in these processes (30). The metabolites identified as elements of the metabolite sets reflect the gene product's function, including possible substrates (glycerolipids and fatty acids), cofactors (haeme) and related compounds (carnitines).
LINGO2
A large set of 17 Metabolon metabolites was significantly enriched at the gene ‘leucine rich repeat and Ig domain containing 2’ (LINGO2). The set included various types of metabolites of the lipid metabolism (fatty acids, carnitines, lysolipid and monoacylglycerol), some amino acids, a nucleotide, one peptide and phenylsulfate. The gene function of LINGO2 is not known yet. A GWAS identified a genome-wide significant association of LINOG2 with BMI (31). The elements of the enriched metabolite set hint that an involvement in the metabolism of fatty acids. This could explain the effect on BMI.
In general, PSEA emphasizes interesting relations between genes and a small set of metabolites out of hundreds. These enrichments can reveal two types of knowledge. First, novel genetic loci can be identified. The ability of PSEA to identify loci other than those identified by GWAS using the same data derives from the consideration of multiple metabolites at a time. Our results demonstrate that several loci could be identified with PSEA but not GWAS on the same data. Second, information about potential gene functions and affected pathways can be extracted from the enriched sets for novel as well as previously known genetic loci. For instance, the PSEA results supported the known gene functions for IL3 and the cytochrome P450 family 4, while the identified sets suggested previously unknown pathways for LINGO2. This knowledge about the association of metabolite sets with specific genes can motivate and direct further analysis.
The computational intensity of the algorithm in combination with computational limitations determined the minimal possible P-value. With 10 000 permutations, the minimal P-value is 10−4, which means that a Bonferroni-correction for >20 000 genes cannot be applied. Therefore, we could not claim statistical significance in the identification step; rather, by terming enrichments with a P-value below the minimal P-value of 10−4 as “promising enrichments”, and validating our results in the TwinsUK, we were then able to use a Bonferroni-corrected multiple testing threshold. This two-stage design has reduced power when compared with an approach that analyses statistical significance in one cohort or from a meta-analysis of both studies, neither of which was computationally possible with the current data. Further studies are needed to replicate our results.
In summary, the present study identified the association of 12 loci with metabolites, which had not been published before. This demonstrates the potential of multi-metabolite analyses. With PSEA, we successfully screened hundreds of metabolites and metabolite sets. The enriched sets carry information on the possible pathways, and the findings hinted at the gene function. Altogether, this knowledge can help to design biological experiments and guide further research on the genetic determination of metabolites.
MATERIALS AND METHODS
Study description and genotyping
Analyses were performed in a two-stage approach consisting of an identification and a validation step. We analysed data of the KORA F4 study from the KORA cohorts (cooperative health research in the region of Augsburg) (32). KORA F4 participants (n = 1814) were genotyped on the Affymetrix 6.0 SNParray. Imputation was performed with Impute v 0.4.2 (reference HapMap phase 2, release 22) (33). Findings identified in the analysis of KORA F4 were validated for data of the TwinsUK study, a British adult twin registry. Participants of the TwinsUK study were genotyped with a combination of different Illumina arrays (HumanHap300, HumanHap510Q, 1M-Duo and 1.2MDuo 1M) and imputed with Impute v2. More details on study description and genotyping are given in the Supplementary Material, Text S2.
Ethics statement
Written informed consent was given by all participants of KORA and TwinsUK. The KORA study, including the protocols for subject recruitment, assessment and the informed consent, was approved by the ethics committee of the Bayerische Landesärztekammer. Ethics approval for the TwinsUK was obtained from the Guy's and St. Thomas’ Hospital Ethics Committee.
