

Background: CD44 on neutrophils is a ligand for E-selectin on endothelial cells.
Results: CD44 forms actin-dependent clusters by binding to ezrin/radixin/moesin (ERM) proteins and ankyrin.
Conclusion: Cytoskeletal interactions regulate CD44 clustering, mobility, and function.
Significance: CD44 organizes at nanometer scale on cell surfaces.
Keywords: Cell Adhesion, Cytoskeleton, Fluorescence Recovery after Photobleaching (FRAP), Fluorescence Resonance Energy Transfer (FRET), Neutrophil, Signal Transduction
Abstract
Interactions of CD44 on neutrophils with E-selectin on activated endothelial cells mediate rolling under flow, a prerequisite for neutrophil arrest and migration into perivascular tissues. How CD44 functions as a rolling ligand despite its weak affinity for E-selectin is unknown. We examined the nanometer scale organization of CD44 on intact cells. CD44 on leukocytes and transfected K562 cells was cross-linked within a 1.14-nm spacer. Depolymerizing actin with latrunculin B reduced cross-linking. Fluorescence resonance energy transfer (FRET) revealed tight co-clustering between CD44 fused to yellow fluorescent protein (YFP) and CD44 fused to cyan fluorescent protein on K562 cells. Latrunculin B reduced FRET-reported co-clustering. Number and brightness analysis confirmed actin-dependent CD44-YFP clusters on living cells. CD44 lacking binding sites for ankyrin and for ezrin/radixin/moesin (ERM) proteins on its cytoplasmic domain (ΔANKΔERM) did not cluster. Unexpectedly, CD44 lacking only the ankyrin-binding site (ΔANK) formed larger but looser clusters. Fluorescence recovery after photobleaching demonstrated increased CD44 mobility by latrunculin B treatment or by deleting the cytoplasmic domain. ΔANKΔERM mobility increased only modestly, suggesting that the cytoplasmic domain engages the cytoskeleton by an additional mechanism. Ex vivo differentiated CD44-deficient neutrophils expressing exogenous CD44 rolled on E-selectin and activated Src kinases after binding anti-CD44 antibody. In contrast, differentiated neutrophils expressing ΔANK had impaired rolling and kinase activation. These data demonstrate that spectrin and actin networks regulate CD44 clustering and suggest that ankyrin enhances CD44-mediated neutrophil rolling and signaling.
Introduction
During inflammation, circulating leukocytes tether to and roll on venular surfaces, arrest, crawl to endothelial cell junctions, and emigrate into underlying tissues (1, 2). Leukocytes roll on P-selectin and E-selectin expressed on activated endothelial cells (3). E-selectin primarily controls rolling velocity in vivo (4). During rolling, E-selectin ligands on neutrophils transduce signals that partially activate integrin αLβ2, which slows rolling through reversible interactions with endothelial cell ligands such as intercellular adhesion molecule-1 (5, 6). Endothelial bound chemokines fully activate integrin αLβ2 to trigger arrest (7).
Murine neutrophils roll on E-selectin through interactions with CD44, E-selectin ligand-1, P-selectin glycoprotein ligand-1 (PSGL-1),2 and at least one unknown O-glycosylated protein (8–12). The lectin domain of E-selectin binds to sialyl Lewis x (NeuAcα2–3Galβ1–4[Fucα1–3]GlcNAcβ1-R), a tetrasaccharide that caps N-glycans on CD44 and E-selectin ligand-1 and core 1-derived O-glycans on PSGL-1 and the unknown glycoprotein(s) (9–11). These glycoproteins cooperate to control neutrophil rolling on E-selectin. On inflammatory T cells, CD44 is the predominant ligand that controls rolling velocities and conversion from rolling to arrest (13). On murine neutrophils, E-selectin engagement of CD44 or PSGL-1 activates Src family kinases (SFKs) and downstream signals that enable integrin αLβ2 to slow rolling (5, 6). Knock-out mice lacking CD44 and PSGL-1 have major defects in recruiting neutrophils or T cells into inflammatory tissues (6, 9).
Despite the documented role of CD44 in mediating rolling on E-selectin, only a small percentage of CD44 solubilized from neutrophils can be precipitated by E-selectin (11). This suggests that CD44 binds to E-selectin with low affinity and/or that most CD44 molecules lack the proper glycosylation to bind to E-selectin. To compensate for these defects, CD44 might organize in membrane domains that facilitate interactions with E-selectin during rolling. Clustering of CD44 for example may increase E-selectin bond numbers and reduce force on individual bonds (3).
Most studies of CD44 have focused on its role as a receptor for hyaluronan. Cytoskeletal regulation of CD44-hyaluronan interactions has received particular attention. In hematopoietic and other cells, the cytoplasmic domain of CD44 interacts with the cytoskeleton through two well characterized adaptors: ezrin/radixin/moesin (ERM) proteins and ankyrin (14). Juxtamembrane basic residues in the CD44 cytoplasmic domain bind to ERM proteins, which bind directly to long actin filaments (15, 16). A more distal 15-residue segment of the CD44 cytoplasmic domain binds to ankyrin, which binds directly to spectrin (17, 18). Spectrin tetramers interact with short actin oligomers, accessory proteins, and phosphatidylinositol lipids to form a submembrane geodesic domelike structure (19). The organization of the ankyrin-spectrin network is thought to assemble proteins and lipids for signaling and other functions. With certain exceptions (18, 20), most studies suggest that disrupting actin filaments or mutating the ERM- or ankyrin-binding sites on CD44 has minimal impact on cell adhesion to hyaluronan under static or flow conditions (21, 22). However, these perturbations can inhibit outside-in signaling by hyaluronan (14, 23). Whether cytoskeletal interactions affect the organization of CD44 on the cell membrane has received much less study. Confocal microscopy of transfected AKR1 lymphoma cells suggests a homogeneous plasma membrane distribution for wild-type (WT) CD44 but a punctate distribution for CD44 lacking the cytoplasmic domain (22). Nanometer scale imaging of CD44 on intact cell membranes has not been reported.
Here we used cross-linking and high resolution imaging to demonstrate that CD44 forms actin-dependent clusters in hematopoietic cells. Disrupting the binding site for ankyrin but not for ERM proteins resulted in larger but looser clusters. Disrupting the ankyrin-binding site also impaired CD44-dependent rolling of neutrophils on E-selectin and CD44-mediated activation of SFKs. These data suggest that ankyrin binding to the cytoplasmic domain of CD44 enhances its interactions with E-selectin.
EXPERIMENTAL PROCEDURES
Reagents
Anti-murine CD44 mAb (clone KM114) was purchased from BD Pharmingen. Anti-C terminus CD44 rabbit polyclonal antibody was purchased from Abcam (Cambridge, MA). Phycoerythrin- or fluorescein isothiocyanate-labeled anti-CD44 mAb (clone IM7) was from eBioscience (San Diego, CA). Anti-GFP mAb (clone B34) was purchased from Covance (Princeton, NJ). Rabbit polyclonal antibodies to SFKs and to SFKs phosphorylated on Tyr-416 were from Cell Signaling Technology (Danvers, MA). The cross-linker bis(sulfosuccinimidyl) suberate (BS3) was from Thermo Scientific (Rockford, IL). Latrunculin B was from Santa Cruz Biotechnology (Dallas, TX). Blebbistatin, poly-l-lysine, and β-estradiol were from Sigma-Aldrich. EasySepTM Mouse Hematopoietic Progenitor Cell Isolation kit was from STEMCELL Technologies (Vancouver, British Columbia, Canada). Stem cell factor, IL-3, and IL-6 were from Shenandoah Biotechnology (Warwick, PA). All restriction endonucleases were from Promega (Madison, WI). Lipofectamine was from Invitrogen. T4 DNA ligase was from New England BioLabs (Ipswich, MA). PfuTurbo DNA polymerase was from Stratagene (La Jolla, CA). Retroviral vector pMSCVpuro was from Clontech. Vector pWay2 containing YFP or CFP was described previously (24). The Hoxb8-ER construct (25) was a gift from Mark P. Kamps (University of California at San Diego, La Jolla, CA). Retroviral vector MSCV-IRES-GFP (pMiG) and the packaging-helper plasmid PCL/Eco (26) were gifts from José Alberola-Ila (Oklahoma Medical Research Foundation, Oklahoma City, OK).
Mice
PSGL-1−/− mice were generated as described (8). CD44−/− mice (B6.129(Cg)) (27) were purchased from The Jackson Laboratory (Bar Harbor, ME). PSGL-1−/− mice were bred with CD44−/− mice to generate PSGL-1−/−/CD44−/− mice. All mice were bred at least 10 generations in the C57/BL6J background. All experiments were performed in compliance with protocols approved by the Institutional Animal Care and Use Committee of the Oklahoma Medical Research Foundation.
