

Abstract
A facile and robust RNA preparation protocol was developed by combining rolling circle transcription (RCT) with RNA cleavage by RNase H. Circular DNA with a complementary sequence was used as the template for promoter-free transcription. With the aid of a 2′-O-methylated DNA, the RCT-generated tandem repeats of the desired RNA sequence were disconnected at the exact end-to-end position to harvest the desired RNA oligomers. Compared with the template DNA, more than 4 × 103 times the amount of small RNA products were obtained when modest cleavage was carried out during transcription. Large amounts of RNA oligomers could easily be obtained by simply increasing the reaction volume.
Keywords: biosynthesis, RNase H, RNA oligonucleotides, rolling circle transcription, site-specific cleavage
Introduction
RNAs play an important role in biology and its related areas. The discovery of small catalytic RNAs (ribozymes) in the 1980s shed light on the hypothesis that life passed through an “RNA world” before proteins and DNA started to serve as the functional components and the genetic carrier, respectively.1,2 In recent years, small RNAs have produced much attention due to their versatile biological activities, such as gene silencing, cell apoptosis, and hormone secretion.3,4,5 For example, more than 20,000 sequences of microRNAs have been identified, some of which are promising candidate regulation factors, vaccine materials, and antivirus factors.6,7,8 Moreover, studies on microRNAs for therapeutic purposes in vivo are now emerging as a research hotspot.6,9,10 Accordingly, a higher caliber is required for synthesizing RNA oligomers in terms of both quality and quantity. Currently, organic synthesis serves as the predominant avenue for preparing RNA oligomers. However, in comparison with DNA, RNA is much more expensive due to the costly RNA phosphoramidite monomer and the low yield caused by special protection/deprotection steps on the 2′-OH of the ribose.11 Usually, commercially supplied RNA is only available in microgram (<100 nmol) quantities, and synthesizing milligram quantities of RNA oligomer is difficult.
“Runoff transcription”, a process based on in vitro transcription using a linear double-stranded DNA as the template, is an alternative approach to synthesize RNA.12 Compared to chemical approaches, it provides higher quantities for synthesizing longer RNA oligomers (>50 nt). Because a double-stranded promoter is essential for RNA polymerase to transcribe with high efficiency, runoff transcription relies on DNAzyme or Ribozyme to remove the redundant RNA encoded by the promoter sequence. However, DNAzymes used for cleavage have limited cutting sites, such as 5′-AU-3′ or 5′-GU-3′ for DNAzyme 10–23, and ribozymes are the RNAs themselves.13,14,15 Another recognized drawback of runoff transcription is that the length of the products are often nonhomogeneous.16 Furthermore, an additional adenine nucleotide is usually attached to the 3′ termini of the RNA products by T7 and Sp6 RNA polymerase after being transcribed to the end of the DNA template.12 To eliminate the redundant nucleotide, a longer template must be used and the redundant part is cleaved later. This additional process makes the protocol tricky and labor intensive. Therefore, protocols based on “runoff transcription” cannot be used in place of chemical synthesis methods.
As a comparable method to runoff transcription, rolling circle transcription (RCT), using small circular single-stranded DNA as the template, has been well investigated in the past two decades.17,18,19 Interestingly, transcription via the rolling circle mechanism could happen in the absence of any canonical promoters and generate transcripts that are tandemly repeated sequences complementary to the circular template.20 Drawbacks of runoff transcription, such as the dependence of the promoter sequence, transcription abortion and the 3′ add-on nucleotide, are circumvented once RCT is employed. In the practical sense, RCT seems to have potential and use for the generation of certain biologically relevant RNAs, but the products are multimers of the desired RNAs. The multimeric transcript must be cut at specific sites to yield a large amount of small RNAs of the desired length. We noticed that RNase H, known for its unique property to hydrolyze RNA strands in a DNA/RNA heteroduplex, could perform site-specific RNA cleavage once the DNA strand is subjected to a well-designed 2′-O-methyl modification.21 By designing the sequence, length, and positions for 2′-O-methylation of the modified DNA, small RNAs may be obtained from RCT products after disconnection by RNase H.
Here, we demonstrate a facile and efficient enzymatic RNA synthesis strategy, dubbed RCT-SSD, which combines RCT with the site-specific disconnection (SSD) of long RNA transcripts. The transcription was carried out by T7 RNA polymerase and the precise cleavage of the transcribed RNA was accomplished by RNase H with the aid of a 2′-O-methylated DNA (Aid-DNA).
Results
The detailed strategy is illustrated in Figure 1. First, the 5′-phosphorated DNA oligomer (cDNA), which is complementary to the RNA sequence to be synthesized, is circularized by a DNA ligase with the help of a splint DNA. The splint is complementary to the two ends of the cDNA. Then, the splint serves as the primer to initiate RCT with T7 RNA polymerase, which can initiate transcription in the absence of its promoter.20 The transcript is the covalently joined tandem repeats of the desired small RNA, and it can be cleaved at the joint positions by RNase H with the aid of a modified DNA. The novel design of the 2′-O-methyl modified DNA presented here was based on several reports.21,22,23 Considering that the long transcript may impede further transcription, RNase H and the Aid-DNA can also be added during transcription.