Genes
PSEA is a gene-based approach, i.e. it is necessary that SNPs are mapped to genes. Only autosomal SNPs were used that had a minor allele frequency >5%, call rate >95% and imputation quality >0.4. SNPs were mapped to genes when they were in the transcribed region of a gene or in the flanking region of 110 kb upstream or 40 kb downstream. These thresholds were chosen as it has been previously shown that 99% of the expected cis-eQTLs are located within this interval (34). This leads to a good coverage of SNPs that possibly affect the gene product. The same mapping of SNPs to genes was also used for gene set enrichment approaches based on GWAS data (35). A SNP was mapped to multiple genes when it was in the transcribed or flanking region of more than one gene. Gene information was downloaded from the UCSC (University of California Santa Cruz) genome browser (http://genome.ucsc.edu/). The SNP gene mapping has been described in detail previously (22). In total, 20 801 genes were analysed. As described above, due to the broad assignment of SNPs to genes proximate genes often overlap in SNPs. Such overlapping genes are named gene group. In our data, the 20 801 genes led to 2319 gene groups.
Metabolite measurement
Metabolites were measured with two technologies, Biocrates and Metabolon, in the same individuals in both the KORA F4 and the TwinsUK studies. Slight differences in final numbers were caused by quality control exclusions.
Biocrates metabolites
A panel of 163 metabolites was measured for individuals of KORA F4 using electro spray ionization tandem mass spectrometry with the AbsoluteIDQTM p150 kit (BIOCRATES Life Sciences AG, Innsbruck, Austria). Details of the measurement methods and quality control were described in previous publications (7,8,14). After quality control, 151 metabolites remained for further analyses. These 151 metabolites can be grouped in 10 metabolite classes and include 14 amino acids, 1 hexose, carnitine species (1 free carnitine, 22 acylcarnitines and 12 hydroxy- and dicarboxyacylcarnitines), 9 sphingomyelins, 5 hydroxysphingomyelins and different forms of phosphatidylcholines (36 diacyl-phosphatidylcholines, 38 acyl–alkyl-phosphatidylcholines and 13 lyso-phosphatidiylcholines). A full list of all metabolites is available in Supplementary Material, Table S1. For 1809 individuals in KORA F4, Biocrates metabolites and genome-wide genotypes were available.
Samples from the TwinsUK cohort that had measurements for metabolites with the same AbsoluteIDQTM p150 kit was used for replication. The metabolites underwent the same quality control as described for KORA F4. All 151 metabolites passed the quality control in TwinsUK as well. Eight hundred and forty-three unrelated individuals with genotypes and valid Biocrates metabolites measurements were used for further analysis.
Metabolon metabolites
A different panel of 295 metabolites was measured with a technique supplied by Metabolon (Metabolon, Inc., Durham, USA). It used ultrahigh-performance liquid-phase chromatography and gas chromatography separation with tandem mass spectrometry (36,37). The measurement method was described in detail in a previous publication (4). One hundred and two metabolites had >10% missing values and were excluded from the analyses. Missing values for the remaining metabolites were imputed with the MICE algorithm (http://cran.r-project.org/web/packages/mice/index.html) that was implemented in R (http://www.r-project.org/). The remaining 193 metabolites spanned different super pathways including amino acids (52), carbohydrates (10), cofactors and vitamins (7), energy (3) and lipid (90) pathway-relevant compounds, nucleotides (9), peptides (11) and xenobiotics (11). The full list of all 193 metabolites together with additional information about the pathways they belong to is given in the Supplementary Material, Table S2. In total, 1768 KORA F4 individuals with valid Metabolon metabolites measurements and genotypes were used for further analysis.
The same technology was applied to measure metabolites from the TwinsUK data. Only metabolites that passed quality control in KORA F4 were regarded. Individuals with >50% missing values were excluded. Four metabolites that were present in KORA F4 had <300 valid measurements in TwinsUK data. According to Suhre et al. (4), 300 is the critical limit of non-missing values to avoid false-positive findings due to small sample size. Therefore, these four metabolites were excluded from further analysis. In the remaining 189 metabolites, the maximal missing rate per metabolite was 65.59%, which is equivalent to 362 valid measurements. To assure that most metabolite sets could be analysed in the replication, no further exclusion criteria for metabolites were applied. No imputation of missing data was performed. After reduction to unrelated and genotyped individuals, 705 individuals remained in the analysis.