Generation of CD44 Constructs
A full-length cDNA encoding the murine standard form of CD44 without the stop codon was annealed between the EcoRI and SmaI sites of the vector pWay2 containing cDNAs encoding CFP or YFP in place of GFP (24). To ensure the monomeric status of CFP and YFP, each cDNA was altered to express an A206K mutation (monomeric CFP or monomeric YFP) (28) before inserting cDNA for CD44. All but the first six residues (NSRRRC) of the CD44 cytoplasmic domain were deleted to generate ΔCD CD44. Juxtamembrane lysines and arginines in the CD44 cytoplasmic domain that confer binding to ERM proteins were replaced with alanines by PCR-based site-directed mutagenesis to generate ΔERM CD44. Τhe 15-residue sequence in the CD44 cytoplasmic domain that binds ankyrin was deleted by overlap extension PCR to generate ΔANK CD44. A mutant lacking both binding sites was made by mutating the residues conferring ERM binding into an ankyrin binding-deficient template to generate ΔANKΔERM CD44. The primers (5′–3′) for mutagenesis were as follows: YFP, 5′-CCTGAGCTACCAGTCCAAACTGAGCAAAGACCCCAACG-3′; CFP, 5′-CCTGAGCACCCAGTCCAAACTGAGCAAAGACCCCAACG-3′; ΔERM or ΔANKΔERM, 5′-GGTCAATAGTGCCGCCGCCTGTGGGCAGGCCGCCGCCCTGGTGATCAACGG-3′; overlapping primers for ΔANK, 5′-GGAAGATCTATGGACAAGTTTTGGTGGCACACAGCTTGGGG-3′, 5′-GACTTGCTGGCCTCCCCGTTGATCACCAGCTTTTTCTTCTGC-3′, 5′-GCAGAAGAAAAAGCTGGTGATCAACGGGGAGGCCAGCAAGTC-3′, and 5′-CGGGAATTCCTACACCCCAATCTTCATGTCCACACTCTGC-3′; overlapping primers for ΔCD, 5′-CCGGAATTCGCCACCATGGACAAGTTTTGG-3′, 5′-CGCCCTTGCTCACCCCACACCTTCTCCTAC-3′, 5′-GTAGGAGAAGGTGTGGGGTGAGCAAGGGCG-3′, and 5′-GCTCTAGACTCGAGTTACTTGTACAGC-3′.
To prepare constructs for retroviral transduction, cDNAs encoding WT or mutant CD44 were cloned into the retroviral vector pMiG containing cDNA for GFP with restriction enzyme sites BglII and EcoRI on 5′- and 3′-ends, respectively. All constructs were confirmed by DNA sequencing.
Transfection
To produce retrovirus, 106 293T cells were seeded in a 10-cm plate 1 day before transfection. Packaging-helper plasmid PCL/Eco containing gag, pol, and env genes (5 μg) and pMiG encoding WT or mutant forms of CD44 (5 μg) were co-transfected into subconfluent 293T cells with a calcium phosphate transfection kit (Invitrogen) in the presence of 25 μg of chloroquine. The viruses were harvested 48 h post-transfection.
Human erythroleukemia K562 cells were maintained in RPMI 1640 medium containing 10% FBS and 1% penicillin/streptomycin/glutamine. For cell transfection, electroporation was used. Briefly, cells (2 × 107/ml) were suspended in serum-free RPMI 1640 medium. Plasmids encoding CD44-YFP and CD44-CFP (15 μg each) were mixed and preincubated with cells for 15 min before adding the cell-DNA mixture into a 0.4-cm-gap electroporation cuvette (Bio-Rad). In some experiments, only cDNA encoding CD44-YFP was added. The instrument was set for 0.3 kV and 1000-microfarad capacitance. Cells were centrifuged immediately after shock. After incubating the pellet at room temperature for 15 min, cells were resuspended in culture medium. Cells were harvested 24 h post-transfection for cross-linking, fluorescence resonance energy transfer (FRET), or number and brightness (N&B) experiments. Alternatively, stably transfected clones were selected in medium containing 800 μg/ml G418. After 2–3 weeks, cells were sorted for matched surface expression of CD44 in the Oklahoma Medical Research Foundation cell sorting facility.
Flow Cytometry
To compare CD44 surface expression with or without latrunculin B treatment, transfected K562 cells were stained with fluorescein isothiocyanate-labeled anti-CD44 mAb (clone IM7) and analyzed as described previously (6). Neutrophil progenitors or stably transfected K562 cells were stained with phycoerythrin-labeled anti-CD44 mAb (clone IM7) and sorted for matched CD44 expression.
Cross-linking
Transiently transfected K562 cells expressing CD44-YFP or murine bone marrow leukocytes were incubated with 2 mm BS3 in Hanks' balanced salt solution without Mg2+ and Ca2+, pH 7.2 on ice for 2 h. The reactions were stopped by adding 20 mm Tris, pH 7.5. Cells were lysed in radioimmune precipitation assay buffer (1% Triton X-100, 125 mm NaCl, 50 mm Tris, pH 8.0, 10 mm EDTA, 10 mm NaF, 10 mm sodium pyrophosphate, 10 mm pepstatin, 50 μg/ml bestatin, 2 mm PMSF, 0.1% SDS, and 0.5% sodium deoxycholate) and analyzed by Western blotting (29). Briefly, SDS-PAGE under reducing conditions was used to separate the proteins. The resolved proteins were transferred to a PVDF membrane and immunostained with anti-GFP mAb (which cross-reacts with YFP and CFP) or anti-C terminus CD44 rabbit polyclonal antibody. The specific bands identified by these antibodies were not detected with control rabbit IgG.
FRET
Transfected K562 cells (106) were seeded on poly-l-lysine-coated glass bottom dishes and fixed with 2% paraformaldehyde for 30 min at room temperature. In some experiments, K562 cells were treated with 5 μm latrunculin B or 10 μm blebbistatin in 0.05% DMSO or with 0.05% DMSO as a control in RPMI 1640 medium with 50 mm HEPES, pH 7.4 at 37 °C for 30 min. Cells were then washed in PBS, pH 7.4 before fixation.
Microscopy was performed using a Zeiss LSM510 Meta confocal microscope. Images were collected with a 63× water immersion objective (numerical aperture, 1.2). YFP was excited at 514 nm, and emission was measured between wavelengths 530 and 600 nm. CFP was excited at 458 nm, and emission was measured between 484 and 516 nm. The images were recorded in a 12-bit mode with a scanning speed of 2.56 μs/pixel. All images were analyzed by IPLab (BD Biosciences).
FRET was measured based on the increase in CFP fluorescence intensity upon photobleaching YFP. Photobleaching of YFP, restricted to an area on the plasma membrane of 8 × 5 pixels (2.1 × 1.3 μm), was performed by illuminating the sample for 1200 iterations (∼11 s) with the 514-nm laser at full power. FRET efficiency was calculated as described previously (24). Briefly, the following equation was used.
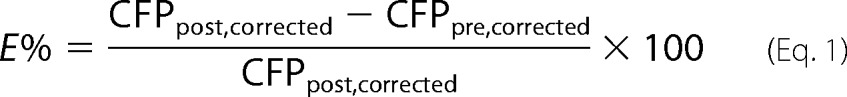 |
The CFPpre, corrected was derived by correcting the YFP bleed-through, and the CFPpost, corrected was derived by correcting the YFP bleed-through and the CFP photobleaching when bleaching YFP as described previously (24). Cells (70–100) with approximately matched expression of YFP and CFP were measured. Co-clustering was derived from the constant K by fitting the E% versus acceptor intensity using the following equation (24, 28).
 |
Curve fitting was performed with Igor Pro (WaveMetrics, Lake Oswego, OR) with 70–100 data points per experiment. For Lyn-YFP-CFP, CFP-T2DN-YFP, and L10-YFP/S15-CFP controls, standard error was determined using standard deviation from the curve fitting. Analysis of the statistical significance of K determined for WT and mutant CD44 was by a two-tailed Student's t test for distributions of equal variance (GraphPad Software) using K values determined from three or more separate experiments (24).
N&B Analysis
N&B experiments were performed with a Zeiss LSM510 Meta confocal microscope coupled with an avalanche photodiode detector (30) or with a Leica SP8 microscope coupled with an HyD detector. K562 cells stably expressing WT or mutant CD44 fused with monomeric YFP or monomeric YFP alone were captured in 35-cm glass bottom microwell dishes (MatTek) coated with poly-l-lysine. Cells were treated with 0.05% DMSO alone or with 5 μm latrunculin B in 0.05% DMSO in RPMI 1640 medium containing 1.5% FBS, 1% gentamycin/penicillin/streptomycin, and 50 mm HEPES, pH 7.4 at 37 °C for 30 min. Cells were then visualized in RPMI 1640 medium without phenol red, 50 mm HEPES, and 1.5% FBS. For the drug-treated group, acquisition was performed with the Zeiss LSM510 Meta confocal microscope in photon-counting mode with a 63× water-immersion objective (numerical aperture, 1.2), and the pixel dwell time and the frame time were 50 μs and 5 s, respectively. YFP was excited at 514 nm, and the laser intensity was 0.5% (0.06 milliwatt). Image stacks were acquired for 30 frames of a 256 × 256 pixel image with 220-nm pixel dimensions in the x and y directions. For the CD44 mutants, a 25× dipping objective (numerical aperture, 0.95) with the Leica system was used, and the pixel dwell time and frame time were 10 μs and 3 s, respectively. YFP was excited at 488 nm, and the laser intensity was 0.5% (0.075 milliwatt). Image stacks were acquired for 100 frames of a 256 × 256 pixel image with 121-nm pixel dimensions in the x and y directions.