Figure 1.

Strategy for rolling circle transcription (RCT) site-specific disconnection (SSD) synthesis. cDNA is circularized to form a circular DNA template, and long RNA strands are generated by RCT consisting of tandem repeats of desired RNA. With the help of Aid-DNA, RNase H disconnects the transcript to generate thousands of copies of the desired RNA.
Synthesis of microRNA-16 by RCT-SSD
To demonstrate the feasibility of our methodology, we synthesized the 22-nt-long microRNA-16 (mir-16, see Table 1 for sequence) that regulates cell cycle progression and the metabolism of the amyloid precursor protein as an example.25,26 In the presence of 1.0 μmol/l of the splint DNA, 1.0 μmol/l of cDNA was ligated to form a circle at 30 °C by T4 DNA ligase (see detailed synthesis steps in the Materials and Methods). As shown in Figure 2a, the ligation products appeared as three major bands. By comparing the circular DNA markers (C72, C66, C60, see Supplementary Material for their sequences), the main product was determined to be the 44-nt-long DNA circle, dimer of the cDNA. The other two bands were assigned as the 66-nt-long circular trimer and the 22-nt-long circular monomer. The weaker band below the three major bands was the 22 nt linear cDNA that failed to be circularized. Obviously, RCT of all of these circular templates with various lengths can generate the same products, which are the tandem repeats of the desired RNA. Accordingly, the mixture of ligation products was used directly for the following RCT without further purification.
Table 1. Sequence of oligomers used in this studya.

Figure 2.

Synthesis of the mir-16 oligomer using rolling circle transcription (RCT) site-specific disconnection (SSD). (a) Ligation products of the cDNA and transcript of the RCT reaction on circular cDNA. C72, C66, and C60 are synthesized circular ssDNA oligomers used as markers; Lane L, RNA ladder. Samples were separated by 14% denaturing PAGE (8 mol/l urea). (b) Analysis of synthesized mir-16. Lane 1, chemically synthesized mir-16; lane 2, RNase H was absent during RCT; lane 3, RNase-H (2.5 U/ml) was present during RCT. Samples were separated by 20% denaturing PAGE (8 mol/l urea). Other conditions used for RCT were: 0.5 μmol/l cDNA, 2.5 U/ml RNA polymerase, 37 °C, 2 hours; other conditions used for SSD were, 125 U/ml RNase H, 1.0 μmol/l Aid-DNA, 37 °C, 2 hours.
After RCT with T7 RNA polymerase at 37 °C for 2 hours, RNAs longer than 1,000 nt were observed (lane RCT in Figure 2a), indicating that long RNA strands consisting of tandem repetitive RNA sequences could be transcribed from the circular template. Interestingly, RNA transcription was initiated from the 3′ end of the splint DNA in the absence of a promoter sequence, and T7 RNA polymerase could displace the synthesized RNA strand (but not the sense DNA strand when using dsDNA as the template) for continuous transcription. Considering that the cDNA can form a DNA/RNA hybrid with the synthesized RNA and cause random RNA digestion by RNase H (data not shown), the cDNA was removed by DNase I treatment before SSD. RNase H-mediated SSD was accomplished using Aid-DNA-16-1 with four 2′-O-methyl-modified nucleotides (Table 1). Aid-DNA-16-1 could form a 12 bp DNA/RNA heteroduplex on the target region of the RNA transcript, which may undergo a sequence-directed cleavage by RNase H. As shown in Figure 2b, a single clear band was observed with the same mobility as that of chemically synthesized mir-16 with 5′-phosphate, indicating that the desired microRNA oligomer was successfully synthesized with high purity. Shibahara et al.24 first demonstrated that a 12-nt-long 2′-O-methyl-modified DNA with seven modified nucleotides (similar as Aid-DNA-16-2, Table 1) could be used for site-specific digestion of RNA. Here, we demonstrated that the number of modified nucleotides could be reduced. DNA with only two modified nucleotides could also be used to mediate site-specific cleavage, although more strict conditions must be applied (data not shown).
During the RCT reaction, the synthesized long RNA strand accumulated around the circular template and may block further RNA synthesis.17 Considering that this block of RNA synthesis may be alleviated if the newly synthesized long RNA strand was removed, small amounts of RNase H (2.5 U/ml) and Aid-DNA-16-1 were added before RCT. As expected, more products were observed (lane 3, in Figure 2b). When much more RNase H was added (125 U/ml), RCT efficiency was reduced (data not shown), likely because the transcribed long RNA strand that hybridized to the cDNA was also quickly digested by RNase H in a non-specific fashion. In 2006, Seyhan et al.27 reported a strategy that directly utilized RCT to prepare shRNA or siRNA. A dumbbell template was used and the transcripts were introduced into cells to function as potent and specific gene inhibitors. The disconnection of the dsRNA was carried out by recombinant Dicer enzyme in vitro and by intracellular processing in vivo. The research work demonstrated the potential of RCT as a general tool to develop advanced protocols. However, only functional RNAs based on duplex structures can be generated with this strategy, while single-stranded small RNA oligomers could not be obtained. Moreover, the template used in Seyhan's protocol must be longer than the desired RNAs. For RCT-SSD, cDNA templates are of the same length as the corresponding RNA oligomers, and small RNA oligomers both in duplex and single-stranded form can be synthesized.