For both Metabolon and Biocrates metabolites, outliers that differed >5 SD from the mean were excluded. The residuals of log-transformed metabolites with adjustment for sex and age were calculated and taken as phenotypic input for PSEA. For Biocrates metabolites, additional adjustments for an internal batch variable accounting for possible measurement differences was applied. After log transformation, most (146) Biocrates metabolites were closer to the normal distribution than the untransformed metabolite concentrations. For Metabolon metabolites, the same was previously shown with log10 transformation (4). For simplicity per panel, the same transformation was applied to all metabolites.
Phenotype set enrichment analysis
The basic strategy of PSEA is shown in Figure 1. The details of the algorithm were described in a previous publication (22). In general, PSEA is a gene-based approach to identify association of phenotype sets with a gene by a permutation test. For each permutation, the phenotypes are permuted over individuals, whereas all phenotypes of a set are permuted in the same way to conserve the correlation structure of phenotypes. The genotypes are not changed. PSEA was applied to Biocrates and Metabolon metabolites separately. To define the phenotype sets, two strategies were used: GGM and single phenotype association, as described below.
GGM-defined metabolite sets (GGM sets)
To define phenotype sets, GGM was applied as a statistical method that estimates the conditional dependence between variables (24). For each pair of metabolites, we estimated the partial-correlation coefficient, which represent the pairwise (regular) Pearson correlation coefficient conditioned for the correlation with all other metabolites in the dataset. This completely data-driven approach was shown to be a valuable tool to identify metabolite networks, which is able to distinguish direct from indirect associations (24,25). Another advantage is that this estimation of metabolite sets is independent from further information like availability of database information. The analysis strategy was applied to the panel of all metabolite measurements in KORA F4 that passed quality control and to all individuals with metabolite measurements and genotypes. Two partial-correlation coefficient threshold levels (0.3 and 0.45) were used, both of which gave a different range of metabolite sets. The sets were not overlapping for each threshold, but sets gained from the higher threshold level were subsets of the sets gained with the lower threshold level.
Single association-defined metabolite sets (SAD sets)
A SAD set is defined per gene with the use of a P-value criterion for the association of single metabolites. The gene association is calculated in the same way as in PSEA and is the minimal association P-value of all SNPs mapped to this gene and the metabolite. All phenotypes for which the P-value for association with the gene was below this P-value criterion are taken in a SAD set. At most, one metabolite set can be identified for each gene. Two runs were made using different P-value levels: 10−4 and 10−6. The specification of SAD sets was based on the discovery cohort KORA F4.
Two-step identification–validation strategy
As described in Results, we performed our analyses with a two-step approach, with an initial identification of “promising enrichments” in KORA F4, and a subsequent validation of those in the TwinsUK study. A phenotype set that showed enrichment at a gene with a permutation P <10−4 was named promisingly enriched and was taken forward for validation in the TwinsUK study. As described above, genes in the so-called gene groups are not independent as the share SNPs. We observed that, within a gene group, the same phenotype sets are often promisingly enriched for several genes. Therefore, the number of independent promising enrichments was introduced. This counts independent gene group enrichments of the same set only once. It was used to correct for multiple testing in the validation stage, and the corrected validation P-value level is 0.05/number of independent promising enrichments.
Permutation strategy
Identification and validation were based on a permutation test with a total of 10 000 permutations. For computational reasons, the permutations were performed in a graded process. In contrast to performing the maximal number of permutations in KORA F4, for all genes the number of permutations was increased in steps. After each step, only those genes are taken forward to the round of permutations with a promising P-value. Initially, 100 permutations were calculated. Only those genes for which at least one phenotype set had P-value ≤ 0.03 were analysed with 1000 permutations. The genes that had P-value ≤ 0.003 were taken for the 10 000 permutations step. The enrichments with P-value < 0.0001 were validated in the TwinsUK study (compare Fig. 1D). The graded permutation strategy considerably reduced the computational effort. As a consequence, the strategy has a reduced statistical power but does not cause more false-positive results.