Image stacks were analyzed with nb_tools, a customized MATLAB program (MathWorks) incorporating functions of the DIPimage toolbox (Delft University of Technology, Delft, The Netherlands) (31). The analysis was carried out cell by cell. Data for a cell were discarded if the variance of all the pixels within the stack did not linearly correlate with their fluorescence intensities. Low frequency fluctuation due to photobleaching was removed by high pass Fourier transform with a cutoff of 3. The apparent brightness (B) and the molecular brightness (ϵ) were calculated as follows.
 |
σ2 and I represent the variance and mean fluorescence intensity of each pixel, respectively. The calculation was restricted to the plasma membrane, avoiding other regions of the cell, by choosing the fluorescence intensity above a threshold. The thresholds were different for each cell depending on its fluorescence intensity, ranging from ∼20 to 50% of the maximum intensity. The mean intensity, mean brightness, and brightness distribution for all selected pixels within the cell were given by the software. The mean molecular brightness, ϵ (quantified in Tables 1 and 2), was derived from the mean brightness, B (histograms in Figs. 3 and 5), according to Equation 3. The value ϵ was used instead of B as molecules with B = 1 are considered immobile (32). The ϵ ± S.E. was evaluated from 5 to 20 cells for each group. The mean expression levels (intensities) were very similar for all CD44 constructs. Plots of molecular brightness versus mean intensity demonstrated no relationship between the two parameters (data not shown). Unlike Raster image scan correlation spectroscopy (33), adjacent pixels in N&B analysis are not correlated. Therefore, the effect of non-linearity of the scanner is minimal. We used monomeric YFP-expressing K562 cells to measure molecular brightness of a small region of interest from the very edge to almost the center of the field of view. This plot revealed no relationship between brightness and the distance of the region of interest from the edge (data not shown). Furthermore, molecular brightness was not measured on the edge in the great majority of the cells.
TABLE 1.
Molecular brightness of transfected K562 cells expressing YFP or CD44-YFP with or without treatment with control DMSO or latrunculin B (Lat B)
The molecular brightness (ϵ) was derived from brightness (B) (see Fig. 3) according to Equation 3. The values for ϵ ± S.E. were calculated from five to 15 cells from two independent experiments. The data were obtained with a Zeiss microscope (see “Experimental Procedures”). The pixel dwell time and the frame time were 50 μs and 5 s, respectively, with a total of 30 frames.
| Monomeric YFP | CD44-YFP |
||
|---|---|---|---|
| DMSO | Lat B | ||
| Molecular brightness | 0.06 ± 0.01 | 0.25 ± 0.03 | 0.16 ± 0.02a |
a p < 0.05 compared with DMSO.
TABLE 2.
Molecular brightness of transfected K562 cells expressing YFP or CD44-YFP constructs
The molecular brightness (ϵ) was derived from brightness (B) (see Fig. 5) according to Equation 3. The values for ϵ ± S.E. were calculated from 10 to 20 cells from two independent experiments. The data were obtained with a Leica microscope (see “Experimental Procedures”). The pixel dwell time and the frame time were 10 μs and 3 s, respectively, with a total of 100 frames.
| Monomeric YFP | CD44-YFP |
||||
|---|---|---|---|---|---|
| WT | ΔANK | ΔERM | ΔANK ΔERM | ||
| Molecular brightness | 0.08 ± 0.01 | 0.23 ± 0.02 | 0.43 ± 0.05a | 0.23 ± 0.01 | 0.17 ± 0.01b |
a p < 0.05 compared with WT.
b p < 0.01 compared with WT.
FIGURE 3.
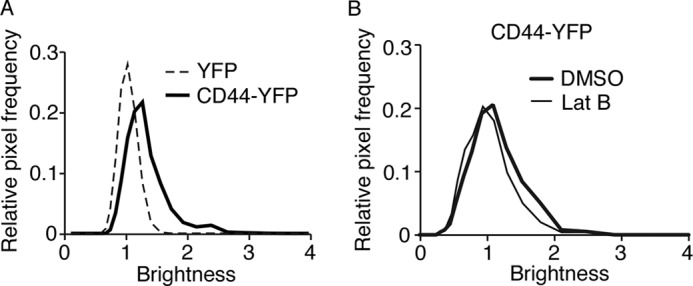
CD44 forms actin-dependent co-clusters on transfected K562 cells as measured by N&B analysis. A, brightness histograms for cells expressing monomeric YFP or CD44-YFP. B, brightness histograms for cells expressing CD44-YFP pretreated with DMSO control or latrunculin B (Lat B). The data are representative of 5–10 cells for each group.
FIGURE 5.
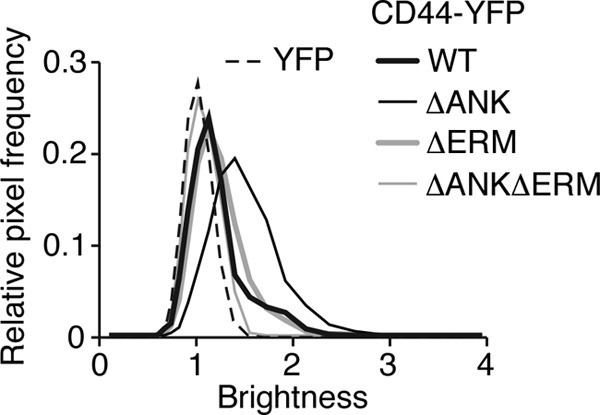
CD44 lacking the binding site for ankyrin forms clusters with more molecules as measured by N&B analysis. Shown are brightness histograms for transfected K562 cells expressing monomeric YFP or the indicated CD44-YFP construct. The data are representative of 5–10 cells for each group.
The spatial sampling for the Zeiss and Leica systems was different, but temporal rather than spatial resolution is key to resolving fluctuations in N&B analysis (32, 34). The temporal resolution is determined by the pixel dwell time, frame time, and number of frames. The pixel dwell time should always be shorter than the characteristic decay time of the fluctuating molecule so that the fluctuations are not averaged (32). Typically, biological molecules have diffusion coefficients of 0.01–30 μm2/s, and for a typical confocal detection volume, the diffusion time is of the order of 0.1–10 ms (34). A pixel dwell time of 50 or 10 μs was used for the Zeiss and Leica systems, respectively; this was much shorter than 0.1 ms. Furthermore, a frame time of 5 or 3 s, respectively, was used; this was much longer than the magnitude of the molecular diffusion time and thus allowed the consecutive samplings of the same pixel to be independent from each other.
Generation of Neutrophils ex Vivo with Conditional Hoxb8 Expression
Bone marrow leukocytes were harvested from 8-week-old female PSGL-1−/−/CD44−/− mice. The generation of differentiated neutrophils from immortalized myeloid precursors was performed as described previously (25). Briefly, lineage-negative hematopoietic progenitors were obtained by negative selection with the EasySep Mouse Hematopoietic Progenitor Cell Isolation kit. The isolated progenitors were prestimulated for 48 h in Iscove's modified Dulbecco's medium containing 50 ng/ml stem cell factor, 25 ng/ml IL-3, and 25 ng/ml IL-6. Approximately 25,000 prestimulated progenitors were infected with Hoxb8-ER retrovirus by spinoculation at 1,500 × g for 1.5 h at 22 °C in the presence of Lipofectamine (1:1,000). The infected progenitors were cultured in Opti-MEM containing 10% FBS, 1% glutamine/penicillin/streptomycin, 50 ng/ml stem cell factor, 30 mm β-mercaptoethanol, and 1 mm β-estradiol. The immortalized neutrophil progenitors were enriched by serial passages of non-adherent cells every 3 days. Immortalized progenitors predominated cultures by day 14, whereas control cultures had reduced proliferation and stopped dividing by day 21. The progenitors were transduced with retrovirus carrying empty vector or vector encoding WT or mutant CD44. Cell sorting was used to select clones with matched surface expression of CD44. Selected progenitors were differentiated into neutrophils by withdrawing estrogen. After 3 days, the mature neutrophils were harvested for flow cytometry or flow chamber assays.