Interestingly, we found that microRNA was obtained even when 1.0 nmol/l of cDNA was used (Figure 3a). When the reaction time was lengthened from 2 to 4 hours, the amount of microRNA product was greatly increased, but no further improvement was observed after a 6-hour incubation, indicating that the reaction was saturated after 4 hours. Considering that the accumulation of pyrophosphate may inhibit the transcription, we added inorganic pyrophosphatase in the RCT reaction to hydrolyze the pyrophosphate and improve the efficiency.28,29 As expected, more microRNA products were obtained from 10 and 100 nmol/l cDNA template (Figure 3b). Quantification was performed by analyzing the intensity of the bands in Figure 3b (see Supplementary Figure S1 in Supplementary Material). Using the chemically synthesized microRNA (lane S, Figure 3b) as a reference, the quantification results showed that ~10 μmol/l microRNA oligomer was present in the final SSD reaction solution using 10 nmol/l cDNA templates in RCT in the presence of inorganic pyrophosphatase. As the RCT reaction solution was diluted a total of four times for DNase I treatment and the SSD reaction, it can be concluded that at least 4 × 103 of microRNA products (compared to DNA template) could be synthesized. This efficiency is greater than that of runoff transcription, which was reported to synthesize 102~103 times of RNA transcript compared to the template.30,31 Other merits of our approach can be summarized as: (i) no smaller by-products; (ii) a short cDNA template with the same length as the RNA product can be used; (iii) the synthesized RNA has the expected length without a 3′ add-on nucleotide; (iv) the RNA product attaches a 5′-phosphate which is the same as natural occurring microRNAs in vivo.32
Figure 3.

MicroRNA synthesized by rolling circle transcription (RCT) site-specific disconnection (SSD) with various concentrations of circular cDNA template. (a) Time course of RCT in the absence of inorganic pyrophosphatase (IPP). (b) Effect of IPP on the amount of RNA products. Lane L, 25 nt RNA marker; Lane S, standard sample of chemically synthesized mir-16 (1 μl, 100 μmol/l). In (b), RCT was processed for 4 hours, with 0.1 μmol/l Aid-DNA-16-1 and 1.0 U/ml IPP.
In the SSD reaction, disconnections at any specific sites of the transcript composed of repetitive sequences will generate microRNA oligomers with the same length. To determine whether the transcript was cleaved at the expected site, a chimeric DNA/RNA oligomer was used as the substrate (Figure 4a). If the cleavage occurred at the correct position, a 17-nt product would be obtained. Otherwise, cleavage products that were longer or shorter than 17 nt would be observed. As shown in Figure 4b, the observed products were the exact length of 17 nt, deviating from 16-nt and 18-nt oligomers, suggesting that the long RNA strand was disconnected at the expected site. Therefore, it can be concluded that the obtained RNA oligomer from RCT-SSD had the correct sequence.
Figure 4.

Determination of the cleavage site by site-specific disconnection (SSD). (a) Sequences of the fluorescein isothiocyanate (FITC)-labelled DNA/RNA chimeric oligomer. The red text indicates the DNA sequence, and the other text indicates the RNA portion that is complementary to the Aid-DNA (purple, italicized, underlined characters indicate the sequence of 2′-O-methyl-modified nucleotides). The expected cleavage site “UG” was shown by the green arrow. (b) Analysis of the cleavage product. Compared to the standard samples, the length of cleavage product was determined to be 17 nt. Other conditions used were 1.0 μmol/l substrate and Aid-DNA-16-1, 125 U/ml RNase H, 37 °C, 40 minutes, 20% denaturing urea polyacrylamide gel electrophoresis (PAGE).
