

Abstract
Brain bioenergetic function declines in some neurodegenerative diseases, this may influence other pathologies and administering bioenergetic intermediates could have therapeutic value. To test how one intermediate, oxaloacetate (OAA) affects brain bioenergetics, insulin signaling, inflammation and neurogenesis, we administered intraperitoneal OAA, 1–2 g/kg once per day for 1–2 weeks, to C57Bl/6 mice. OAA altered levels, distributions or post-translational modifications of mRNA and proteins (proliferator-activated receptor-gamma coactivator 1α, PGC1 related co-activator, nuclear respiratory factor 1, transcription factor A of the mitochondria, cytochrome oxidase subunit 4 isoform 1, cAMP-response element binding, p38 MAPK and adenosine monophosphate-activated protein kinase) in ways that should promote mitochondrial biogenesis. OAA increased Akt, mammalian target of rapamycin and P70S6K phosphorylation. OAA lowered nuclear factor κB nucleus-to-cytoplasm ratios and CCL11 mRNA. Hippocampal vascular endothelial growth factor mRNA, doublecortin mRNA, doublecortin protein, doublecortin-positive neuron counts and neurite length increased in OAA-treated mice. 1H-MRS showed OAA increased brain lactate, GABA and glutathione thereby demonstrating metabolic changes are detectable in vivo. In mice, OAA promotes brain mitochondrial biogenesis, activates the insulin signaling pathway, reduces neuroinflammation and activates hippocampal neurogenesis.
INTRODUCTION
Declining bioenergetic function is recognized as a cause, component or consequence of brain aging and neurodegeneration (1). Respiratory chain enzyme activities, specifically those with mitochondrial DNA (mtDNA)-encoded components, fall with advancing age (2). In normal brain aging, this may induce presumably compensatory changes such as increased brain mtDNA levels (3). For age-related diseases such as Alzheimer's disease (AD), this compensation is less obvious (4). In AD, PCR-amplifiable mtDNA levels decrease as do mRNA and protein levels of peroxisome proliferator-activated receptor-gamma coactivator 1α (PGC1α), a transcriptional co-activator that promotes mitochondrial biogenesis (5–8). Observations such as these identify mitochondria as therapeutic targets for brain aging, as well as for neurodegenerative diseases that show mitochondrial dysfunction (9).
‘Mitochondrial medicine’ approaches are being considered for the treatment of neurodegenerative diseases (10). One approach is the induction of mitochondrial biogenesis, which could benefit bioenergetic capacity by upregulating aerobic infrastructures. Bioenergetic capacity, though, does not pertain just to mitochondria since ATP and reducing equivalents are also generated in the cytoplasm. For this reason, we have proposed a new term, bioenergetic medicine, which applies to strategies specifically designed to increase cell energy levels regardless of whether they directly or indirectly target mitochondria (11).
One bioenergetic medicine agent we find especially interesting is oxaloacetate (OAA). OAA reportedly reduces hyperglycemia in type II diabetes (12), and extends longevity in C. elegans (13). Largely because OAA participates in the de-amination of glutamate to α-ketoglutarate, some have tested OAA in seizure, stroke and traumatic brain injury rodent models and found therapeutic benefits (14–17).
OAA is an intermediate in several metabolic pathways including the Krebs cycle, gluconeogenesis and glyceroneogenesis. In the cytosol, reduction of OAA to malate oxidizes NADH to NAD+, which should stimulate glycolysis flux, and malate itself can provide carbon skeletons to the mitochondria. Further, OAA is sequentially decarboxylated and phosphorylated in the cytosol by the enzyme phosphoenolpyruvate carboxykinase (PEPCK-C), and overexpressing PEPCK-C in mouse muscle increases muscle mitochondria, endurance and longevity (18).
In this study, we administered OAA to mice in order to test its impact on genes, proteins and pathways that monitor, regulate and mediate brain bioenergetic fluxes. We also ascertained the effect of OAA on brain insulin signaling, inflammation and hippocampal neurogenesis as relationships exist between these parameters and bioenergetics (19,20). We further probed for changes in metabolic fluxes by measuring in vivo levels of some key neurochemicals.
RESULTS
Stability of OAA in solution
To test the stability of OAA in solution, we measured the pyruvate concentration that resulted after exposing the solution to ambient air for 2 h at room temperature. After 2 h, ∼15% of the OAA had decarboxylated to pyruvate (data not shown). Since our OAA solutions were prepared fresh and immediately administered, unless the OAA decarboxylation rate vastly accelerated following intraperitoneal (IP) injection OAA was the main component of our treatments.
Effect of OAA on genes and proteins related to mitochondrial biogenesis and bioenergetics
Mitochondrial biogenesis is regulated through the PGC1-family proteins (21). These co-transcriptional activators, which promote mitochondrial biogenesis, include PGC1α, PGC1β and PGC1 related co-activator (PRC). Brain PGC1α mRNA levels were 58% higher in the 2 g/kg/day group than they were in the control group, and for the combined OAA group (all mice treated with OAA, whether at 1 or 2 g/kg/day), it was 37% higher (Fig. 1A). Although brain PGC1α protein levels did not increase to a statistically significant extent (Supplementary Material, Table S1 and Fig. S1A), PGC1α protein within cells redistributed such that its nucleus:cytoplasm ratio increased; the nucleus:cytoplasm ratio approximately doubled in both the 1 and 2 g/kg/day groups (Fig. 1B, Supplementary Material, Fig. S1A and Table S1).
Figure 1.

Effect on the PGC1-family. (A) PGC1α mRNA was higher in the 2 g/kg/day and combined OAA groups. (B) The PGC1α nucleus:cytoplasm ratio was higher in the 1 g/kg/day, 2 g/kg/day, and combined OAA groups. (C) PRC mRNA was higher in the 2 g/kg/day OAA group. (D) On post hoc analysis, the PRC nucleus:cytoplasm ratio was higher in the 2 g/kg/day OAA group. (E) PGC1β mRNA levels were comparable. (F) PGC1β protein levels were comparable. Values shown are relative group means ± SEM. *P < 0.05; **P < 0.005; #ANOVA comparison was not significant, but the post hoc LSD test between the specified treatment group and the control group was significant at P < 0.05.
Brain PRC showed similar changes. PRC mRNA was 47% higher in the mice receiving 2 g/kg/day than it was in the control mice (Fig. 1C). For the PRC nucleus:cytoplasm ratio, while the analysis of variance (ANOVA) value was not significant (P = 0.09), a post hoc analysis suggested the PRC nucleus:cytoplasm ratio was higher (by 70%) in the 2 g/kg/day group (Fig. 1D, Supplementary Material, Fig. S1A). This difference was primarily driven by a relative decrease in the cytosolic PRC protein level (Supplementary Material, Table S1 and Fig. S1A).
No statistically significant changes were seen in brain PGC1β mRNA levels, PGC1β protein levels or PGC1β protein distribution (Fig. 1E and F, Supplementary Material, Table S1). Despite this, mice treated with OAA showed PGC1β trends that reflected the PGC1α and PRC changes. For OAA-treated mice, the mean mRNA levels were ∼1.25 (versus 1.00 in the control group), and the mean nucleus:cytoplasm ratio in the 2 g/kg group was 2.00 (versus 1.00 in the control group).
Other evidence suggested OAA-stimulated brain mitochondrial biogenesis. For the combined OAA group, mRNA levels of nuclear respiratory factor 1 (NRF1), a PGC1α-co-activated transcription factor that promotes the expression of nuclear genes that encode mitochondrial proteins (21), increased by 20% (Fig. 2A). Measurements of transcription factor A of the mitochondria (TFAM), a gene whose expression is driven by the PGC1α–NRF1 complex (21), validated this change. Although the TFAM mRNA level ANOVA was not significant (P = 0.06), post hoc analysis suggested TFAM mRNA was higher (by 30%) in the 2 g/kg/day OAA group (Fig. 2B). OAA treatment also increased cytochrome oxidase subunit 4 isoform 1 (COX4I1) mRNA and protein levels. COX4I1 mRNA rose ∼45% in the OAA-treated groups (Fig. 2C). The COX4I1 protein was 60% higher in the 1 g/kg/day group, and 30% higher in the 2 g/kg/day group (Fig. 2D, Supplementary Material, Fig. S1B).