Fluorescence Recovery after Photobleaching (FRAP)
K562 cells stably expressing WT or mutant CD44-YFP were captured onto glass bottom dishes coated with poly-l-lysine. Cells were imaged in RPMI 1640 medium without phenol red, 50 mm HEPES, and 1.5% FBS using a Zeiss LSM510 Meta confocal microscope. FRAP was performed as described previously (35). Briefly, the regions of interest (6.72 × 3.36 μm) were photobleached for ∼9 s at maximum 514-nm laser power. Subsequently, time lapse images were collected at 2% laser power until the bleached signal reached a stable level. FRAP curves from four independent trials with five cells per trial were derived by fitting the normalized fluorescence at each time point versus time into a one-phase association model plugged into the Prism software. Fmax, which represents the mobile fraction of the molecule in the bleached region, and τ½, which is the time to recover half of the maximum fluorescence and is inversely correlated to the diffusion coefficient, were derived from this curve. Mean ± S.E. was used to present Fmax and τ½ in Table 3.
TABLE 3.
Kinetic analysis of FRAP in transfected K562 cells expressing CD44-YFP constructs with or without treatment with control DMSO, latrunculin B (Lat B), or blebbistatin
FRAP curves (see Fig. 6) from four independent trials with five cell measurements per trial were fitted to a function for monoexponential recovery of fluorescence to derive τ½ and Fmax. The data represent the mean ± S.E. The data were obtained with a Zeiss microscope (see “Experimental Procedures”). The experiments in the upper three rows were performed in parallel at one time, whereas the experiments in the bottom five rows were performed in parallel at a different time. For each of the two groupings, the cells for each experimental condition were grown at the same density. The different τ½ values for WT-DMSO (row 1) and WT (row 4) may reflect different cell cycle conditions, which can alter the mobilities of membrane proteins (52, 53).
| τ½a | Fmaxb | |
|---|---|---|
| s | ||
| WT-DMSO | 59 ± 6 | 0.65 ± 0.03 |
| WT-Lat B | 39 ± 3c | 0.77 ± 0.02c |
| WT-blebbistatin | 49 ± 5 | 0.64 ± 0.03 |
| WT | 86 ± 9 | 0.66 ± 0.03 |
| ΔCD | 50 ± 6d | 0.75 ± 0.03d |
| ΔANK | 61 ± 6d | 0.73 ± 0.03 |
| ΔERM | 57 ± 6d | 0.69 ± 0.03 |
| ΔANKΔERM | 62 ± 5d | 0.73 ± 0.03 |
a τ½ indicates half-time for maximum fluorescence recovery.
b Fmax indicates the maximum fluorescence recovery.
c p < 0.05 compared with WT-DMSO.
d p < 0.05 compared with WT.
Flow Chamber Assay
Neutrophil rolling on immobilized E-selectin was measured as described previously (6, 36) except that human E-selectin Fc chimera was captured on an anti-human Fc mAb immobilized on 35-cm dishes. To measure resistance to detachment, neutrophils were allowed to accumulate at a wall shear stress of 1 dyne/cm2. After introducing cell-free buffer, the wall shear stress was increased every 30 s, and the numbers and velocities of rolling cells were measured.
CD44 Activation of SFKs in Differentiated Neutrophils
Activation of SFKs was measured as described previously (6) with minor modifications. Briefly, F(ab′)2 fragments of anti-CD44 mAb or isotype control mAb were immobilized on 48-well plates. Differentiated neutrophils expressing WT or mutant CD44 were suspended in Hanks' balanced salt solution with Ca2+ and Mg2+ containing 0.5% human serum albumin. Neutrophils added to the wells were incubated for 10 min at room temperature. Cells were lysed immediately with radioimmune precipitation assay buffer, and the lysate was analyzed with Western blotting using rabbit antibody against phospho-SFK (Tyr-416). The same membrane was stripped and reprobed with antibody against SFK. The activation level for SFK induced by engaging CD44 was derived from the ratio of the percentage of activated SFK in cells incubated on anti-CD44 mAb to that in cells incubated on isotype control mAb. Statistical difference was determined with a two-tailed Student's t test with Prism 6 (GraphPad Software).
RESULTS
CD44 Forms Actin-dependent Clusters on Leukocytes and Transfected K562 Cells
We used cross-linking to examine the organization of CD44 on cell surfaces. BS3 is a membrane-impermeable compound with an amine-reactive N-hydroxysulfosuccinimide ester at each end of a 1.14-nm spacer. It cross-links only nearby proteins located within the length of the spacer. We studied native CD44 on murine leukocytes (Fig. 1, A–C). We also studied murine CD44 fused to YFP (CD44-YFP; see schematic in Fig. 2A) on transfected human K562 cells (Fig. 1, D–F). Previous publications demonstrated that fusing GFP or its variants (CFP and YFP) to the C terminus of CD44 does not affect binding to extracellular hyaluronan (37) or to intracellular ERM proteins (38). After incubation with BS3 or buffer control, the cells were lysed and analyzed by Western blotting with anti-CD44 antibody (Fig. 1, B and C) or with anti-GFP antibody, which cross-reacts with YFP (Fig. 1, E and F). BS3 generated a cross-linked species with an apparent Mr ∼3 times greater than monomeric CD44 on leukocytes (Fig. 1B) or monomeric CD44-YFP on K562 cells (Fig. 1E). The cross-linked product was significantly decreased in cells pretreated with latrunculin B, which depolymerizes actin by sequestering G-actin and preventing F-actin assembly (39) (Fig. 1, B, C, E, and F). Control DMSO vehicle did not alter cross-linking (Fig. 1, B and C). Latrunculin B did not alter global surface expression of CD44 (Fig. 1A) or CD44-YFP (Fig. 1D). These data suggest that CD44 closely associates with itself and/or other proteins in an actin-dependent manner.
FIGURE 1.

CD44 can be cross-linked on leukocytes or transfected K562 cells. Leukocytes expressing native CD44 (A–C) or transfected K562 cells expressing CD44-YFP (D–F) were pretreated with buffer alone, DMSO, or latrunculin B (Lat B) in DMSO. A, flow cytometry of leukocytes incubated with phycoerythrin (PE)-labeled isotype control mAb or anti-CD44 mAb. B, leukocytes were incubated with BS3 or buffer control. Cell lysates were probed by Western blotting (WB) with anti-CD44 polyclonal antibody under reducing conditions. C, relative CD44 oligomer/monomer ratio in buffer-, DMSO-, and latrunculin B-treated leukocytes measured by densitometry. D, flow cytometry of transfected K562 cells incubated with fluorescein isothiocyanate (FITC)-labeled isotype control mAb or anti-CD44 mAb. Transfected K562 cells were incubated with BS3 or buffer control. Cell lysates were probed by Western blotting with anti-GFP antibody under reducing conditions. F, relative CD44-YFP oligomer/monomer ratio in DMSO- and latrunculin B-treated K562 cells measured by densitometry. The data in B and E are representative of three experiments. The data in C and F represent the mean of three experiments. Error bars represent S.E. **, p < 0.01 by a two-tailed Student's t test.
FIGURE 2.

CD44 forms actin-dependent co-clusters on transfected K562 cells as measured by FRET. A, schematic diagram of YFP/CFP fusion proteins. B–I, transfected K562 cells expressing the indicated donor/acceptor pairs were analyzed by FRET. Cells in F–H were pretreated with DMSO control, latrunculin B (Lat B), or blebbistatin as shown. The FRET efficiency is plotted against acceptor intensity in cells where the donor-to-acceptor ratio was ∼1:1. The red line is the fitted curve derived from Equation 2. The blue dotted lines represent the 95% confidence intervals for the fit. The top of each plot shows the residual differences for the fitted curve. The values for K are shown in E and I. Error bars represent S.E. The data are derived from three independent experiments. ***, p < 0.001 by a two-tailed Student's t test. AA, amino acids.
To determine whether CD44 associates with itself, we measured FRET in K562 cells co-transfected with equal amounts of cDNA encoding CD44 fused to monomeric YFP or CFP (Fig. 2A). FRET was measured from the increase in CFP fluorescence intensity after photobleaching the YFP signal in a small region of the plasma membrane. FRET is strongest when acceptor and donor are within 2 nm of each other. We also transfected cells with cDNAs encoding YFP/CFP fusion proteins as FRET controls (24) (Fig. 2A). The positive control Lyn-CFP-YFP anchors to the plasma membrane with CFP and YFP separated by only two amino acids. The negative control CFP-T2DN-YFP is expressed in the cytoplasm with CFP and YFP separated by 236 amino acids. In the other negative control, L10-YFP was co-expressed with S15-CFP. L10-YFP is YFP fused to the N-terminal 10 residues of Lck, which targets YFP to the Triton-X-100-insoluble membrane fraction representing lipid rafts. S15-CFP is CFP fused to the N-terminal 15 residues of Src, which targets CFP to the Triton-X-100-soluble membrane fraction.