Separation and purification of synthesized RNA oligomers
As only 0.1 μmol/l Aid-DNA-16-1 (~1/100 of the synthesized RNA) was used during the SSD reaction as shown in Figure 3b, the synthesized microRNA oligomer could be used directly in some cases. To satisfy higher purity standards, the synthesized RNA oligomers were purified using HPLC equipped with C18 reverse-phase column (Figure 5a). The purified oligomer featured a desirable integrity as no small fragments appeared on the electrophoretogram (Figure 5b). Calculated from the band intensity, we estimated that the purification procedure provided an 87% recovery rate. Stem-loop reverse transcription quantitative polymerase chain reaction was also applied to precisely quantify the purified products (see Supplementary Material for detailed steps and results). Under optimized conditions and in an 80 μl reaction, 7.7 nmol (55 μg) mir-16 was synthesized, which was comparable to the medium scale (1.7 OD) of the chemical synthesis.33 For a reaction volume of 0.5 ml, 31.7 nmol (7.2 OD) mir-16 was obtained. Obviously, much larger scales can be easily realized by using a larger reaction volume. The total cost of preparing a large amount (>10 nmol) of a specific small RNA oligomer by RCT-SSD was much lower than that of chemical protocols. The molecular weight of the synthesized RNA oligomer was measured by electrospray ionization mass spectrometry (ESI-MS). As shown in Figure 6a, a variety of molecular ion signals in the region between m/z 500 and m/z 1,120 were obtained for the RNA oligomer. Deconvolution was applied using Thermo Xcalibur software and the result was shown in Figure 6b. The molecular weight of the RNA oligomer was measured as 7,135.17 Da, while the calculated molecular weight of the mir-16 oligomer was 7,144.3 Da. The error between tested and theoretical values was in a reasonable range; thus, we can conclude that the RNA oligomer synthesized by RCT-SDD was of the desired length and sequence.
Figure 5.

Purification of the RNA oligomer by high-performance liquid chromatography (HPLC) and vacuum freeze drying. (a) Elution profile obtained from reverse-phase HPLC. (b) Denaturing urea PAGE analysis of the purified RNA oligomer. L indicates the 25 nt RNA marker.
Figure 6.
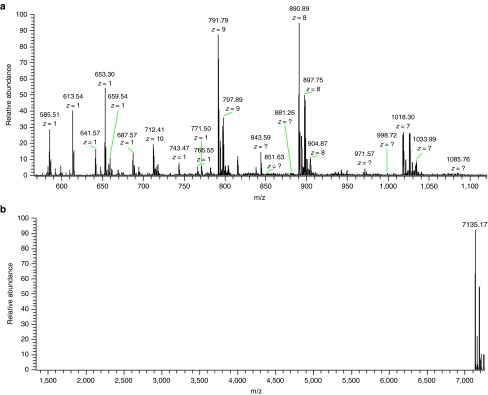
Mass spectrometry analysis of the synthesized RNA by ESI-MS. ESI-MS spectrum of the purified RNA oligomer. (a) Negative ion mass spectrum for microRNA-16 analysis. (b) The negative ion mass spectrum was deconvolved by Thermo Xcalibur.
Generality of the RCT-SSD synthesizing protocol
To test the generality of RCT-SSD, three other microRNAs, mir-168a, mir-7i, and mir-159a (see Table 1 for the sequences) were synthesized (Figure 7a). mir-168a oligomers were observed by PAGE, but the other two microRNAs were not. The reason for the missing bands for the mir-7i and mir-159a oligomers may be ascribed to their lower GC content, which caused difficulty in staining with SYBR Green II. As an alternative approach, HPLC was used to analyze the synthesized microRNA oligomers. Three synthesized microRNAs were detected and separated from the Aid-DNAs (see Figure 7b–d). The amount estimated from HPLC indicated that the RCT-SSD approach could be used as a general approach to synthesize small RNA with high yields and high purity. Compared with other “Runoff transcription” approaches, our strategy has the merits of directly producing RNAs of interest of an exact length.34,35
Figure 7.
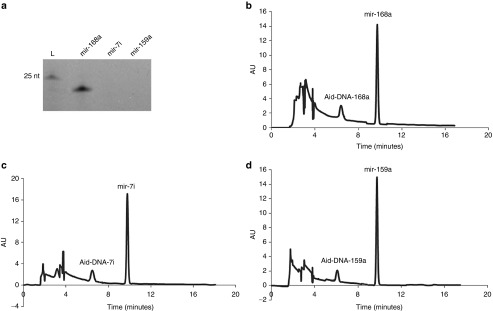
Analysis of synthesized microRNA oligomers by denaturing PAGE and reverse-phase high-performance liquid chromatography (HPLC). (a) Analysis of microRNA oligomers by 20% urea denaturing PAGE (stained with SYBR Green II). (b) Elution profile of mir-168a. (c) Elution profile of mir-7i. (d) Elution profile of mir-159a. The HPLC conditions were the same as those used for the mir-16 analysis.
Synthesis of the hammerhead ribozyme by RCT-SSD
Ribozyme is a small RNA motif that cleaves single-stranded RNAs at a specific phosphodiester bond. It functions to inhibit gene expression through sequence-specific cleavage of target RNAs has been studied intensively. Until now, eight classes of natural ribozymes have been discovered.36 Traditionally, these catalytic RNAs are produced either by transcription of DNA templates with promoter sequences or by stepwise chemical synthesis on a solid support. Here, RCT-SSD was utilized to prepare a desired ribozyme oligomer.