Figure 2.

Effect on other mitochondrial biogenesis-related parameters. (A) NRF1 mRNA was higher in the combined OAA group. (B) On post hoc analysis, TFAM mRNA was higher in the 2 g/kg/day group. (C) COX4I1 mRNA was higher in the 1 g/kg/day, 2 g/kg/day and combined OAA groups. (C) Cytoplasmic COX4I1 protein was higher in the 1 g/kg/day, 2 g/kg/day and combined OAA groups. Values shown are relative group means ± SEM. *P < 0.05; **P < 0.005; #ANOVA comparison was not significant, but the post hoc LSD test between the specified treatment group and the control group was significant at P < 0.05.
Significant changes in other mitochondrial biogenesis-related gene expression were not observed in the brains we studied. Nuclear respiratory factor 2 (NRF2), NADH Dehydrogenase Ubiquinone Fe-S Protein 2 (NDUFS2), NADH dehydrogenase subunit 2 (ND2) and cytochrome oxidase subunit 2 (COX2) mRNA levels were comparable across the groups (Supplementary Material, Table S2). TFAM protein levels were unchanged (Supplementary Material, Table S1). mtDNA levels were also comparable between the groups (Table 1).
Table 1.
Relative mtDNA copy numbers
| Control (±SEM) | 1 g/kg (±SEM) | 2 g/kg (±SEM) | Combined (±SEM) | |
|---|---|---|---|---|
| ND2/18s rRNA | 1.00 ± 0.18 | 1.05 ± 0.20 | 1.09 ± 0.24 | 1.07 ± 0.15 |
| 16s /18s rRNA | 1.00 ± 0.20 | 1.21 ± 0.26 | 1.11 ± 0.27 | 1.16 ± 0.18 |
PGC1α is regulated by other proteins that are sensitive to bioenergetic states. HIF1α facilitates glycolysis flux, generally counters aerobic flux and may negatively regulate PGC1α (22). Brain HIF1α protein levels did not increase and no intracellular redistribution was observed (Supplementary Material, Table S1). Silent information regulator of transcription 1 (SIRT1) is a deacetylase that activates PGC1α (23). SIRT1 mRNA levels did not change (Supplementary Material, Table S1). No statistically significant differences in SIRT1 protein levels or distribution were seen, although cytoplasmic SIRT1 protein perhaps trended lower in the OAA-treated mice (P = 0.08 between the control and both the 2 g/kg/day and combined OAA groups) (Supplementary Material, Table S1).
Adenosine monophosphate-activated protein kinase (AMPK) monitors cell AMP:ATP, low AMP:ATP ratios and increased cytosolic calcium promote AMPK phosphorylation at Thr172, and Thr172 phosphorylation positively correlates with its kinase activity (24). PGC1α is an AMPK target, and AMPK-mediated PGC1α phosphorylation activates PGC1α (25). We observed increased brain AMPK Thr172 phosphorylation in both the 1 g/kg/day OAA (3-fold increase) and combined OAA groups (2-fold increase) (Fig. 3A and B, Supplementary Material, Fig. S1C). Similarly, the p38 MAPK phosphorylates PGC1α and this too is an activating phosphorylation (26). We detected evidence of brain p38 MAPK activation in the 1 g/kg/day OAA group as p38 phosphorylation, which positively correlates with its activation state, increased by 92% when normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Fig. 3D, Supplementary Material, Fig. S1C). p38 phosphorylation was not significantly increased after normalizing levels to total MAPK, perhaps because total p38 also trended higher in the 1 g/kg/day OAA group (Fig. 3C, Supplementary Material, Fig. S1C and Table S1). Finally, phosphorylation at Ser133 of cAMP-response element binding (CREB) protein, a modification that directly correlates with its activation state (27), occurred within the brain (Fig. 3E and F, Supplementary Material, Fig. S1C). CREB is known to stimulate PGC1α gene expression (28).
Figure 3.

Effect on proteins that regulate PGC1α. (A) Normalized to total AMPK, AMPK was more phosphorylated at Thr172 in the 1 g/kg/day and combined OAA groups. (B) Normalized to GAPDH, AMPK was more phosphorylated at Thr172 in the 1 g/kg/day and combined OAA groups. (C) Normalized to total p38 protein, levels of p38 phosphorylation at Thr180/Tyr182 were comparable across groups. (D) Normalized to GAPDH protein, p38 phosphorylation was greater at Thr180/Tyr182 in the 1 g/kg/day OAA group. (E) Normalized to total CREB, CREB was more phosphorylated at Ser133 in the 1 g/kg/day, 2 g/kg/day and combined OAA groups. (F) When normalized to GAPDH, CREB was more phosphorylated at Ser133 in the combined OAA group. Although the ANOVA was not significant, on post hoc analysis CREB phosphorylation was higher in the 2 g/kg/day OAA group than in the control group. Values shown are relative group means ± SEM. *P < 0.05; **P < 0.005; #ANOVA comparison was not significant, but the post hoc LSD test between the specified treatment group and the control group was significant at P < 0.05.
Effect of OAA on insulin signaling pathway proteins
A tissue's bioenergetic status influences its sensitivity to insulin, or at least the activation state of its insulin signaling pathway (19). We therefore assessed OAA's impact on that pathway in the brains of our OAA-treated and untreated mice. Part of the signal produced when insulin binds its receptor is transmitted via Akt, which is activated through phosphorylation of its Ser473 site (29). Akt Ser473 phosphorylation increased in a dose-dependent manner; with the 2 g/kg/day treatment there was more than a 3-fold increase (Fig. 4A and B, Supplementary Material, Fig. S1D). Total Akt levels were unchanged (Supplementary Material, Table S1 and Fig. S1D). The insulin pathway also includes mammalian target of rapamycin (mTOR), and the activity of the pathway positively correlates with the amount of mTOR phosphorylation at its Ser2448 site (30). mTOR Ser2448 phosphorylation increased; at the 2 g/kg/day dose, the phospho-Ser2448 mTOR/total mTOR ratio rose almost 3-fold (Fig. 4C and D, Supplementary Material, Fig. S1D). The amount of total mTOR protein and its nucleus:cytoplasm distribution were unchanged (Supplementary Material, Table S1 and Fig. S1D). Thr389 phosphorylation of P70S6K, a protein which is likely both a downstream target of mTOR and a protein that phosphorylates mTOR (31), also increased in OAA-treated mice (by 81% in the 1 g/kg/day group and by 70% in the combined OAA group) (Fig. 4E and F, Supplementary Material, Fig. S1D), while total P70S6K protein levels remained comparable across the groups (Supplementary Material, Table S1 and Fig. S1D).
Figure 4.

Effect on proteins involved in insulin signaling. (A) Normalized to total Akt, Akt was more phosphorylated at Ser473 in the 2 g/kg/day and combined OAA groups. (B) Normalized to GAPDH, Akt was more phosphorylated at Ser473 in the 1 g/kg/day, 2 g/kg/day and combined OAA groups. (C) Normalized to total mTOR protein, mTOR was more phosphorylated at Ser2448 in the 2 g/kg/day and combined OAA groups. (D) Normalized to GAPDH, mTOR was more phosphorylated at Ser2448 in the 2 g/kg/day and combined OAA groups. (E) Normalized to total P70S6K, P70S6K was more phosphorylated at Thr389 in the 1 g/kg/day and combined OAA groups. (F) Normalized to GAPDH, levels of P70S6K phosphorylation at Thr389 were comparable between the groups. Values shown are relative group means ± SEM. *P < 0.05; **P < 0.005.