We assessed co-clustering of the donor and acceptor by fitting E% to a saturable binding model based on the relative concentration of acceptor while keeping the donor-to-acceptor ratio constant throughout the measurements (see “Experimental Procedures”). The parameter K resolved from the curve fit indicates the degree of co-clustering of donors and acceptors independent of acceptor/donor concentration. K is analogous to a dissociation constant for interactions between the donor and acceptor: the lower the K, the higher the level of co-clustering. The positive control Lyn-CFP-YFP exhibited a low K, whereas the negative controls CFP-T2DN-YFP and L10-YFP/S15-CFP each exhibited a much higher K (Fig. 2, B, C, and E). The CD44-YFP/CFP pair manifested a low K close to that of the positive control (Fig. 2, D and E), indicating a high degree of co-clustering. For membrane proteins, a low K also indicates a higher probability for donors and acceptors to co-cluster, which we predict will cause each to be in the same cluster as the cluster size increases. Treating cells with latrunculin B, but not DMSO vehicle, markedly increased K for CD44-YFP/CFP (Fig. 2, F, G, and I). In contrast, treating cells with blebbistatin, which inhibits energy-dependent myosin II ATPase activity and reduces actomyosin tension without depolymerizing actin filaments (40), had no effect on K (Fig. 2, H and I). These data demonstrate that self-association of CD44 requires intact actin filaments but not actomyosin tension.
To measure clustering of CD44 based on protein mobility, we used N&B analysis, which is based on the statistics of fluorescence fluctuation. The brightness (B) histogram of CD44-YFP in K562 cells shifted to the right relative to the histogram of monomeric YFP expressed in K562 cells (Fig. 3A). The molecular brightness (ϵ) of CD44-YFP was ∼3 times greater than that of YFP (Table 1), indicating that CD44 moved as a cluster. These data are consistent with the low K for CD44-YFP/CFP in FRET experiments and suggest that many clusters contain two CD44 proteins and some contain three or more. Treating cells with latrunculin B significantly reduced the size of CD44 co-clusters (Fig. 3B and Table 1). These observations are consistent with the FRET data and indicate that mobile CD44 co-clusters in living cells in an actin-dependent manner.
Disrupting Binding Sites for Cytoskeletal Adaptors on the CD44 Cytoplasmic Domain Alters the Size and Tightness of CD44 Clusters
Two cytoskeletal adaptors, ERM proteins and ankyrin, are known to bind to the CD44 cytoplasmic domain (14). To determine whether these interactions regulate CD44 clustering, we made an ankyrin binding-deficient CD44 (ΔANK) by deleting the 15-residue ankyrin-binding sequence (Fig. 4A). We made an ERM binding-deficient CD44 (ΔERM) by mutating two clusters of basic residues required for binding (Fig. 4A). We also made CD44 lacking binding sites for both adaptors (ΔANKΔERM). The C terminus of each CD44 mutant was fused to monomeric YFP or CFP (Fig. 4A).
FIGURE 4.

Disrupting the binding sites for ERM proteins and ankyrin on CD44 reduces co-clustering as measured by FRET. A, schematic diagram of CD44-YFP/CFP fusion proteins. B–F, transfected K562 cells expressing the indicated donor/acceptor pairs were analyzed by FRET. The FRET efficiency is plotted against acceptor intensity in cells where the donor-to-acceptor ratio was ∼1:1. The red line is the fitted curve derived from Equation 2. The blue dotted lines represent the 95% confidence intervals for the fit. The top of each plot shows the residual differences for the fitted curve. The values for K are shown in F. Error bars represent S.E. The data are derived from three independent experiments. *, p < 0.05 by a two-tailed Student's t test.
We first used FRET to compare the membrane organization of WT with that of mutant CD44-YFP/CFP pairs expressed in transfected K562 cells. Both the ΔANK and ΔERM single mutants retained a low K like that of WT CD44, whereas the K of the ΔANKΔERM mutant markedly increased (Fig. 4, B–F). These results suggest that disrupting both binding sites decreases the extent of CD44 co-clusters. We then used N&B analysis to examine clustering of WT or mutant CD44-YFP proteins in transfected K562 cells. Surprisingly, the brightness (B) histogram of ΔANK shifted to the right relative to histograms of the other CD44 constructs (Fig. 5) with a resultant increase in the molecular brightness (ϵ) (Table 2). This suggests that the ΔANK mutant formed clusters with more molecules (greater brightness). These clusters may be less tight; the K may not change because FRET is less sensitive as donor-acceptor separation increases from 2 to 8 nm. Thus, binding to ankyrin may prevent some CD44 clusters from merging into larger, looser aggregates by other linkages to filamentous actin, including ERM proteins.
Disrupting Interactions with the Actin Cytoskeleton Increases the Mobility of CD44 on Transfected K562 Cells
We used FRAP to measure the mobility of WT or mutant CD44-YFP proteins on transfected K562 cells. Treating cells with latrunculin B, but not with blebbistatin, increased the mobile fraction (Fmax) and the rate of fluorescence recovery (inversely proportional to τ½) (Fig. 6A and Table 3). Deleting all but the first six residues of the cytoplasmic domain of CD44 (ΔCD) caused similar effects (Fig. 6B and Table 3). Disrupting either or both binding sites for ankyrin and ERM proteins slightly decreased τ½ but had minimal effects on Fmax (Fig. 6, C–E, and Table 3). These results demonstrate that actin filaments but not actomyosin tension restrain the mobility of CD44. Interactions of the cytoplasmic domain with ankyrin, ERM proteins, and perhaps other cytoskeletal adaptors contribute to this restraint.
FIGURE 6.
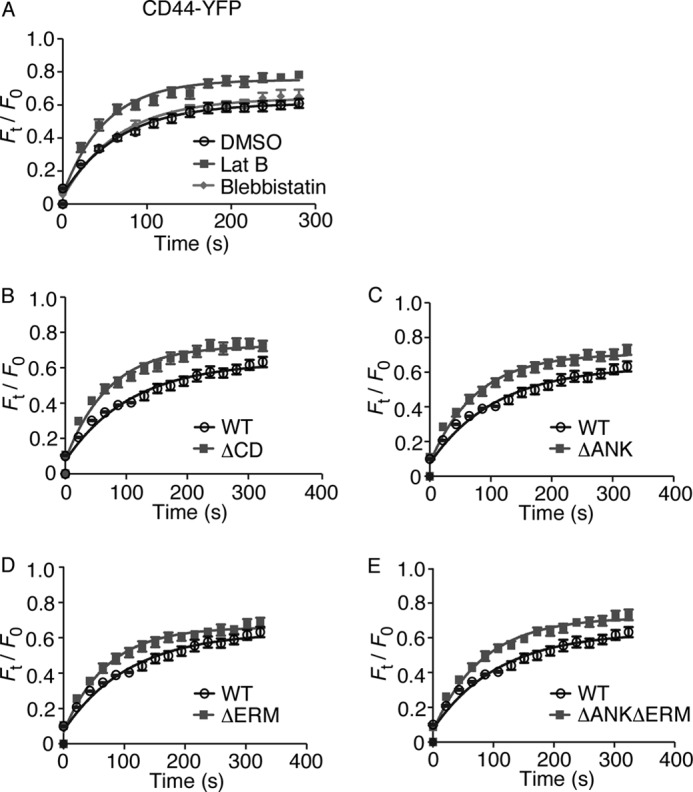
Disrupting interactions with the actin cytoskeleton increases the mobility of CD44. Shown are plots (A–E) of normalized fluorescence intensity versus time after photobleaching a small area of plasma membrane (6.72 × 3.36 μm) in transfected K562 cells expressing the indicated CD44-YFP construct. Cells in A were pretreated with DMSO control, latrunculin B (Lat B), or blebbistatin. FRAP curves from four independent trials with five cells per trial were fitted to a function for monoexponential recovery of fluorescence to derive τ½ and Fmax as reported in Table 3. The data represent the mean. Error bars represent S.E.