A hammerhead ribozyme (HHR) sequence that catalyzes the cleavage of ErbB-2 (human epidermal growth factor receptor) mRNA was used as an example (Figure 8a). ErbB-2 is a hallmark of dysfunctional cells and is overexpressed in breast, ovarian and stomach cancer.37 The sequences of the RNA substrate, the cDNA, and the splint DNA for HHR synthesis were listed in Table 1. The cDNA used as the template (cDNA-Ribo) was not perfectly complementary to the ribozyme sequence. The reason is that, for the rolling circle mechanism, transcription started at any site of the circular template and could deliver the same products that consist of tandemly repeated small RNA oligomers. Hence, the novelly designed circular cDNA (cDNA-Ribo) was used as the template, where the first nucleotide incorporated by T7 RNA polymerase is “G”, which is conducive to enhancing the transcription efficiency.12 The detailed design strategy of the ribozyme cDNA was illustrated in Supplementary Figure S3 (Supplementary Material). The unpaired portion of the arms (Figure 8a, 7 nt at 5′ end, 5 nt at 3′ end) shared the same sequence as two ends of mir-16; therefore, long tandemly repeated transcription products of HHR can be cut by Aid-DNA-16-1. Using this design, different HHRs could be synthesized with one type of Aid-DNA to reduce the cost. As shown in Figure 8b, the synthesized HHR featured an expected length (51 nt), while a smeared pattern was observed below the main band. The smeared pattern can be ascribed to the self-cleaved HHR during RCT and the subsequent reactions.38 To eliminate the undesired oligomers, purification was performed. The purified oligomer was obtained with high purity and integrity (Figure 8b) and was directly used to cleave the RNA substrate to check its validity and function. As shown in Figure 8c, the FITC-labelled RNA substrate was degraded over time. After 60-minute incubation, more than 90% of the substrate RNA was degraded. The cleavage product featured an expected length (11 nt), suggesting that the synthesized ribozyme has the correct catalytic and binding sequence. To prepare an RNA oligomer used as a ribozyme, chemical synthesis protocols may be inadequate due to the low yield of synthesizing long RNA oligomers. As an alternative approach to prepare RNA oligomers longer than 50 nt, RCT-SSD guaranteed the accuracy of the synthesized sequence with RNA polymerase that incorporated nucleotides strictly according to the base-paring principle.39
Figure 8.

Preparation of the hammerhead ribozyme by rolling circle transcription (RCT)site-specific disconnection (SSD). (a) Sequence of the hammerhead ribozyme prepared by RCT-SSD and the complex structure of the ribozyme/substrate. ErbB-2 mRNA (red) was used as the substrate for the ribozyme and was labeled with FITC at 5′ end. Two arms of the hammerhead ribozyme were designed to be complementary to the RNA substrate. The unpaired portion of the arms (blue) shared the same sequence with mir-16, providing a universal sequence that facilitated the synthesis of other ribozymes using the same Aid-DNA. (b) Analysis of synthesized and purified ribozyme oligomer products. (c) Time course of RNA cleavage by the hammerhead ribozyme synthesized by RCT-SSD. Reaction conditions: 1.0 μmol/l RNA substrate, 10 mmol/l MgCl2, 50 °C.
In conclusion, we have developed a novel protocol for synthesizing small RNA oligomers, where RCT and SSD were combined for inexpensive and simple performance. The cleavage by RNase H facilitates the RCT reaction and greatly promotes transcription efficiency. Our approach is especially suitable for a molecular biology laboratory to prepare a relatively large amount of small RNA oligomers. Further studies to analyze the sequence dependence of RCT and site-specific cleavage of SSD are in progress.
Discussion
RCT-SSD is designed to be used as a universal protocol for preparing small RNAs. T7 RNA polymerase is responsible for synthesizing long RNA transcript, while RNase H takes charge of cleaving the transcript to engender desired small RNAs. Normally, RNA polymerase requires a specific double-stranded DNA sequence, named “promoter,” to initiate transcription. For rolling circle mechanism, however, promoter sequence is unnecessary and transcription can be started on the single-stranded DNA circle.17,18 The circular DNA may mimic the “bubble”-like structure that can be found in the “open transcription complex” formed by DNA template and RNA polymerase. The “bubble” is flanked by double-stranded DNA, which serves as the binding site for RNA polymerase.40 Transcription starts in the bubble region; however, the initiation stage is relatively slow due to the de no synthesis of RNA primer.12 In this study, DNA circle is prepared by ligating two ends of single-stranded DNA with the help of a splint DNA. The splint can form a duplex structure with the circularized single-stranded DNA, which provides the binding site for RNA polymerase. Nonetheless, different from traditional initiation, the transcription can start immediately from the 3′-OH of the splint. Therefore, the rate of transcription initiation may be significantly improved.
RNase H is able to cleave RNA strand in DNA/RNA heteroduplex. As the product of rolling circle transcription, the transcript is a long RNA strand consisting of repeated unit of desired small RNA. To direct the site-specific cleavage, Aid-DNA has to anneal to the target site. Usually, the RNA molecules tend to form stable secondary or tertiary structures, which cause the difficulty for Aid-DNA to bind the target site. Thus, a denature step is employed to destroy the high dimensional structures and facilitate the formation of RNA/Aid-DNA duplex. If the secondary structure of synthesized RNA repeats is too stable to be hybridized by a 12 nt long Aid-DNA, a longer Aid-DNA with more 2′-O-methyl modifications may be helpful.