In the analysis of SAD sets, the number of sets tested for enrichment was much higher than in the analysis of GGM sets. To reduce the computational effort, we introduced an intermediate validation step to the graded permutation scheme (compare Fig. 1E). All genes at which a SAD set had a P-value ≤ 0.003 after 1000 permutations in KORA F4 were validated in the TwinsUK study with 1000 permutations. Only those genes for which a SAD set gained a P-value ≤ 0.003 in PSEA on TwinsUK with 1000 permutations (intermediate validation) were analysed in KORA F4 with 10 000 permutations.
SUPPLEMENTARY MATERIAL
FUNDING
The work of C.G. and J.S.R. was supported by a grant of the RFBR (Russian Foundation for Basic Research)-Helmholtz Joint Research Group. S.-Y.S. is supported by a Post-Doctoral Research Fellowship from the Oak Foundation. J.K. is supported by a grant from the German Helmholtz Postdoc Programme. K.S. is supported by ‘Biomedical Research Program’ funds at Weill Cornell Medical College in Qatar, a program funded by the Qatar Foundation.
Supplementary Material
ACKNOWLEDGEMENTS
KORA: We thank Dr. Werner Römisch-Margl, Dr Cornelia Prehn, Julia Scarpa and Katharina Sckell for metabolomics measurements performed at the Helmholtz Zentrum München, Genome Analysis Center, Metabolomics Core Facility.
The KORA Augsburg studies were financed by the Helmholtz Zentrum München, German Research Center for Environmental Health, Neuherberg, Germany and supported by grants from the German Federal Ministry of Education and Research (BMBF). Part of this work was financed by the German National Genome Research Network (NGFN). Our research was supported within the Munich Center of Health Sciences (MC Health) as part of LMUinnovativ. Moreover, the research leading to these results has received funding from the European Union's Seventh Framework Programme (FP7-Health-F5-2012) under grant agreement n° 305280 (MIMOmics). This study was supported in part by a grant from the German Federal Ministry of Education and Research (BMBF Förderkennzeichen 01GI0922) to Deutsches Zentrum für Diabetes Forschung (D.Z.D.) (German Center for Diabetes Research DZD e.V.).
TwinsUK: We thank the staff from the Genotyping Facilities at the Wellcome Trust Sanger Institute for sample preparation, Quality Control and Genotyping led by Leena Peltonen and Panos Deloukas; Le Centre National de Génotypage, France, led by Mark Lathrop, for genotyping; Duke University, North Carolina, USA, led by David Goldstein, for genotyping; and the Finnish Institute of Molecular Medicine, Finnish Genome Center, University of Helsinki, led by Aarno Palotie.