Disrupting the Binding Site for Ankyrin but Not for ERM Proteins Impairs CD44-mediated Neutrophil Rolling on E-selectin
PSGL-1−/−/CD44−/− neutrophils roll faster than PSGL-1−/− neutrophils on E-selectin, establishing the contribution of CD44 as a “rolling ligand” for E-selectin (6, 9, 10). To examine whether CD44 interactions with the cytoskeleton affect this function, we transduced retroviruses encoding cDNAs for WT or mutant CD44 into conditionally immortalized myeloid progenitors from PSGL-1−/−/CD44−/− mice. Clones of these progenitors were then differentiated into neutrophils ex vivo. These cells retain the properties of neutrophils differentiated in vivo, including the glycosylation of selectin ligands as demonstrated here. Cell sorting was used to normalize the expression of the CD44 constructs (Fig. 7A). We perfused the neutrophils over immobilized E-selectin and used video microscopy and image analysis software to monitor the numbers of rolling cells and their velocities at a wall shear stress of 1 dyne/cm2 (Fig. 7, B and C) and their resistance to detachment in response to increasing wall shear stress (Fig. 7, D and E). Differentiated PSGL-1−/−/CD44−/− neutrophils expressing exogenous WT CD44 rolled in greater numbers and with slower velocities than mock-transduced PSGL-1−/−/CD44−/− neutrophils (Fig. 7, B–E), confirming a CD44-dependent contribution to rolling. Unlike cells expressing WT CD44, neutrophils expressing the ΔANK mutant rolled in smaller numbers and with higher velocities. Neutrophils expressing the ΔANKΔERM mutant also rolled in smaller numbers but with velocities closer to that of WT than ΔANK. Neutrophils expressing the ΔERM mutant rolled like neutrophils expressing WT CD44. These data suggest that larger but looser CD44 clusters as in the ΔANK mutant impair rolling on E-selectin. Interestingly, mutating the binding sites for both adaptors led to a milder rolling defect despite the more complete inhibition of clustering.
FIGURE 7.

Disrupting the binding site for ankyrin but not for ERM proteins impairs CD44-mediated neutrophil rolling on E-selectin. A, flow cytometric analysis of transduced ex vivo differentiated PSGL-1−/−/CD44−/− neutrophils expressing the indicated CD44 construct incubated with phycoerythrin (PE)-labeled isotype control mAb or anti-CD44 mAb. Cell number (B) or velocity (C) of transduced neutrophils rolling on E-selectin at a wall shear stress of 1 dyne/cm2 is shown. Cell number (D) or velocity (E) of transduced neutrophils rolling on E-selectin in response to stepwise increases in wall shear stress is shown. Note that no rolling ΔANK cells remained at 25 dynes/cm2. The data represent the mean of at least three experiments. Error bars represent S.E. *, p < 0.05; ***, p < 0.001.
Disrupting the Binding Site for Ankyrin but Not for ERM Proteins Impairs CD44-mediated Signaling in Neutrophils
E-selectin engagement of PSGL-1 or CD44 on neutrophils triggers a signaling pathway that partially activates integrin αLβ2. The earliest identified step in this pathway is tyrosine phosphorylation of the SFKs Hck, Lyn, and Fgr (6, 41). To examine whether CD44 interactions with the cytoskeleton affect this function, we incubated differentiated PSGL-1−/−/CD44−/− neutrophils expressing exogenous WT or mutant CD44 on immobilized F(ab′)2 fragments of anti-CD44 or isotype control mAb. We used anti-CD44 mAb as a surrogate for E-selectin that binds only to CD44. Cell lysates were analyzed by Western blotting with antibodies against total SFKs (cross-reactive with Hck, Lyn, and Fgr) or activated phospho-SFKs (SFKs bearing phosphotyrosine 416 in the catalytic domain). Anti-CD44 mAb, but not control mAb, activated SFKs on neutrophils expressing WT or ΔERM CD44 but not on neutrophils expressing ΔANK CD44 (Fig. 8).
FIGURE 8.
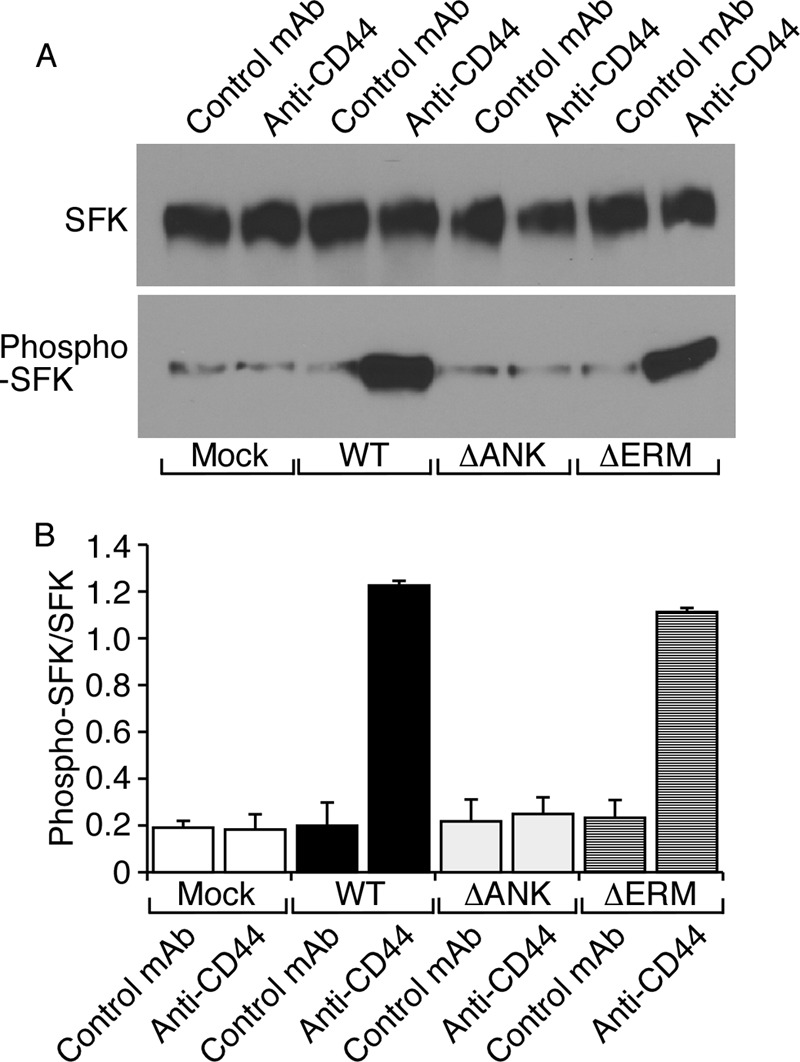
Disrupting the binding site for ankyrin but not for ERM proteins impairs CD44-mediated signaling in neutrophils. Ex vivo differentiated neutrophils expressing the indicated CD44 construct were incubated on immobilized F(ab′)2 fragments of isotype control or anti-CD44 mAb. Lysates were probed by Western blotting with antibodies to total SFK or phospho-SFK. A, representative Western blot. B, the ratio of phospho-SFK to total SFK was measured by densitometry. The data represent the mean of three experiments. Error bars represent S.E.
DISCUSSION
Our results provide the first description of the nanometer scale organization of CD44 on the plasma membrane of intact cells. CD44 formed actin-dependent clusters that required the cytoplasmic domain probably through interactions with ERM proteins, ankyrin, and other adaptors. At least some clusters were very tight. They could be cross-linked within a 1.14-nm spacer, and they were detected by FRET, which is maximal at distances less than 2 nm. Most CD44 clusters detected by N&B analysis (nanometer scale motion) appeared to be doublets or triplets. FRAP documented larger scale, actin-dependent restraint on CD44 movement. Neither clustering nor restraint of CD44 required actomyosin tension.
Many studies have reported binding of ERM proteins and ankyrin to separate regions of the CD44 cytoplasmic domain (14–18). Whether this binding affects CD44 membrane topography has been unclear. Our FRET data demonstrated that both binding sites must be disrupted to reduce CD44 clustering (Fig. 9). Surprisingly, N&B analysis indicated that disrupting only the ankyrin-binding site increases CD44 brightness, suggesting larger clusters. The apparent discrepancy of the FRET and N&B data could be explained by differences in cluster spacing. Small CD44 clusters may be separated by less than 2 nm at which FRET is maximal. Larger ΔANK clusters may be separated by 2–8 nm at which FRET efficiency declines rapidly (Fig. 9). Overall FRET efficiency may not change because some smaller clusters remain where FRET is efficient (Fig. 9). If this interpretation is correct, ankyrin may link subsets of CD44 to the spectrin network, restraining its interaction with other cytoskeletal components. Loss of this restraint results in larger but looser CD44 clusters.
FIGURE 9.

Model for cytoskeletal regulation of CD44 membrane organization. Interactions of the cytoplasmic domain with ERM proteins, ankyrin, and perhaps other adaptors link CD44 to spectrin and actin networks. These interactions cause CD44 to form tight clusters, most of which probably comprise two to three molecules. Disrupting the ERM-binding site does not detectably alter clustering, although spacing between molecules might increase slightly. Disrupting the ankyrin-binding site allows some CD44 molecules to coalesce into larger but looser clusters. Disrupting both binding sites markedly reduces clustering. See “Discussion” for details. d, distance.