Effect of OAA on brain inflammation
To assess the effects of OAA administration on brain inflammation, we measured the mRNA levels for two inflammation-related genes, tumor necrosis factor α (TNFα) and C–C motif chemokine 11 (CCL11) (32,33). While hippocampal TNFα levels were comparable across the groups, hippocampal CCL11 mRNA levels were 44% lower in the group receiving 2 g/kg/day than they were in the control group (Fig. 5A and B). The amount of nuclear factor κB (NFκB) protein, a transcription factor that translocates to the nucleus when inflammation is present (34), was lower in the nucleus of the combined OAA treatment group (by 50%), and in both OAA treatment groups the nucleus-to-cytoplasm ratio was ∼70% lower than it was in the control group (Fig. 5C and D, Supplementary Material, Fig. S1E).
Figure 5.

Effect on inflammation. (A) TNFα mRNA levels were comparable. (B) CCL11 mRNA was lower in the 2 g/kg/day OAA group. (C) Nuclear NFκB protein was lower in the combined OAA group. Although the ANOVA was not significant, on post hoc analysis nuclear NFκB protein was lower in the 1 g/kg/day OAA group. (D) The NFκB nucleus:cytoplasm ratio was lower in the 1 g/kg/day, 2 g/kg/day and combined OAA groups. Values shown are relative group means ± SEM. *P < 0.05; **P < 0.005; #ANOVA comparison was not significant, but the post hoc LSD test between the specified treatment group and the control group was significant at P < 0.05.
Effect of OAA on neurogenesis
Neurogenesis persists in the adult mouse brain (35), and one site that maintains neurogenesis throughout even late human adulthood is the subgranular layer of the hippocampal dentate gyrus (36,37). It was previously reported in mice that CCL11 retards subgranular layer neurogenesis (33). Because CCL11 gene expression was reduced in the hippocampus of OAA-treated mice, and because Akt and mTOR activation promotes cell growth (29,38), we evaluated hippocampal neurogenesis. We measured hippocampal mRNA levels for two genes recognized to promote neurogenesis, brain-derived neurotrophic factor (BDNF) and vascular endothelial growth factor A (VEGF) (39,40). Although BDNF gene expression was unchanged, VEGF gene expression was 25% higher in the combined OAA group than it was in the control group (Fig. 6A and B). The VEGF protein level itself was not elevated (Supplementary Material, Table S1), but it is important to note mRNA was measured from a hippocampus-enriched region, while protein was measured from hemisphere tissue. We further measured mRNA levels for doublecortin (DCX), a gene whose protein product indicates neuron immaturity (41). DCX can be used as a surrogate measure of new neuron formation, with higher levels suggesting increased neurogenesis (42). Relative to the control group, DCX mRNA was increased by 81% in the 1 g/kg/day OAA group and by 48% in the 2 g/kg/day group (Fig. 6C).
Figure 6.
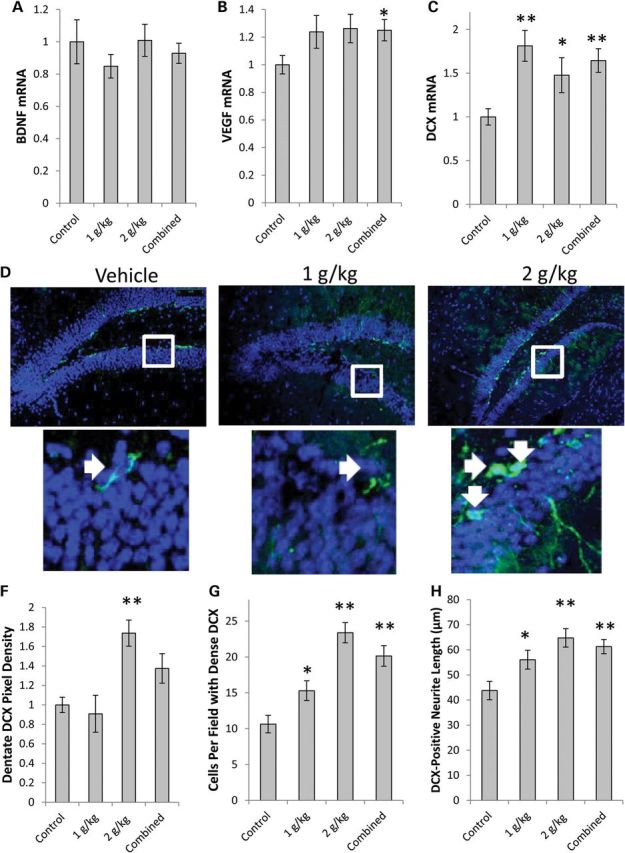
Effect on growth factors and neurogenesis. (A) BDNF mRNA was comparable. (B) VEGF mRNA was higher in the combined OAA group. (C) DCX mRNA was higher in the 1 g/kg/day, 2 g/kg/day and combined OAA groups. (D) Representative confocal microscopy pictures of the hippocampal dentate gyrus. The top image used the ×10 objective, the bottom image represents an enlarged view of the boxed area from the 10× picture. (E) Dentate gyrus DCX pixel density was higher in the 2 g/kg/day OAA group. (F) Mice in the 1 g/kg/day, 2 g/kg/day and combined OAA groups had more intensely DCX-positive neurons. (G) Mice in the 1 g/kg/day, 2 g/kg/day and combined OAA groups had longer DCX-positive neurites. Values shown are relative group means ± SEM. *P < 0.05; **P < 0.005.
Next, we prepared sections of the remaining hippocampus (the right hippocampus). We were able to identify a relatively intact dentate gyrus in sections from 10 control, seven 1 g/kg/day, and nine 2 g/kg/day OAA-treated mice. These sections were stained with a primary antibody to DCX (Fig. 6D). Relative to the control group, DCX pixel density in the dentate gyrus was 73% higher in the 2 g/kg/day OAA group (Fig. 6E). The number of DCX-positive neurons was increased by 44% in the 1 g/kg/day group and by 120% in the 2 g/kg/day group (Fig. 6F). Relative to the control group, DCX-positive neurites were 28% longer in the 1 g/kg/day OAA group, and 48% longer in the 2 g/kg/day OAA group (Fig. 6G).
Effect of OAA on in vivo levels of key neurochemicals
Proton magnetic resonance spectroscopy (1H-MRS) was performed on mice before receiving any treatment, and on the same mice after they received 2 g/kg/day OAA IP for 1 week (the 1 g/kg/day dose was not studied). OAA induced changes in several brain biochemical intermediates. After 1 week of OAA, brain lactate, GABA and glutathione (GSH) levels were respectively 21, 15, and 27% higher than they were on the pre-treatment scans (Fig. 7A–C). Non-significant trends toward increased glutamate (6% mean increase, P = 0.14) and glutamine (13% mean increase, P = 0.07) were observed, while aspartate was essentially unchanged (1% mean decrease after treatment, P = 0.95) (Fig. 7D–F).
Figure 7.

Pre- and post-OAA brain MRS. (A) Lactate was higher post-treatment. (B) GABA was higher post-treatment. (C) Glutathione was higher post-treatment. (D) Glutamate did not change. (E) Glutamine did not change. (F) Alanine did not change.
DISCUSSION
We administered OAA to mice and tested its ability to alter brain bioenergetic infrastructures, insulin signaling, inflammation and neurogenesis. OAA increased levels of a mitochondrial protein and positively altered proteins that up-regulate mitochondrial mass, activated the insulin signaling pathway, reduced two inflammation-related parameters and increased new neuron formation in the hippocampus. Figure 8 summarizes these effects and their relationships.
Figure 8.