The TwinsUK study was funded by the Wellcome Trust; European Community's Seventh Framework Programme (FP7/2007-2013/grant agreement HEALTH-F2-2008-201865-GEFOS) and (FP7/2007-2013), ENGAGE project grant agreement HEALTH-F4-2007-201413 and the FP-5 GenomEUtwin Project (QLG2-CT-2002-01254). The study also receives support from the Dept of Health via the National Institute for Health Research (NIHR) comprehensive Biomedical Research Centre award to Guy's & St Thomas’ NHS Foundation Trust in partnership with King's College London. TDS is an NIHR senior Investigator. The project also received support from a Biotechnology and Biological Sciences Research Council (BBSRC) project grant (G20234). The authors acknowledge the funding and support of the National Eye Institute via an NIH/CIDR genotyping project (PI: Terri Young). Genotyping was also performed by CIDR as part of an NEI/NIH project grant.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Suhre K., Gieger C. Genetic variation in metabolic phenotypes: study designs and applications. Nat. Rev. Genet. 2012;13:759–769. doi: 10.1038/nrg3314. [DOI] [PubMed] [Google Scholar]
- 2.Adamski J., Suhre K. Metabolomics platforms for genome wide association studies-linking the genome to the metabolome. Curr. Opin. Biotech. 2013;24:39–47. doi: 10.1016/j.copbio.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 3.Krug S., Kastenmüller G., Stückler F., Rist M.J., Skurk T., Sailer M., Raffler J., Römisch-Margl W., Adamski J., Prehn C., et al. The dynamic range of the human metabolome revealed by challenges. FASEB J. 2012;26:2607–2619. doi: 10.1096/fj.11-198093. [DOI] [PubMed] [Google Scholar]
- 4.Suhre K., Shin S.Y., Petersen A.K., Mohney R.P., Meredith D., Wägele B., Altmaier E., Deloukas P., Erdmann J., Grundberg E., et al. Human metabolic individuality in biomedical and pharmaceutical research. Nature. 2011;477:54–60. doi: 10.1038/nature10354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kettunen J., Tukiainen T., Sarin A.P., Ortega-Alonso A., Tikkanen E., Lyytikainen L.P., Kangas A.J., Soininen P., Wurtz P., Silander K., et al. Genome-wide association study identifies multiple loci influencing human serum metabolite levels. Nat. Genet. 2012;44:269–276. doi: 10.1038/ng.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nicholson G., Rantalainen M., Li J.V., Maher A.D., Malmodin D., Ahmadi K.R., Faber J.H., Barrett A., Min J.L., Rayner N.W., et al. A genome-wide metabolic QTL analysis in Europeans implicates two loci shaped by recent positive selection. PLoS Genet. 2011;7:e1002270. doi: 10.1371/journal.pgen.1002270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gieger C., Geistlinger L., Altmaier E., Hrabe de Angelis M., Kronenberg F., Meitinger T., Mewes H.W., Wichmann H.E., Weinberger K.M., Adamski J., et al. Genetics meets metabolomics: a genome-wide association study of metabolite profiles in human serum. PLoS Genet. 2008;4:e1000282. doi: 10.1371/journal.pgen.1000282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Illig T., Gieger C., Zhai G., Römisch-Margl W., Wang-Sattler R., Prehn C., Altmaier E., Kastenmüller G., Kato B.S., Mewes H.W., et al. A genome-wide perspective of genetic variation in human metabolism. Nat. Genet. 2010;42:137–141. doi: 10.1038/ng.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tanaka T., Shen J., Abecasis G.R., Kisialiou A., Ordovas J.M., Guralnik J.M., Singleton A., Bandinelli S., Cherubini A., Arnett D., et al. Genome-wide association study of plasma polyunsaturated fatty acids in the InCHIANTI Study. PLoS Genet. 2009;5:e1000338. doi: 10.1371/journal.pgen.1000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hicks A.A., Pramstaller P.P., Johansson A., Vitart V., Rudan I., Ugocsai P., Aulchenko Y., Franklin C.S., Liebisch G., Erdmann J., et al. Genetic determinants of circulating sphingolipid concentrations in European populations. PLoS Genet. 2009;5:e1000672. doi: 10.1371/journal.pgen.1000672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Suhre K., Wallaschofski H., Raffler J., Friedrich N., Haring R., Michael K., Wasner C., Krebs A., Kronenberg F., Chang D., et al. A genome-wide association study of metabolic traits in human urine. Nat. Genet. 2011;43:565–569. doi: 10.1038/ng.837. [DOI] [PubMed] [Google Scholar]