Deleting both ERM- and ankyrin-binding sites markedly reduced clustering as measured by FRET and N&B. This suggests that ERM proteins and ankyrin are the major adaptors linking CD44 to the actin cytoskeleton. As assessed by FRAP, however, deleting the entire cytoplasmic domain was required to increase CD44 mobility to the level observed in cells treated with latrunculin B. This supports the concept that the cytoplasmic domain binds to at least one more adaptor. One candidate is a C-terminal binding site for PDZ domain-containing proteins (14), which link to actin through ERM proteins (42, 43). Unlike the FRAP data for CD44, FRAP analysis of PSGL-1 revealed that mutating the ERM-binding site in the cytoplasmic domain increases mobility to the level of latrunculin B-treated cells (35). Thus, PSGL-1 may recognize fewer cytoskeletal adaptors than CD44. This might partially explain why PSGL-1 is concentrated on microvilli of leukocytes (44), whereas CD44 is mostly on non-microvillar regions of the plasma membrane (45).
Co-immunoprecipitation experiments have suggested that the transmembrane domain mediates self-association of CD44 in Triton X-100-insoluble membrane fractions representing lipid rafts (46–48). Our data demonstrate that CD44 requires the cytoplasmic domain to cluster in intact cells. CD44 may also associate with lipid rafts, which are in turn regulated by the cytoskeleton (24, 49).
Because of its weak binding to E-selectin (11), we hypothesized that CD44 must cluster to mediate optimal neutrophil rolling on E-selectin. CD44-dependent rolling was similar in PSGL-1−/−/CD44−/− neutrophils reconstituted with WT or ΔERM CD44, consistent with the minimal effect of the ERM binding mutation on clustering. However, we observed only mild rolling defects in ΔANKΔERM neutrophils despite reduced CD44 clustering. Moreover, ΔANK neutrophils had greater rolling defects perhaps because of larger but looser clusters spaced at greater intervals. These data suggest that clustering per se does not adequately explain the ability of CD44 to mediate neutrophil rolling on E-selectin.
Cross-linking CD44 activates SFKs and other signaling steps in multiple cell types (14, 23). CD44 co-immunoprecipitates with SFKs perhaps in part through co-association with detergent-insoluble membrane domains (17, 50, 51). We observed that immobilized anti-CD44 mAb activates SFKs in ex vivo differentiated neutrophils expressing WT or ΔERM CD44 but not in neutrophils expressing ΔANK CD44. Our data are consistent with the failure of ΔANK CD44 to associate with SFKs in a prostate cell line (17). Signaling might require a direct interaction of CD44 with ankyrin to organize signaling components, including SFKs, at the membrane. Alternatively or in addition, the ankyrin-binding site of CD44 may bind directly to SFKs. These mechanisms may explain how E-selectin triggers CD44-dependent activation of β2 integrins on rolling neutrophils (6). PSGL-1 also requires its cytoplasmic domain to activate β2 integrins as neutrophils roll on P- or E-selectin (6, 29). However, the PSGL-1 cytoplasmic domain is not known to bind ankyrin, and it might transduce signals by a different mechanism.
The mutations/deletions in the cytoplasmic domain of CD44 may affect functions that we did not examine. However, our approach combining CD44 mutants and latrunculin B treatments provides strong evidence for actin-dependent clustering of CD44 through interactions of the cytoplasmic domain with ERM proteins, ankyrin, and perhaps other adaptors. How these nanometer scale domains affect CD44 function in leukocytes and other cells is a promising area for further study.
Acknowledgments
We thank Gurunadh Chichili for initial advice on FRET experiments, Ben Fowler for assistance with microscopy, Benjamin Smith (University of Oklahoma Samuel Roberts Noble Microscopy Laboratory) for assistance with N&B data collection, and Peter Nagy (University of Debrecen, Debrecen, Hungary) for generously providing his customized nb_tools and for advice on N&B data analysis.
This work was supported, in whole or in part, by National Institutes of Health Grants HL03463 and HL085607.
- PSGL-1
- P-selectin glycoprotein ligand-1
- CFP
- cyan fluorescent protein
- ERM
- ezrin/radixin/moesin
- FRAP
- fluorescence recovery after photobleaching
- N&B
- number and brightness
- SFK
- Src family kinase
- ANK
- ankyrin
- BS3
- bis(sulfosuccinimidyl) suberate.
REFERENCES
- 1. Ley K., Laudanna C., Cybulsky M. I., Nourshargh S. (2007) Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat. Rev. Immunol. 7, 678–689 [DOI] [PubMed] [Google Scholar]
- 2. Nourshargh S., Hordijk P. L., Sixt M. (2010) Breaching multiple barriers: leukocyte motility through venular walls and the interstitium. Nat. Rev. Mol. Cell Biol. 11, 366–378 [DOI] [PubMed] [Google Scholar]
- 3. McEver R. P., Zhu C. (2010) Rolling cell adhesion. Annu. Rev. Cell Dev. Biol. 26, 363–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kunkel E. J., Ley K. (1996) Distinct phenotype of E-selectin-deficient mice—E-selectin is required for slow leukocyte rolling in vivo. Circ. Res. 79, 1196–1204 [DOI] [PubMed] [Google Scholar]
- 5. Zarbock A., Lowell C. A., Ley K. (2007) Spleen tyrosine kinase Syk is necessary for E-selectin-induced αLβ2 integrin-mediated rolling on intercellular adhesion molecule-1. Immunity 26, 773–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yago T., Shao B., Miner J. J., Yao L., Klopocki A. G., Maeda K., Coggeshall K. M., McEver R. P. (2010) E-selectin engages PSGL-1 and CD44 through a common signaling pathway to induce integrin αLβ2-mediated slow leukocyte rolling. Blood 116, 485–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lefort C. T., Ley K. (2012) Neutrophil arrest by LFA-1 activation. Front. Immunol. 3, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xia L., Sperandio M., Yago T., McDaniel J. M., Cummings R. D., Pearson-White S., Ley K., McEver R. P. (2002) P-selectin glycoprotein ligand-1-deficient mice have impaired leukocyte tethering to E-selectin under flow. J. Clin. Investig. 109, 939–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Katayama Y., Hidalgo A., Chang J., Peired A., Frenette P. S. (2005) CD44 is a physiological E-selectin ligand on neutrophils. J. Exp. Med. 201, 1183–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hidalgo A., Peired A. J., Wild M. K., Vestweber D., Frenette P. S. (2007) Complete identification of E-selectin ligands on neutrophils reveals distinct functions of PSGL-1, ESL-1, and CD44. Immunity 26, 477–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yago T., Fu J., McDaniel J. M., Miner J. J., McEver R. P., Xia L. (2010) Core 1-derived O-glycans are essential E-selectin ligands on neutrophils. Proc. Natl. Acad. Sci. U.S.A. 107, 9204–9209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sreeramkumar V., Leiva M., Stadtmann A., Pitaval C., Ortega-Rodríguez I., Wild M. K., Lee B., Zarbock A., Hidalgo A. (2013) Coordinated and unique functions of the E-selectin ligand ESL-1 during inflammatory and hematopoietic recruitment in mice. Blood 122, 3993–4001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nácher M., Blázquez A. B., Shao B., Matesanz A., Prophete C., Berin M. C., Frenette P. S., Hidalgo A. (2011) Physiological contribution of CD44 as a ligand for E-selectin during inflammatory T-cell recruitment. Am. J. Pathol. 178, 2437–2446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Thorne R. F., Legg J. W., Isacke C. M. (2004) The role of the CD44 transmembrane and cytoplasmic domains in co-ordinating adhesive and signalling events. J. Cell Sci. 117, 373–380 [DOI] [PubMed] [Google Scholar]
- 15. Yonemura S., Hirao M., Doi Y., Takahashi N., Kondo T., Tsukita S. (1998) Ezrin/radixin/moesin (ERM) proteins bind to a positively charged amino acid cluster in the juxta-membrane cytoplasmic domain of CD44, CD43, and ICAM-2. J. Cell Biol. 140, 885–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Legg J. W., Isacke C. M. (1998) Identification and functional analysis of the ezrin-binding site in the hyaluronan receptor, CD44. Curr. Biol. 8, 705–708 [DOI] [PubMed] [Google Scholar]
- 17. Zhu D., Bourguignon L. Y. (1998) The ankyrin-binding domain of CD44s is involved in regulating hyaluronic acid-mediated functions and prostate tumor cell transformation. Cell Motil. Cytoskeleton 39, 209–222 [DOI] [PubMed] [Google Scholar]