We designed the Aid-DNA sequence for specific RNA cleavage with unmodified deoxynucleotides at both 3′ and 5′ end. Lima et al. reported that a minimum of five contiguous unmodified deoxynucleotides at the 3′-end were required for effective RNA cleavage by RNase H.23 Although the binding affinity of RNase H towards the heteroduplex portion with 2′-O-methyl modification is approximately equal to the unmodified portion, the catalytic activity is virtually absent at the modified site. Accordingly, the presence of three unmodified deoxynucleotides at the 5′-end in Aid-DNA-16-1 did not show detectable nonspecific cleavage, although the single-stranded part of the RNA may be cleaved at the site two or three deoxynucleotides away from the 5′-end of Aid-DNA-16-1 when more RNase H was present (data not shown). Cleavage of the single-stranded part of RNA extending from heteroduplex by RNase H was also documented. 23,24 As a result, we found for the first time that the site-specific cleavage could be obtained even when three natural deoxynucleotides were present at the 5′-end. This design can reduce the cost of the modified DNA for site-specific RNA cleavage.
In preparation of the hammerhead ribozyme using RCT-SSD, by-products supposed to be generated by unexpected self-cleavage were observed (Figure 8b). Traditionally, ribozymes for laboratory use are synthesized by using runoff transcription, where self-cleavage of product ribozyme rarely takes place.41 The unexpected self-cleavage found in RCT-SSD can be ascribed to the fact that the transcript is tandemly repeated units of ribozyme sequence, whose catalytic motif tends to interact with RNA sequence more readily than ribozyme in separated conditions. To reduce the unexpected self-cleavage, more Aid-DNA was introduced and the formation of higher order structures conducive to self-cleavage was prevented as expected.
RCT-SSD protocol is capable of preparing functional RNA sequences with high efficiency. Certainly, longer RNA oligomers (>100 nt), which cannot be chemically synthesized are also promising products for this strategy. Preparation of small interfering RNA (siRNA) could easily be realized by utilizing two circular DNA templates separately, while four oligodeoxynucleotides must be synthesized for each siRNA duplex by runoff transcription. Free from the sophisticated steps in chemical synthesis protocols, our protocol is promising to be used as a feasible and robust RNA synthesis method to obtain desired RNAs with high purity and high quantity.
Materials and methods
Ligation reaction. The ligation reaction for circularizing cDNA using splint DNA was performed in 10 μl of reaction mixture containing 1× ligation buffer (40 mmol/l Tris-HCl, 10 mmol/l MgCl2, 10 mmol/l DTT, and 5 mmol/l ATP, pH 7.8), 5 U T4 DNA ligase (New England Biolabs, Ipswich, MA), 1.0 μmol/l cDNA and 1.0 μmol/l splint DNA. Before the ligase was added, the mixture was incubated at 95 °C for 7 minutes and slowly cooled to room temperature. Then, the ligase was added and the reaction was carried out at 37 °C for 40 minutes, followed by inactivation of T4 DNA ligase at 65 °C for 10 minutes. The ligation product was subjected to gradient dilution to obtain circular templates of various concentrations.
RCT reaction. An aliquot (10 μl) of ligation products of varying concentrations was mixed with 40 U T7 RNA polymerase (New England Biolabs), 0.05 U RNase H (Thermo Scientific, Pittsburgh, PA), 20 U RNase inhibitor (Thermo Scientific) in 20 μl of 1× transcription buffer (40 mmol/l Tris-HCl (pH 7.9 at 25 °C), 10 mmol/l NaCl, 6 mmol/l MgCl2, 10 mmol/l DTT, and 2 mmol/l spermidine) containing 0.1 μmol/l Aid-DNA and 0.5 mmol/l NTPs (Thermo Scientific). The mixture was incubated at 37 °C for 4 hours or as otherwise noted. Inorganic pyrophosphatase (0.02 U) purchased from New England Biolabs, was applied to selected RCT reactions.
SSD reaction. At the end of the RCT reaction, the transcription product was subjected to DNase digestion to eliminate the DNA template. The reaction (20 μl) consisted of 10 μl transcription products, 20 U RNase inhibitor (which does not inhibit RNase H), 5 U DNase I (Takara Bio, Shiga, Japan) and 2.0 μl 10× DNase I buffer. The mixture was incubated at 37 °C for 1 hour followed by inactivation of the DNase at 80 °C for 10 minutes. An aliquot (10 μl) of the digestion product was incorporated in 10 μl 1× RNase H buffer containing 20 mmol/l Tris-HCl (pH 7.8), 40 mmol/l KCl, 8 mmol/l MgCl2 and 1 mmol/l dithiothreitol (DTT), and 0.1 μmol/l Aid-DNA. The reaction mixture was heated to 65 °C for 7 minutes and slowly cooled to room temperature. Then, 2.5 U RNase H (Thermo Scientific) was added and the mixture was incubated at 37 °C for 40 minutes, followed by inactivation of the RNase H at 65 °C for 10 minutes. The products were analyzed by 20% denaturing urea PAGE followed by SYBR Green II (Life Technologies, Carlsbad, CA) staining.