- 12.Demirkan A., van Duijn C.M., Ugocsai P., Isaacs A., Pramstaller P.P., Liebisch G., Wilson J.F., Johansson A., Rudan I., Aulchenko Y.S., et al. Genome-wide association study identifies novel loci associated with circulating phospho- and sphingolipid concentrations. PLoS Genet. 2012;8:e1002490. doi: 10.1371/journal.pgen.1002490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tukiainen T., Kettunen J., Kangas A.J., Lyytikainen L.P., Soininen P., Sarin A.P., Tikkanen E., O'Reilly P.F., Savolainen M.J., Kaski K., et al. Detailed metabolic and genetic characterization reveals new associations for 30 known lipid loci. Hum. Mol. Genet. 2012;21:1444–1455. doi: 10.1093/hmg/ddr581. [DOI] [PubMed] [Google Scholar]
- 14.Ried J.S., Baurecht H., Stückler F., Krumsiek J., Gieger C., Heinrich J., Kabesch M., Prehn C., Peters A., Rodriguez E., et al. Integrative genetic and metabolite profiling analysis suggests altered phosphatidylcholine metabolism in asthma. Allergy. 2013;68:629–636. doi: 10.1111/all.12110. [DOI] [PubMed] [Google Scholar]
- 15.Shriner D. Moving toward system genetics through multiple trait analysis in genome-wide association studies. Front. Genet. 2012;3:1. doi: 10.3389/fgene.2012.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang Q., Wu H., Guo C.Y., Fox C.S. Analyze multivariate phenotypes in genetic association studies by combining univariate association tests. Genet. Epidemiol. 2010;34:444–454. doi: 10.1002/gepi.20497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang J., Johnson A.D., O'Donnell C.J. PRIMe: a method for characterization and evaluation of pleiotropic regions from multiple genome-wide association studies. Bioinformatics. 2011;27:1201–1206. doi: 10.1093/bioinformatics/btr116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gupta M., Cheung C.L., Hsu Y.H., Demissie S., Cupples L.A., Kiel D.P., Karasik D. Identification of homogeneous genetic architecture of multiple genetically correlated traits by block clustering of genome-wide associations. J. Bone. Miner. Res. 2011;26:1261–1271. doi: 10.1002/jbmr.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ferreira M.A., Purcell S.M. A multivariate test of association. Bioinformatics. 2009;25:132–133. doi: 10.1093/bioinformatics/btn563. [DOI] [PubMed] [Google Scholar]
- 20.Denny J.C., Ritchie M.D., Basford M.A., Pulley J.M., Bastarache L., Brown-Gentry K., Wang D., Masys D.R., Roden D.M., Crawford D.C. PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinformatics. 2010;26:1205–1210. doi: 10.1093/bioinformatics/btq126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stephens M. A unified framework for association analysis with multiple related phenotypes. PLoS ONE. 2013;8:e65245. doi: 10.1371/journal.pone.0065245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ried J.S., Döring A., Oexle K., Meisinger C., Winkelmann J., Klopp N., Meitinger T., Peters A., Suhre K., Wichmann H.E., et al. PSEA: phenotype set enrichment analysis – a new method for analysis of multiple phenotypes. Genet. Epidemiol. 2012;36:244–252. doi: 10.1002/gepi.21617. [DOI] [PubMed] [Google Scholar]