- 18. Lokeshwar V. B., Fregien N., Bourguignon L. Y. (1994) Ankyrin-binding domain of CD44(GP85) is required for the expression of hyaluronic acid-mediated adhesion function. J. Cell Biol. 126, 1099–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bennett V., Healy J. (2009) Membrane domains based on ankyrin and spectrin associated with cell-cell interactions. Cold Spring Harb. Perspect. Biol. 1, a003012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brown K. L., Birkenhead D., Lai J. C., Li L., Li R., Johnson P. (2005) Regulation of hyaluronan binding by F-actin and colocalization of CD44 and phosphorylated ezrin/radixin/moesin (ERM) proteins in myeloid cells. Exp. Cell Res. 303, 400–414 [DOI] [PubMed] [Google Scholar]
- 21. Lesley J., English N., Charles C., Hyman R. (2000) The role of the CD44 cytoplasmic and transmembrane domains in constitutive and inducible hyaluronan binding. Eur. J. Immunol. 30, 245–253 [DOI] [PubMed] [Google Scholar]
- 22. Gal I., Lesley J., Ko W., Gonda A., Stoop R., Hyman R., Mikecz K. (2003) Role of the extracellular and cytoplasmic domains of CD44 in the rolling interaction of lymphoid cells with hyaluronan under physiologic flow. J. Biol. Chem. 278, 11150–11158 [DOI] [PubMed] [Google Scholar]
- 23. Ponta H., Sherman L., Herrlich P. A. (2003) CD44: from adhesion molecules to signalling regulators. Nat. Rev. Mol. Cell Biol. 4, 33–45 [DOI] [PubMed] [Google Scholar]
- 24. Chichili G. R., Rodgers W. (2007) Clustering of membrane raft proteins by the actin cytoskeleton. J. Biol. Chem. 282, 36682–36691 [DOI] [PubMed] [Google Scholar]
- 25. Wang G. G., Calvo K. R., Pasillas M. P., Sykes D. B., Häcker H., Kamps M. P. (2006) Quantitative production of macrophages or neutrophils ex vivo using conditional Hoxb8. Nat. Methods 3, 287–293 [DOI] [PubMed] [Google Scholar]
- 26. Gimferrer I., Hu T., Simmons A., Wang C., Souabni A., Busslinger M., Bender T. P., Hernandez-Hoyos G., Alberola-Ila J. (2011) Regulation of GATA-3 expression during CD4 lineage differentiation. J. Immunol. 186, 3892–3898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Protin U., Schweighoffer T., Jochum W., Hilberg F. (1999) CD44-deficient mice develop normally with changes in subpopulations and recirculation of lymphocyte subsets. J. Immunol. 163, 4917–4923 [PubMed] [Google Scholar]
- 28. Zacharias D. A., Violin J. D., Newton A. C., Tsien R. Y. (2002) Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science 296, 913–916 [DOI] [PubMed] [Google Scholar]
- 29. Miner J. J., Xia L., Yago T., Kappelmayer J., Liu Z., Klopocki A. G., Shao B., McDaniel J. M., Setiadi H., Schmidtke D. W., McEver R. P. (2008) Separable requirements for cytoplasmic domain of PSGL-1 in leukocyte rolling and signaling under flow. Blood 112, 2035–2045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Abdisalaam S., Davis A. J., Chen D. J., Alexandrakis G. (2014) Scanning fluorescence correlation spectroscopy techniques to quantify the kinetics of DNA double strand break repair proteins after γ-irradiation and bleomycin treatment. Nucleic Acids Res. 42, e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nagy P., Claus J., Jovin T. M., Arndt-Jovin D. J. (2010) Distribution of resting and ligand-bound ErbB1 and ErbB2 receptor tyrosine kinases in living cells using number and brightness analysis. Proc. Natl. Acad. Sci. U.S.A. 107, 16524–16529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Digman M. A., Dalal R., Horwitz A. F., Gratton E. (2008) Mapping the number of molecules and brightness in the laser scanning microscope. Biophys. J. 94, 2320–2332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rossow M. J., Sasaki J. M., Digman M. A., Gratton E. (2010) Raster image correlation spectroscopy in live cells. Nat. Protoc. 5, 1761–1774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hellriegel C., Caiolfa V. R., Corti V., Sidenius N., Zamai M. (2011) Number and brightness image analysis reveals ATF-induced dimerization kinetics of uPAR in the cell membrane. FASEB J. 25, 2883–2897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shao B., Yago T., Coghill P. A., Klopocki A. G., Mehta-D'souza P., Schmidtke D. W., Rodgers W., McEver R. P. (2012) Signal-dependent slow leukocyte rolling does not require cytoskeletal anchorage of P-selectin glycoprotein ligand-1 (PSGL-1) or integrin αLβ2. J. Biol. Chem. 287, 19585–19598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yago T., Leppänen A., Qiu H., Marcus W. D., Nollert M. U., Zhu C., Cummings R. D., McEver R. P. (2002) Distinct molecular and cellular contributions to stabilizing selectin-mediated rolling under flow. J. Cell Biol. 158, 787–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yang C., Cao M., Liu H., He Y., Xu J., Du Y., Liu Y., Wang W., Cui L., Hu J., Gao F. (2012) The high and low molecular weight forms of hyaluronan have distinct effects on CD44 clustering. J. Biol. Chem. 287, 43094–43107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Legg J. W., Lewis C. A., Parsons M., Ng T., Isacke C. M. (2002) A novel PKC-regulated mechanism controls CD44 ezrin association and directional cell motility. Nat. Cell Biol. 4, 399–407 [DOI] [PubMed] [Google Scholar]
- 39. Coué M., Brenner S. L., Spector I., Korn E. D. (1987) Inhibition of actin polymerization by latrunculin A. FEBS Lett. 213, 316–318 [DOI] [PubMed] [Google Scholar]
- 40. Kovács M., Tóth J., Hetényi C., Málnási-Csizmadia A., Sellers J. R. (2004) Mechanism of blebbistatin inhibition of myosin II. J. Biol. Chem. 279, 35557–35563 [DOI] [PubMed] [Google Scholar]
- 41. Zarbock A., Abram C. L., Hundt M., Altman A., Lowell C. A., Ley K. (2008) PSGL-1 engagement by E-selectin signals through Src kinase Fgr and ITAM adapters DAP12 and FcRγ to induce slow leukocyte rolling. J. Exp. Med. 205, 2339–2347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fehon R. G., McClatchey A. I., Bretscher A. (2010) Organizing the cell cortex: the role of ERM proteins. Nat. Rev. Mol. Cell Biol. 11, 276–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Levi M. (2003) Role of PDZ domain-containing proteins and ERM proteins in regulation of renal function and dysfunction. J. Am. Soc. Nephrol. 14, 1949–1951 [DOI] [PubMed] [Google Scholar]
- 44. Moore K. L., Patel K. D., Bruehl R. E., Li F., Johnson D. A., Lichenstein H. S., Cummings R. D., Bainton D. F., McEver R. P. (1995) P-selectin glycoprotein ligand-1 mediates rolling of human neutrophils on P-selectin. J. Cell Biol. 128, 661–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. von Andrian U. H., Hasslen S. R., Nelson R. D., Erlandsen S. L., Butcher E. C. (1995) A central role for microvillous receptor presentation in leukocyte adhesion under flow. Cell 82, 989–999 [DOI] [PubMed] [Google Scholar]
- 46. Neame S. J., Uff C. R., Sheikh H., Wheatley S. C., Isacke C. M. (1995) CD44 exhibits a cell type dependent interaction with Triton X-100 insoluble, lipid rich, plasma membrane domains. J. Cell Sci. 108, 3127–3135 [DOI] [PubMed] [Google Scholar]
- 47. Perschl A., Lesley J., English N., Hyman R., Trowbridge I. S. (1995) Transmembrane domain of CD44 is required for its detergent insolubility in fibroblasts. J. Cell Sci. 108, 1033–1041 [DOI] [PubMed] [Google Scholar]
- 48. Li R., Walker J. R., Johnson P. (1998) Chimeric CD4/CD44 molecules associate with CD44 via the transmembrane region and reduce hyaluronan binding in T cell lines. Eur. J. Immunol. 28, 1745–1754 [DOI] [PubMed] [Google Scholar]
- 49. Goswami D., Gowrishankar K., Bilgrami S., Ghosh S., Raghupathy R., Chadda R., Vishwakarma R., Rao M., Mayor S. (2008) Nanoclusters of GPI-anchored proteins are formed by cortical actin-driven activity. Cell 135, 1085–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ilangumaran S., Briol A., Hoessli D. C. (1998) CD44 selectively associates with active Src family protein tyrosine kinases Lck and Fyn in glycosphingolipid-rich plasma membrane domains of human peripheral blood lymphocytes. Blood 91, 3901–3908 [PubMed] [Google Scholar]
- 51. Lefebvre D. C., Lai J. C., Maeshima N., Ford J. L., Wong A. S., Cross J. L., Johnson P. (2010) CD44 interacts directly with Lck in a zinc-dependent manner. Mol. Immunol. 47, 1882–1889 [DOI] [PubMed] [Google Scholar]
- 52. de Laat S. W., van der Saag P. T., Elson E. L., Schlessinger J. (1980) Lateral diffusion of membrane lipids and proteins during the cell cycle of neuroblastoma cells. Proc. Natl. Acad. Sci. U.S.A. 77, 1526–1528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Damjanovich S., Edidin M., Szollosi J., Tron T. (1994) Mobility and Proximity in Biological Membranes, CRC Press, Boca Raton, FL [Google Scholar]