OAA effects and inter-effect relationships. OAA, a bioenergetic intermediate, affects bioenergetic flux. This produces a number of molecular changes. CREB phosphorylation and CREB activity increase, which in turn promotes the expression of PGC1 family member genes. AMPK and p38 MAP phosphorylation increase, and these activated kinases enhance PGC1α co-activator function. PGC1-induced co-activation of the NRF1 transcription factor stimulates COX4I1 production, while PGC1-induced co-activation of the ERRα transcription factor stimulates VEGF gene expression (61). OAA-induced flux changes also stimulate the pro-growth insulin signaling pathway and reduce inflammation. The pro-growth effects of increased insulin pathway signaling and increased VEGF, in conjunction with a more favorable bioenergetic status and less inflammation, cooperatively stimulate hippocampal neurogenesis.
While some bioenergetic intermediates such as lactate or pyruvate can enhance respiration, these metabolites lie at the end of the glycolysis pathway and increasing levels can reduce glycolysis flux (43). OAA is not a glycolysis intermediate, and is less likely to inhibit glycolysis. Oxidation of OAA to malate converts NADH to NAD+, which may enhance glycolysis flux, increase mitochondrial mass and mimic caloric restriction (44,45). Malate, OAA's redox partner, crosses mitochondrial membranes and can have anaplerotic or cataplerotic effects on the Krebs cycle (46).
OAA induced changes that suggest an overall pro-mitochondrial effect. Levels of COX4I1 protein, a nuclear-encoded cytochrome oxidase subunit, increased. While this by itself does not prove increased mitochondrial biogenesis, elevated mRNA copy numbers for two PGC1 family members (PGC1α and PRC) were observed. PGC1α and PRC protein also shifted to the nucleus. These changes would arguably support mitochondrial biogenesis. Increased TFAM and NRF1 mRNA levels are consistent with this view. CREB, a transcription factor that promotes PGC1α expression (28), was more extensively phosphorylated at a residue that reflects its activity. AMPK and p38 MAPK proteins were more extensively phosphorylated at residues that correlate with their activation state, and AMPK and p38 MAPK in turn activate PGC1α (25,26). For these reasons, we conclude that mitochondrial biogenesis was at least to some extent activated. This effect could prove useful for treating brain diseases in which mitochondrial mass parameters and PGC1α levels decline, as they do in AD (7,8).
It is nevertheless important to note the most straightforward pro-mitochondrial biogenesis changes we report occurred at the mRNA level. In some cases, increases in specific mRNA molecules were accompanied only by inter-compartmental shifts of the corresponding protein; in other cases, no corresponding changes in the corresponding protein (either at the total level or in the distribution pattern) were observed. These outcomes, we believe, at least partly reflect the complexities of documenting mitochondrial biogenesis. For instance, we previously found mitochondria in different tissues, when stimulated to increase their mitochondrial mass, can selectively up-regulate only the components that are functionally needed at that time (47). Further to this point, while the brains of elderly mice certainly do manifest evidence of bioenergetic stress (2), the brains of the young mice used in this study presumably lacked bioenergetic stress and may have limited the up-regulation of mitochondrial components (including specific proteins and mtDNA). Also, the function of proteins that mediate mitochondrial biogenesis may be influenced more by post-translational modifications than by absolute levels, and our study was not designed to comprehensively characterize the post-translational modifications of all the proteins we analyzed. Finally, from an anatomic perspective, our gene expression studies utilized a hippocampus-enriched region, while our protein and mtDNA studies excluded the hippocampus. Overall, therefore, we believe the pattern of changes observed support the conclusion that mitochondrial biogenesis signaling was at least activated, and for some endpoints (for example, COX protein) successfully executed.
Insulin signaling is influenced by bioenergetics (19). In at least some tissues, impaired mitochondrial function causes insulin resistance (48). In AD subjects, brain insulin pathway signaling is markedly reduced, and for this reason some investigators consider AD a form of diabetes (49). OAA enhanced the brain's insulin signaling pathway as evidenced by an increase in Akt Ser473 phosphorylation, mTOR Ser2448 phosphorylation and P70S6K Thr389 phosphorylation. We expect this reflects intracellular effects rather than a direct impact on insulin receptors. This property could prove useful for treating brain diseases in which the insulin signaling pathway is de-activated.
Neuroinflammation occurs in AD and other neurodegenerative diseases (50). In various tissues, inflammation has been linked to damaged mitochondria or declines in mitochondrial function (51–53). We hypothesized, therefore, that promoting mitochondrial function would reduce inflammatory markers. We did not detect a statistically significant change in TNFα mRNA, perhaps because in young mice microglial TNFα expression is already very low. mRNA levels of CCL11, though, a chemokine produced by macrophages and neurons (54), were lower in OAA-treated mice.
Consistent with this, nuclear protein lysates from OAA-treated mice contained less NFκB protein, as well as lower NFκB nuclear:cytoplasm ratios. NFκB, a transcription factor, is restrained within the cytoplasm until activated by inflammatory cytokines and other cell-stress events (55). When activated, NFκB's restraint is removed and it moves to the nucleus. Since NFκB promotes the CCL11 gene (56), reduced NFκB may account for lower CCL11 expression. OAA, therefore, could prove useful for treating brain diseases in which neuroinflammation occurs.
In humans and other mammals, stem cells in the subgranular layer of the hippocampal dentate gyrus generate new neurons throughout adulthood (35–37,57). The rate of neurogenesis declines to some extent with advancing age (35,36). It is speculated this rate may further fall in AD, but few studies have tried to address this question (58,59). We found that protein and mRNA levels of DCX increased in the hippocampi of OAA-treated mice, the number of intensely DCX-positive neurons rose, and the length of DCX-positive neurites increased. We did not ascertain the mechanistic basis for these pro-neurogenesis changes, but it is worth noting CCL11 levels inversely correlate with hippocampal neurogenesis (33) and OAA treatment lowered hippocampal CCL11 gene expression. In addition, OAA treatment enhanced the brain insulin signaling pathway, which is a pro-growth pathway. Finally, VEGF mRNA levels were higher in hippocampi from OAA-treated mice. VEGF is mostly recognized for its role in angiogenesis, but it also promotes neurogenesis (40,60). VEGF expression is regulated by HIF1α (61), a transcription factor which did not appear altered by OAA, but also by PGC1α (62), which OAA seemed to positively effect.
While relationships between hippocampal neurogenesis and memory are not entirely understood (63), in aged rodents promoting hippocampal neurogenesis associates with improved performance on memory tasks (35). Therefore, this effect may prove useful for treating AD, a disorder clinically characterized by hippocampal atrophy and memory decline.
1H-MRS revealed increased lactate levels several hours after an OAA IP injection, which could indicate an increased glycolysis flux. The amount of glutathione also increased. The mechanisms underlying the glutathione increase are unclear, but could be consistent with less consumption or more production. GABA increased and trends toward increased glutamate and glutamine were observed. This stands in possible contrast to a previous study in which rats administered OAA underwent 1H-MRS analysis. That study reported OAA attenuated an increase in brain glutamate that occurred following a middle cerebral artery occlusion procedure (14). The explanation provided for the glutamate reduction seen in that study was that OAA likely reacted with glutamate to generate α-ketoglutarate and aspartate. Discrepant results between our study and the other MRS study may reflect experimental differences. Overall, though, our MRS results suggest this technology can be used to monitor target engagement in the brains of living organisms.
The brain is an anatomically and metabolically heterogeneous construct. Neurons are respiration-dependent and rely primarily on mitochondria to generate ATP (64). Astrocytes rely more on glycolysis, especially in their filopodial and lamellipodial extensions (65). The bioenergetics of these two cell types are presumably linked, as glutamatergic transmission at neuron synapses likely activates astrocyte glycolysis and lactate produced through astrocyte glycolysis probably fuels neuron respiration (66). Evidence of bioenergetic interplay between oligodendrocytes and neurons was also recently demonstrated (67).