Measuring the molecular weight of synthesized RNA oligomer by ESI-MS. ESI-MS analysis was performed using a Thermo LTQ ESI-MS (Thermo Fisher Scientific, Waltham, MA) equipped with an electrospray ionization source. Nitrogen was used as a nebulizer gas. An electrospray was generated by applying a voltage of 4.0 kV across the stainless steel electrospray needle and the heated capillary. The heated capillary was maintained at 250 °C. Negative ion mass spectra were acquired over the range from 600 to 1,500 mass-to-charge ratio (m/z) at a scan rate of 5 seconds/decade. The purified RNA oligomer was dissolved in a 10 μl solution containing 50% acetonitrile (v/v). The results were obtained and processed with Thermo Xcalibur (Thermo Fisher Scientific).
Preparation of the HHR using RCT-SSD. The HHR was synthesized and purified according to the protocol for microRNAs preparation. The ligation reaction was carried out in a 10 μl volume with 1.0 μmol/l cDNA and 1.0 μmol/l splint DNA. An aliquot (10 μl) of the ligation product was introduced into the transcription system in a 20 μl volume, and the mixture was incubated at 37 °C for 4 hours. An aliquot (10 μl) of the transcription products were subjected to DNase treatment at 37 °C for 1 hour, followed by inactivation of the DNase at 80 °C for 10 minutes. Then, 10 μl of the products were added to the SSD system containing 0.1 μmol/l Aid-DNA-16-1 and 2.5 U RNase H. Before RNase H was added, the reaction mixture was heated to 65 °C for 7 minutes and slowly cooled to room temperature. Then, the mixture with RNase H was kept at 37 °C for 40 minutes, followed by inactivation of the RNase H at 65 °C for 10 minutes. The products were analyzed using 20% denaturing urea PAGE followed by SYBR Green II staining.
Cleavage of the RNA substrate by the RCT-SSD synthesized HHR. A typical ribozyme-catalyzed RNA cleavage reaction was performed as follows. The substrate RNA and the enzymatically synthesized ribozyme were mixed with buffer (10 mmol/l KCl, 10 mmol/l (NH4)2SO4, and 20 mmol/l Tris-HCl, pH 7.5) in a final volume of 20 μl containing a particular amount of ribozyme and 1.0 μmol/l RNA substrate. The mixture was heated to 90 °C for 1.0 minutes, and cooled to 37 °C over 20 minutes to allow annealing between the substrate and the catalyst. Next, MgCl2 was added to a final concentration of 10 mmol/l to start the cleavage reaction at 50 °C. After a set time interval, an aliquot of 10 μl of the solution was pipetted and mixed with 2.0 μl of 6× loading buffer (60 mmol/l ethylenediaminetetraacetic acid, 10 mmol/l Tris-HCl (pH 7.6), 0.03% bromophenol blue, 0.03% xylene cyanol FF, 60% glycerol) to terminate the reaction. The resulting mixture (12 μl) was subjected to electrophoresis on a 20% denaturing polyacrylamide gel containing 8.0 mol/l urea. Imaging and quantification of the digested RNA was carried out on a Bio-Rad molecular imager Gel Doc XR+ imaging system (Bio-Rad, Hercules, CA). The fluorescence of FITC moiety attached to 5′-end of RNA substrate (Table 1) was detected using an excitation wavelength of 473 nm and an emission wavelength of 520 nm.
SUPPLEMENTARY MATERIAL Figure S1. Quantification of microRNA products using a Bio-Rad gel imaging system. Figure S2. Quantification of microRNA by RT-qPCR. Figure S3. Illustration of the design of cDNA-Ribo for circularization. Table S1. Sequence of oligomers used in preparing circular DNA markers. Table S2. Sequence of oligomers used in cleavage of chimeric oligomer. Table S3. Sequence of oligomers used in stem-loop RT-qPCR.
Acknowledgments
This work was supported by the “Fund for Distinguished Young Scholars” of the Shandong province (JQ201204), the Program for Changjiang Scholars and Innovative Research Team in University (IRT1188), and the “National Youth Qianren Plan”. Funding for open access charges: National Youth Qianren Plan.