- 23.Inouye M., Ripatti S., Kettunen J., Lyytikainen L.P., Oksala N., Laurila P.P., Kangas A.J., Soininen P., Savolainen M.J., Viikari J., et al. Novel Loci for metabolic networks and multi-tissue expression studies reveal genes for atherosclerosis. PLoS Genet. 2012;8:e1002907. doi: 10.1371/journal.pgen.1002907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krumsiek J., Suhre K., Illig T., Adamski J., Theis F.J. Gaussian graphical modeling reconstructs pathway reactions from high-throughput metabolomics data. BMC Syst. Biol. 2011;5:21. doi: 10.1186/1752-0509-5-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krumsiek J., Suhre K., Evans A.M., Mitchell M.W., Mohney R.P., Milburn M.V., Wägele B., Römisch-Margl W., Illig T., Adamski J., et al. Mining the unknown: a systems approach to metabolite identification combining genetic and metabolic information. PLoS Genet. 2012;8:e1003005. doi: 10.1371/journal.pgen.1003005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andrew T., Hart D.J., Snieder H., de Lange M., Spector T.D., MacGregor A.J. Are twins and singletons comparable? A study of disease-related and lifestyle characteristics in adult women. Twin Res. 2001;4:464–477. doi: 10.1375/1369052012803. [DOI] [PubMed] [Google Scholar]
- 27.Lopez A.F., Dyson P.G., To L.B., Elliott M.J., Milton S.E., Russell J.A., Juttner C.A., Yang Y.C., Clark S.C., Vadas M.A. Recombinant human interleukin-3 stimulation of hematopoiesis in humans: loss of responsiveness with differentiation in the neutrophilic myeloid series. Blood. 1988;72:1797–1804. [PubMed] [Google Scholar]
- 28.Bauer D.E., Hatzivassiliou G., Zhao F., Andreadis C., Thompson C.B. ATP citrate lyase is an important component of cell growth and transformation. Oncogene. 2005;24:6314–6322. doi: 10.1038/sj.onc.1208773. [DOI] [PubMed] [Google Scholar]
- 29.Deberardinis R.J., Lum J.J., Thompson C.B. Phosphatidylinositol 3-kinase-dependent modulation of carnitine palmitoyltransferase 1A expression regulates lipid metabolism during hematopoietic cell growth. J. Biol. Chem. 2006;281:37372–33780. doi: 10.1074/jbc.M608372200. [DOI] [PubMed] [Google Scholar]
- 30.Chaudhary K.R., Batchu S.N., Seubert J.M. Cytochrome P450 enzymes and the heart. IUBMB Life. 2009;61:954–960. doi: 10.1002/iub.241. [DOI] [PubMed] [Google Scholar]
- 31.Speliotes E.K., Willer C.J., Berndt S.I., Monda K.L., Thorleifsson G., Jackson A.U., Lango Allen H., Lindgren C.M., Luan J., Magi R., et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat. Genet. 2010;42:937–948. doi: 10.1038/ng.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wichmann H.-E., Gieger C., Illig T. KORA-gen-resource for population genetics, controls and a broad spectrum of disease phenotypes. Gesundheitswesen. 2005;67:S26–S30. doi: 10.1055/s-2005-858226. [DOI] [PubMed] [Google Scholar]
- 33.Marchini J., Howie B. Genotype imputation for genome-wide association studies. Nat. Rev. Genet. 2010;11:499–511. doi: 10.1038/nrg2796. [DOI] [PubMed] [Google Scholar]
- 34.Veyrieras J.B., Kudaravalli S., Kim S.Y., Dermitzakis E.T., Gilad Y., Stephens M., Pritchard J.K. High-resolution mapping of expression QTLs yields insight into human gene regulation. PLoS Genet. 2008;4:e1000214. doi: 10.1371/journal.pgen.1000214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Serge A.V., Groop L., Mootha V.K., Daly M.J., Altshuler D. Common inherited variation in mitochondrial genes is not enriched for association with type 2 diabetes or related glycemic traits. PLoS Genet. 2010;6:e1001058. doi: 10.1371/journal.pgen.1001058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Evans A.M., DeHaven C.D., Barrett T., Mitchell M., Milgram E. Integrated, nontargeted ultrahigh performance liquid chromatography/electrospray ionization tandem mass spectrometry platform for the identification and relative quantification of the small-molecule complement of biological systems. Anal. Chem. 2009;81:6656–6667. doi: 10.1021/ac901536h. [DOI] [PubMed] [Google Scholar]
- 37.Ohta T., Masutomi N., Tsutsui N., Sakairi T., Mitchell M., Milburn M.V., Ryals J.A., Beebe K.D., Guo L. Untargeted metabolomic profiling as an evaluative tool of fenofibrate-induced toxicology in Fischer 344 male rats. Toxicol. Pathol. 2009;37:521–535. doi: 10.1177/0192623309336152. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.