Our gene expression studies utilized a hippocampus-enriched region, while our protein and mtDNA studies excluded the hippocampus. This could account for occasional observed discordance between our gene expression and protein data. We cannot exclude the possibility that pyruvate, the decarboxylation product of OAA, mediated some effects. However, because pyruvate levels increased only to a limited extent after OAA was placed into solution, we expect OAA was primarily responsible. We cannot exclude the possibility that observed effects were an indirect consequence of a biochemical change arising outside the brain (68), but due to the extent of the observed brain changes we think indirect effects could account for only a limited part of the observed changes.
This study was intended to pre-clinically evaluate a new potential treatment for AD and other neurodegenerative diseases. AD and other neurodegenerative diseases, though, are chronic disorders and the treatment duration in our study lasted only 2 weeks. Should clinical studies show a beneficial effect, chronic treatment would likely be required. If so, the route through which OAA is delivered could impact this intervention's feasibility. Our study also has implications for the recently defined bioenergetic medicine field (11), which emphasizes that bioenergetic intermediates can be used to manipulate bioenergetic fluxes and this, in turn, can have protean molecular effects. In this respect, it should be noted that altering the concentrations of other bioenergetic intermediates, for example increasing levels of beta-hydroxybutyrate through ketogenic diets or by simply administering medium chain triglycerides, has been proposed and studied for the treatment of AD (69–71).
We do not know what blood and brain levels resulted from the IP injections we administered. Addressing this will facilitate the planning of rational clinical trials that test the efficacy of OAA in human subjects with AD and other neurodegenerative diseases. Overall, though, as systemically administered OAA appears to activate brain bioenergetics-related proteins, enhance the insulin signaling pathway, reduce neuroinflammation and stimulate neurogenesis, further therapeutic development is indicated.
MATERIALS AND METHODS
Mice and OAA treatments
The University of Kansas Medical Center's Institutional Animal Care and Use Committee approved these experiments. Male, 5-month-old C57Bl/6 mice from the Jackson Laboratory were accommodated to our vivarium for 1 week, housed up to five per cage in clear plastic cages on a 12 : 12 h light:dark schedule and given ad libitum access to standard chow and water.
We tested the stability of our OAA solutions by using a colorimetric-based kit (Eton Bioscience) that measures the pyruvate concentration of pyruvate, the decarboxylation product of OAA, in solution. Immediately prior to use, OAA solutions were prepared by dissolving OAA in phosphate-buffered saline (PBS) and pH-adjusted with NaOH. For 1 g/kg/day OAA injections, 600 mg of OAA was suspended in 10 ml of PBS (0.45 m) and pH-adjusted to 7.0. For 2 g/kg/day OAA injections, 1.2 g of OAA was suspended in 10 ml of PBS (0.9 m) and pH-adjusted to 7.0. A target pH of 7.0 was used because over time the pH of the OAA solutions drifted toward more alkaline levels. The vehicle-treated group received PBS. For each group, the total volume received via IP injection was 500 µl. 1 and 2 g/kg doses were selected based on a previous study that reported 1 g/kg IP OAA injections affect mouse seizure threshold (16), and our own preliminary studies that showed C57Bl/6 mice routinely survived 2 g/kg but not 3 g/kg OAA IP injections. For all studies except the 1H-MRS studies, the treatment period lasted 2 weeks; for 1H-MRS studies the treatment period lasted 1 week.
Immunochemistry, gene expression and mtDNA levels
Mice analyzed for immunochemistry, gene expression and mtDNA levels were treated for 2 weeks. Each group contained 15 mice, and all 45 mice completed the 2-week treatment period. At the end of the treatment period, mice were euthanized by decapitation 2 h after receiving their final injection. Brains were rapidly removed. The left hemisphere was immediately placed into RNA-Later Solution (Ambion), and kept overnight at 4o. The following morning, the hippocampus was identified, isolated, returned to RNA-Later solution and stored at −80°. This region was subsequently used to isolate RNA. The remainder of the left hemisphere was placed into a separate tube containing RNA-Later solution and stored at −80°. This tissue was later used to prepare protein lysates. For the right hemisphere, we estimated the approximate location of the hippocampus, removed that region and placed it in 4% paraformaldehyde overnight at 4°. This section of fixed tissue was later used for immunohistochemistry as described below. The remaining right hemisphere tissue was placed in RNA-Later solution and stored at −80°. A piece of this tissue was later used to isolate DNA.
Protein lysates were prepared from the left hemisphere cortices using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific), according to the manufacturer's instructions. Protein concentrations were measured using a BCA protein assay kit (Thermo Scientific). Protein aliquots were electrophoresed within 4–20% Criterion TGX Tris-glycine polyacrylamide gels (Bio-Rad) and transferred to nitrocellulose membranes (Invitrogen). Blots were stained with primary antibodies to the proteins listed in Supplementary Material, Table S3. To ensure equivalent protein loading, blots with cytosolic lysates were stained with an antibody to GAPDH, while blots with nuclear lysates were stained using an antibody to histone deacetylase 1 (HDAC1) or histone 3 (H3). Primary antibody binding was detected using horseradish peroxidase-conjugated secondary antibodies (1 : 2000 dilution; Cell Signaling Technology) and SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Scientific). Densitometry was performed using a ChemiDoc XRS with Quantity One software (Bio-Rad).
Total RNA was prepared from the left hippocampus-containing region using TRI Reagent (Life Technologies). Reverse transcription was performed on total RNA (1 μg) using an iScript RT qPCR master mix (Bio-Rad). Amplifications were performed using an Applied Biosystems 7300 PCR System. Quantitative real-time, reverse-transcription PCR (qPCR) used iTaq universal probes supermix (Bio-Rad) and TaqMan Gene Expression Assays (Applied Biosystems) to quantify mRNA levels for the following genes: PGC1α, PGC1β, PRC, NRF1, NRF2, TFAM, SIRT1, CREB, NDUFS2, COX4I1, COX2, ND1, TNFα, CCL11, BDNF, VEGF and DCX. Actin expression served as a reference.
To quantify brain mtDNA, ∼30 mg of hemisphere tissue was placed in 600 µl of lysis buffer (10 mm Tris–HCl pH 8.0, 1 mm EDTA, 0.1% SDS) and Dounce homogenized using 10 strokes. Homogenized tissue was transferred to a microcentrifuge tube, 60 µl of proteinase K (Qiagen) was added and the tubes were vortexed and incubated at 55° for 2 h. Samples were vortexed every 30 min during the 2 h incubation period. Samples were next centrifuged at 8000g for 15 min, and the supernatants were transferred to new tubes. Six hundred microliters of phenol:chloroform:isoamyl alcohol (Sigma) was added and the tubes were vortexed and then centrifuged at 8000g for 15 min. Supernatants were transferred to new tubes, 500 µl of chloroform (Sigma) added and samples were vortexed and centrifuged at 8000g for 15 min. The supernatants were transferred to new tubes, 40 µl of 3 m sodium acetate and 440 µl of 100% isopropanol were added, and samples were incubated at −20°C overnight. Samples were next centrifuged at 8000g for 15 min, the supernatants were discarded and the pellets were washed with 750 µl of 70% ethanol and centrifuged at 8000g for 8 min. Supernatants were discarded, and the pellets were washed with 500 µl of 70% ethanol and centrifuged at 8000g for 8 min. The pellets were gently air dried and re-suspended in 100 µl of nuclease-free water. To obtain relative mtDNA:nuclear DNA ratios, TaqMan Gene Expression Assays for two mtDNA-encoded genes, ND2 and 16s ribosomal RNA (rRNA), and the nuclear 18s rRNA gene were used. Ratios were determined using the comparative ΔΔCT method, in which ND2:18s rRNA and 16s rRNA:18s rRNA ratios were calculated.
Immunohistochemistry
Tissue was fixed in 4% paraformaldehyde overnight and cryopreserved in sequential incubations overnight in 10% sucrose, 20% sucrose and finally 30% sucrose at 4°. Tissue was flash-frozen in Optimum Cutting Temperature compound. Ten micrometer sections were obtained using a cryostat set to −25°, and tissue sections were transferred to Superfrost Plus microscope slides (Fisher Scientific). Antigen retrieval was completed using sodium citrate buffer (10 mm plus 0.05% Tween-20, pH 6.0) for 10 min at 100°C. Slides were equilibrated to room temperature for 20 min and washed in PBS three times. Slides were then stained with the DCX primary antibody (Abcam ab18723, 1:500), an Alexa Flour 488 secondary antibody (Abcam ab150073, 1:500) and ProLong Gold Antifade reagent with DAPI (Invitrogen, Life Technologies).
Images were captured using a Leica TCS SPE confocal microscope platform with an LAS AF interface at 10X. Each section that was subsequently found to contain recognizable dentate gyrus was analyzed. All images were captured using the same parameters. Image analysis was completed using LAS AF lite software.
In vivo 1H-MRS
1H-MRS scans were obtained at 9.4 Tesla (Varian Inova) on 8 isoflurane-anesthetized mice before and after a 1 week 2 g/kg/day IP OAA course. Post-treatment brain scans were acquired 3.5 h after the final OAA injection. Scanning methods were adapted from Harris et al. (72). Briefly, mice were placed on a heating pad in the scanning cradle and core temperature was maintained at 37°C. Isoflurane was adjusted (1–2%) to maintain a respiration rate of 110–160 per min. Gradient echo multi-slice images were used to position the mouse's head in the magnet isocenter (TR = 65 ms, TE = 2.8 ms), followed by rapid acquisition with relaxation enhancement T2-weighted images (TR = 4000 ms, TE = 0.01 ms, echo train length = 16, slices = 17, slice thickness = 0.5 mm) used to position the voxel for 1H-MRS. MR spectra were acquired from a 2.2 × 1.2 × 2.0 mm voxel over the left hippocampus using a SPECIAL sequence with TE = 3 ms and TR = 4000 ms (73). Spectra were analyzed with LCModel software, using the unsuppressed water signal for each scan to calculate absolute metabolite concentrations as previously described (74).
Statistics
Data were summarized by means and standard errors. To compare means between three groups, we used one-way ANOVA followed by Fisher's least significant difference (LSD) post hoc testing. For non-MRS analyses, to compare means between two groups we used two-way, unpaired Student's t-tests, with P-values of <0.05 considered significant. For MRS analyses, data were compared using two-way, paired t-tests. Statistical tests were performed using SPSS 18.0 (SPSS Inc). P-values <0.05 were considered statistically significant.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by NS077852, the University of Kansas Alzheimer's Disease Center (P30AG035982), the Frank and Evangeline Thompson Alzheimer's Therapy Program, the Hugh and Betty Libby Foundation, the Morgan Family Foundation, the Greater Kansas City Automobile Dealers Association and the Gene and Marge Sweeney Chair.
Supplementary Material
ACKNOWLEDGEMENTS
Experimental work was facilitated by the University of Kansas Alzheimer's Disease Center's Neuroimaging Core and Mitochondrial Genomics and Metabolism Core.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Swerdlow R.H. Brain aging, Alzheimer's disease, and mitochondria. Biochim. Biophys. Acta. 2011;1812:1630–1639. doi: 10.1016/j.bbadis.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Navarro A., Boveris A. The mitochondrial energy transduction system and the aging process. Am. J. Physiol. Cell Physiol. 2007;292:C670–C686. doi: 10.1152/ajpcell.00213.2006. [DOI] [PubMed] [Google Scholar]
- 3.Barrientos A., Casademont J., Cardellach F., Estivill X., Urbano-Marquez A., Nunes V. Reduced steady-state levels of mitochondrial RNA and increased mitochondrial DNA amount in human brain with aging. Brain Res. Mol. Brain Res. 1997;52:284–289. doi: 10.1016/s0169-328x(97)00278-7. [DOI] [PubMed] [Google Scholar]
- 4.Swerdlow R.H. Mitochondria and cell bioenergetics: increasingly recognized components and a possible etiologic cause of Alzheimer's disease. Antioxid. Redox Signal. 2012;16:1434–1455. doi: 10.1089/ars.2011.4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hirai K., Aliev G., Nunomura A., Fujioka H., Russell R.L., Atwood C.S., Johnson A.B., Kress Y., Vinters H.V., Tabaton M., et al. Mitochondrial abnormalities in Alzheimer's disease. J. Neurosci. 2001;21:3017–3023. doi: 10.1523/JNEUROSCI.21-09-03017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de la Monte S.M., Luong T., Neely T.R., Robinson D., Wands J.R. Mitochondrial DNA damage as a mechanism of cell loss in Alzheimer's disease. Lab Invest. 2000;80:1323–1335. doi: 10.1038/labinvest.3780140. [DOI] [PubMed] [Google Scholar]
- 7.Qin W., Haroutunian V., Katsel P., Cardozo C.P., Ho L., Buxbaum J.D., Pasinetti G.M. PGC-1alpha expression decreases in the Alzheimer disease brain as a function of dementia. Arch. Neurol. 2009;66:352–361. doi: 10.1001/archneurol.2008.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sheng B., Wang X., Su B., Lee H.G., Casadesus G., Perry G., Zhu X. Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer's disease. J. Neurochem. 2012;120:419–429. doi: 10.1111/j.1471-4159.2011.07581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Swerdlow R.H. Treating neurodegeneration by modifying mitochondria: potential solutions to a ‘complex’ problem. Antioxid. Redox Signal. 2007;9:1591–1603. doi: 10.1089/ars.2007.1676. [DOI] [PubMed] [Google Scholar]
- 10.Swerdlow R.H. Role and treatment of mitochondrial DNA-related mitochondrial dysfunction in sporadic neurodegenerative diseases. Curr. Pharm. Des. 2011;17:3356–3373. doi: 10.2174/138161211798072535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Swerdlow R.H. Bioenergetic medicine. Br. J. Pharmacol. 2014;171:1854–1869. doi: 10.1111/bph.12394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoshikawa K. Studies on the anti-diabetic effect of sodium oxaloacetate. Tohoku J. Exp. Med. 1968;96:127–141. doi: 10.1620/tjem.96.127. [DOI] [PubMed] [Google Scholar]
- 13.Williams D.S., Cash A., Hamadani L., Diemer T. Oxaloacetate supplementation increases lifespan in Caenorhabditis elegans through an AMPK/FOXO-dependent pathway. Aging Cell. 2009;8:765–768. doi: 10.1111/j.1474-9726.2009.00527.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Campos F., Sobrino T., Ramos-Cabrer P., Argibay B., Agulla J., Perez-Mato M., Rodriguez-Gonzalez R., Brea D., Castillo J. Neuroprotection by glutamate oxaloacetate transaminase in ischemic stroke: an experimental study. J. Cereb. Blood Flow Metab. 2011;31:1378–1386. doi: 10.1038/jcbfm.2011.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nagy D., Marosi M., Kis Z., Farkas T., Rakos G., Vecsei L., Teichberg V.I., Toldi J. Oxaloacetate decreases the infarct size and attenuates the reduction in evoked responses after photothrombotic focal ischemia in the rat cortex. Cell Mol. Neurobiol. 2009;29:827–835. doi: 10.1007/s10571-009-9364-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamamoto H.A., Mohanan P.V. Effect of alpha-ketoglutarate and oxaloacetate on brain mitochondrial DNA damage and seizures induced by kainic acid in mice. Toxicol. Lett. 2003;143:115–122. doi: 10.1016/s0378-4274(03)00114-0. [DOI] [PubMed] [Google Scholar]
- 17.Zlotnik A., Sinelnikov I., Gruenbaum B.F., Gruenbaum S.E., Dubilet M., Dubilet E., Leibowitz A., Ohayon S., Regev A., Boyko M., et al. Effect of glutamate and blood glutamate scavengers oxaloacetate and pyruvate on neurological outcome and pathohistology of the hippocampus after traumatic brain injury in rats. Anesthesiology. 2012;116:73–83. doi: 10.1097/ALN.0b013e31823d7731. [DOI] [PubMed] [Google Scholar]
- 18.Hakimi P., Yang J., Casadesus G., Massillon D., Tolentino-Silva F., Nye C.K., Cabrera M.E., Hagen D.R., Utter C.B., Baghdy Y., et al. Overexpression of the cytosolic form of phosphoenolpyruvate carboxykinase (GTP) in skeletal muscle repatterns energy metabolism in the mouse. J. Biol. Chem. 2007;282:32844–32855. doi: 10.1074/jbc.M706127200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morino K., Petersen K.F., Shulman G.I. Molecular mechanisms of insulin resistance in humans and their potential links with mitochondrial dysfunction. Diabetes. 2006;55(Suppl. 2):S9–S15. doi: 10.2337/db06-S002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Voloboueva L.A., Giffard R.G. Inflammation, mitochondria, and the inhibition of adult neurogenesis. J. Neurosci. Res. 2011;89:1989–1996. doi: 10.1002/jnr.22768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scarpulla R.C. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev. 2008;88:611–638. doi: 10.1152/physrev.00025.2007. [DOI] [PubMed] [Google Scholar]
- 22.Semenza G.L. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148:399–408. doi: 10.1016/j.cell.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nemoto S., Fergusson M.M., Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha} J. Biol. Chem. 2005;280:16456–16460. doi: 10.1074/jbc.M501485200. [DOI] [PubMed] [Google Scholar]
- 24.Canto C., Auwerx J. AMP-activated protein kinase and its downstream transcriptional pathways. Cell Mol. Life Sci. 2010;67:3407–3423. doi: 10.1007/s00018-010-0454-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Canto C., Auwerx J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009;20:98–105. doi: 10.1097/MOL.0b013e328328d0a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Puigserver P., Rhee J., Lin J., Wu Z., Yoon J.C., Zhang C.Y., Krauss S., Mootha V.K., Lowell B.B., Spiegelman B.M. Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARgamma coactivator-1. Mol. Cell. 2001;8:971–982. doi: 10.1016/s1097-2765(01)00390-2. [DOI] [PubMed] [Google Scholar]
- 27.Altarejos J.Y., Montminy M. CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nat. Rev. Mol. Cell Biol. 2011;12:141–151. doi: 10.1038/nrm3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu Z., Huang X., Feng Y., Handschin C., Gullicksen P.S., Bare O., Labow M., Spiegelman B., Stevenson S.C. Transducer of regulated CREB-binding proteins (TORCs) induce PGC-1alpha transcription and mitochondrial biogenesis in muscle cells. Proc. Natl Acad. Sci. USA. 2006;103:14379–14384. doi: 10.1073/pnas.0606714103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Manning B.D., Cantley L.C. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reynolds T.H., Bodine S.C., Lawrence J.C., Jr Control of Ser2448 phosphorylation in the mammalian target of rapamycin by insulin and skeletal muscle load. J. Biol. Chem. 2002;277:17657–17662. doi: 10.1074/jbc.M201142200. [DOI] [PubMed] [Google Scholar]
- 31.Chiang G.G., Abraham R.T. Phosphorylation of mammalian target of rapamycin (mTOR) at Ser-2448 is mediated by p70S6 kinase. J. Biol. Chem. 2005;280:25485–25490. doi: 10.1074/jbc.M501707200. [DOI] [PubMed] [Google Scholar]
- 32.McCoy M.K., Tansey M.G. TNF signaling inhibition in the CNS: implications for normal brain function and neurodegenerative disease. J. Neuroinflammat. 2008;5:45. doi: 10.1186/1742-2094-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Villeda S.A., Luo J., Mosher K.I., Zou B., Britschgi M., Bieri G., Stan T.M., Fainberg N., Ding Z., Eggel A., et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature. 2011;477:90–94. doi: 10.1038/nature10357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schreck R., Albermann K., Baeuerle P.A. Nuclear factor kappa B: an oxidative stress-responsive transcription factor of eukaryotic cells (a review) Free Radic. Res. Commun. 1992;17:221–237. doi: 10.3109/10715769209079515. [DOI] [PubMed] [Google Scholar]
- 35.van Praag H., Shubert T., Zhao C., Gage F.H. Exercise enhances learning and hippocampal neurogenesis in aged mice. J. Neurosci. 2005;25:8680–8685. doi: 10.1523/JNEUROSCI.1731-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spalding K.L., Bergmann O., Alkass K., Bernard S., Salehpour M., Huttner H.B., Bostrom E., Westerlund I., Vial C., Buchholz B.A., et al. Dynamics of hippocampal neurogenesis in adult humans. Cell. 2013;153:1219–1227. doi: 10.1016/j.cell.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eriksson P.S., Perfilieva E., Bjork-Eriksson T., Alborn A.M., Nordborg C., Peterson D.A., Gage F.H. Neurogenesis in the adult human hippocampus. Nat. Med. 1998;4:1313–1317. doi: 10.1038/3305. [DOI] [PubMed] [Google Scholar]
- 38.Wullschleger S., Loewith R., Hall M.N. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 39.Cotman C.W., Berchtold N.C. Exercise: a behavioral intervention to enhance brain health and plasticity. Trends Neurosci. 2002;25:295–301. doi: 10.1016/s0166-2236(02)02143-4. [DOI] [PubMed] [Google Scholar]
- 40.Jin K., Zhu Y., Sun Y., Mao X.O., Xie L., Greenberg D.A. Vascular endothelial growth factor (VEGF) stimulates neurogenesis in vitro and in vivo. Proc. Natl Acad. Sci. USA. 2002;99:11946–11950. doi: 10.1073/pnas.182296499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Couillard-Despres S., Winner B., Schaubeck S., Aigner R., Vroemen M., Weidner N., Bogdahn U., Winkler J., Kuhn H.G., Aigner L. Doublecortin expression levels in adult brain reflect neurogenesis. Eur. J. Neurosci. 2005;21:1–14. doi: 10.1111/j.1460-9568.2004.03813.x. [DOI] [PubMed] [Google Scholar]
- 42.von Bohlen Und Halbach O. Immunohistological markers for staging neurogenesis in adult hippocampus. Cell Tissue Res. 2007;329:409–420. doi: 10.1007/s00441-007-0432-4. [DOI] [PubMed] [Google Scholar]
- 43.Williamson J.R., Jones E.A. Inhibition of glycoysis by pyruvate in relation to the accumulation of citric acid cycle intermediates in the perfused rat heart. Nature. 1964;203:1171–1173. doi: 10.1038/2031171a0. [DOI] [PubMed] [Google Scholar]
- 44.Lin S.J., Guarente L. Nicotinamide adenine dinucleotide, a metabolic regulator of transcription, longevity and disease. Curr. Opin. Cell Biol. 2003;15:241–246. doi: 10.1016/s0955-0674(03)00006-1. [DOI] [PubMed] [Google Scholar]
- 45.Yang H., Yang T., Baur J.A., Perez E., Matsui T., Carmona J.J., Lamming D.W., Souza-Pinto N.C., Bohr V.A., Rosenzweig A., et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell. 2007;130:1095–1107. doi: 10.1016/j.cell.2007.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Easlon E., Tsang F., Skinner C., Wang C., Lin S.J. The malate-aspartate NADH shuttle components are novel metabolic longevity regulators required for calorie restriction-mediated life span extension in yeast. Genes Dev. 2008;22:931–944. doi: 10.1101/gad.1648308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.E L., Lu J., Burns J.M., Swerdlow R.H. Effect of exercise on mouse liver and brain bioenergetic infrastructures. Exp. Physiol. 2013;98:207–219. doi: 10.1113/expphysiol.2012.066688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Petersen K.F., Dufour S., Befroy D., Garcia R., Shulman G.I. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N. Engl. J. Med. 2004;350:664–671. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Steen E., Terry B.M., Rivera E.J., Cannon J.L., Neely T.R., Tavares R., Xu X.J., Wands J.R., de la Monte S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer's disease—is this type 3 diabetes? J. Alzheimers Dis. 2005;7:63–80. doi: 10.3233/jad-2005-7107. [DOI] [PubMed] [Google Scholar]
- 50.Witte M.E., Geurts J.J., de Vries H.E., van der Valk P., van Horssen J. Mitochondrial dysfunction: a potential link between neuroinflammation and neurodegeneration? Mitochondrion. 2010;10:411–418. doi: 10.1016/j.mito.2010.05.014. [DOI] [PubMed] [Google Scholar]
- 51.Manfredi A.A., Rovere-Querini P. The mitochondrion—a Trojan horse that kicks off inflammation? N. Engl. J. Med. 2010;362:2132–2134. doi: 10.1056/NEJMcibr1003521. [DOI] [PubMed] [Google Scholar]
- 52.Oka T., Hikoso S., Yamaguchi O., Taneike M., Takeda T., Tamai T., Oyabu J., Murakawa T., Nakayama H., Nishida K., et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. 2012;485:251–255. doi: 10.1038/nature10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou R., Yazdi A.S., Menu P., Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 54.Adzemovic M.Z., Ockinger J., Zeitelhofer M., Hochmeister S., Beyeen A.D., Paulson A., Gillett A., Thessen Hedreul M., Covacu R., Lassmann H., et al. Expression of Ccl11 associates with immune response modulation and protection against neuroinflammation in rats. PLoS ONE. 2012;7:e39794. doi: 10.1371/journal.pone.0039794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen L.F., Greene W.C. Shaping the nuclear action of NF-kappaB. Nat. Rev. Mol. Cell Biol. 2004;5:392–401. doi: 10.1038/nrm1368. [DOI] [PubMed] [Google Scholar]
- 56.Clarke D.L., Sutcliffe A., Deacon K., Bradbury D., Corbett L., Knox A.J. PKCbetaII augments NF-kappaB-dependent transcription at the CCL11 promoter via p300/CBP-associated factor recruitment and histone H4 acetylation. J. Immunol. 2008;181:3503–3514. doi: 10.4049/jimmunol.181.5.3503. [DOI] [PubMed] [Google Scholar]
- 57.Benarroch E.E. Adult neurogenesis in the dentate gyrus: general concepts and potential implications. Neurology. 2013;81:1443–1452. doi: 10.1212/WNL.0b013e3182a9a156. [DOI] [PubMed] [Google Scholar]
- 58.Lazarov O., Marr R.A. Of mice and men: neurogenesis, cognition and Alzheimer's disease. Front. Aging Neurosci. 2013;5:43. doi: 10.3389/fnagi.2013.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lazarov O., Marr R.A. Neurogenesis and Alzheimer's disease: at the crossroads. Exp. Neurol. 2010;223:267–281. doi: 10.1016/j.expneurol.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fabel K., Fabel K., Tam B., Kaufer D., Baiker A., Simmons N., Kuo C.J., Palmer T.D. VEGF is necessary for exercise-induced adult hippocampal neurogenesis. Eur. J. Neurosci. 2003;18:2803–2812. doi: 10.1111/j.1460-9568.2003.03041.x. [DOI] [PubMed] [Google Scholar]
- 61.Pugh C.W., Ratcliffe P.J. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat. Med. 2003;9:677–684. doi: 10.1038/nm0603-677. [DOI] [PubMed] [Google Scholar]
- 62.Arany Z., Foo S.Y., Ma Y., Ruas J.L., Bommi-Reddy A., Girnun G., Cooper M., Laznik D., Chinsomboon J., Rangwala S.M., et al. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1alpha. Nature. 2008;451:1008–1012. doi: 10.1038/nature06613. [DOI] [PubMed] [Google Scholar]
- 63.Akers K.G., Martinez-Canabal A., Restivo L., Yiu A.P., De Cristofaro A., Hsiang H.L., Wheeler A.L., Guskjolen A., Niibori Y., Shoji H., et al. Hippocampal neurogenesis regulates forgetting during adulthood and infancy. Science. 2014;344:598–602. doi: 10.1126/science.1248903. [DOI] [PubMed] [Google Scholar]
- 64.Barros L.F. Metabolic signaling by lactate in the brain. Trends Neurosci. 2013;36:396–404. doi: 10.1016/j.tins.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 65.Hertz L., Peng L., Dienel G.A. Energy metabolism in astrocytes: high rate of oxidative metabolism and spatiotemporal dependence on glycolysis/glycogenolysis. J. Cereb. Blood Flow Metab. 2007;27:219–249. doi: 10.1038/sj.jcbfm.9600343. [DOI] [PubMed] [Google Scholar]
- 66.Pellerin L., Magistretti P.J. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc. Natl Acad. Sci. USA. 1994;91:10625–10629. doi: 10.1073/pnas.91.22.10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee Y., Morrison B.M., Li Y., Lengacher S., Farah M.H., Hoffman P.N., Liu Y., Tsingalia A., Jin L., Zhang P.W., et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature. 2012;487:443–448. doi: 10.1038/nature11314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Teichberg V.I., Cohen-Kashi-Malina K., Cooper I., Zlotnik A. Homeostasis of glutamate in brain fluids: an accelerated brain-to-blood efflux of excess glutamate is produced by blood glutamate scavenging and offers protection from neuropathologies. Neuroscience. 2009;158:301–308. doi: 10.1016/j.neuroscience.2008.02.075. [DOI] [PubMed] [Google Scholar]
- 69.Swerdlow R., Marcus D.M., Landman J., Harooni M., Freedman M.L. Brain glucose and ketone body metabolism in patients with Alzheimer's disease. Clin. Res. 1989;37:461A. [Google Scholar]
- 70.Henderson S.T., Vogel J.L., Barr L.J., Garvin F., Jones J.J., Costantini L.C. Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer's disease: a randomized, double-blind, placebo-controlled, multicenter trial. Nutr. Metab. (Lond.) 2009;6:31. doi: 10.1186/1743-7075-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Krikorian R., Shidler M.D., Dangelo K., Couch S.C., Benoit S.C., Clegg D.J. Dietary ketosis enhances memory in mild cognitive impairment. Neurobiol. Aging. 2012;33:425. doi: 10.1016/j.neurobiolaging.2010.10.006. e419–e427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Harris J.L., Yeh H.W., Choi I.Y., Lee P., Berman N.E., Swerdlow R.H., Craciunas S.C., Brooks W.M. Altered neurochemical profile after traumatic brain injury: (1)H-MRS biomarkers of pathological mechanisms. J. Cereb. Blood Flow Metab. 2012;32:2122–2134. doi: 10.1038/jcbfm.2012.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mlynarik V., Gambarota G., Frenkel H., Gruetter R. Localized short-echo-time proton MR spectroscopy with full signal-intensity acquisition. Magn. Reson. Med. 2006;56:965–970. doi: 10.1002/mrm.21043. [DOI] [PubMed] [Google Scholar]
- 74.Harris J.L., Yeh H.W., Swerdlow R.H., Choi I.Y., Lee P., Brooks W.M. High-field proton magnetic resonance spectroscopy reveals metabolic effects of normal brain aging. Neurobiol. Aging. 2014;35:1686–1694. doi: 10.1016/j.neurobiolaging.2014.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.