Supplementary Material
References
- Kruger K, Grabowski PJ, Zaug AJ, Sands J, Gottschling DE, Cech TR. Self-splicing RNA: autoexcision and autocyclization of the ribosomal RNA intervening sequence of Tetrahymena. Cell. 1982;31:147–157. doi: 10.1016/0092-8674(82)90414-7. [DOI] [PubMed] [Google Scholar]
- Crick FH. The origin of the genetic code. J Mol Biol. 1968;38:367–379. doi: 10.1016/0022-2836(68)90392-6. [DOI] [PubMed] [Google Scholar]
- Bushati N, Cohen SM. microRNA functions. Annu Rev Cell Dev Biol. 2007;23:175–205. doi: 10.1146/annurev.cellbio.23.090506.123406. [DOI] [PubMed] [Google Scholar]
- Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- Harada M, Luo X, Murohara T, Yang B, Dobrev D, Nattel S. MicroRNA regulation and cardiac calcium signaling: role in cardiac disease and therapeutic potential. Circ Res. 2014;114:689–705. doi: 10.1161/CIRCRESAHA.114.301798. [DOI] [PubMed] [Google Scholar]
- Wong SS, Webby RJ. An mRNA vaccine for influenza. Nat Biotechnol. 2012;30:1202–1204. doi: 10.1038/nbt.2439. [DOI] [PubMed] [Google Scholar]
- Zhang GL, Li YX, Zheng SQ, Liu M, Li X, Tang H. Suppression of hepatitis B virus replication by microRNA-199a-3p and microRNA-210. Antiviral Res. 2010;88:169–175. doi: 10.1016/j.antiviral.2010.08.008. [DOI] [PubMed] [Google Scholar]
- van Rooij E, Kauppinen S. Development of microRNA therapeutics is coming of age. EMBO Mol Med. 2014;6:851–864. doi: 10.15252/emmm.201100899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Crawford M, Mao Y, Lee RJ, Davis IC, Elton TS.et al. (2013Therapeutic delivery of microRNA-29b by cationic lipoplexes for lung cancer Mol Ther Nucleic Acids 2e84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall WS, Kaiser RJ. Recent advances in the high-speed solid phase synthesis of RNA. Curr Opin Chem Biol. 2004;8:222–229. doi: 10.1016/j.cbpa.2004.04.012. [DOI] [PubMed] [Google Scholar]
- Milligan JF, Groebe DR, Witherell GW, Uhlenbeck OC. Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res. 1987;15:8783–8798. doi: 10.1093/nar/15.21.8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudna JA, Cech TR. The chemical repertoire of natural ribozymes. Nature. 2002;418:222–228. doi: 10.1038/418222a. [DOI] [PubMed] [Google Scholar]
- Silverman SK. Deoxyribozymes: DNA catalysts for bioorganic chemistry. Org Biomol Chem. 2004;2:2701–2706. doi: 10.1039/B411910J. [DOI] [PubMed] [Google Scholar]
- Carrigan MA, Ricardo A, Ang DN, Benner SA. Quantitative analysis of a RNA-cleaving DNA catalyst obtained via in vitro selection. Biochemistry. 2004;43:11446–11459. doi: 10.1021/bi049898l. [DOI] [PubMed] [Google Scholar]
- Krupp G. RNA synthesis: strategies for the use of bacteriophage RNA polymerases. Gene. 1988;72:75–89. doi: 10.1016/0378-1119(88)90129-1. [DOI] [PubMed] [Google Scholar]
- Daubendiek SL, Ryan K, Kool ET. Rolling-circle RNA synthesis: circular oligonucleotides as efficient substrates for T7 RNA polymerase. J Am Chem Soc. 1995;117:7818–7819. doi: 10.1021/ja00134a032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diegelman AM, Daubendiek SL, Kool ET. Generation of RNA ladders by rolling circle transcription of small circular oligodeoxyribonucleotides. Biotechniques. 1998;25:754–758. doi: 10.2144/98255bm01. [DOI] [PubMed] [Google Scholar]
- Diegelman AM, Kool ET. Generation of circular RNAs and trans-cleaving catalytic RNAs by rolling transcription of circular DNA oligonucleotides encoding hairpin ribozymes. Nucleic Acids Res. 1998;26:3235–3241. doi: 10.1093/nar/26.13.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daubendiek SL, Kool ET. Generation of catalytic RNAs by rolling transcription of synthetic DNA nanocircles. Nat Biotechnol. 1997;15:273–277. doi: 10.1038/nbt0397-273. [DOI] [PubMed] [Google Scholar]
- Inoue H, Hayase Y, Iwai S, Ohtsuka E. Sequence-dependent hydrolysis of RNA using modified oligonucleotide splints and RNase H. FEBS Lett. 1987;215:327–330. doi: 10.1016/0014-5793(87)80171-0. [DOI] [PubMed] [Google Scholar]
- Lima WF, Crooke ST. Binding affinity and specificity of Escherichia coli RNase H1: impact on the kinetics of catalysis of antisense oligonucleotide-RNA hybrids. Biochemistry. 1997;36:390–398. doi: 10.1021/bi962230p. [DOI] [PubMed] [Google Scholar]
- Lima WF, Crooke ST. Cleavage of single strand RNA adjacent to RNA-DNA duplex regions by Escherichia coli RNase H1. J Biol Chem. 1997;272:27513–27516. doi: 10.1074/jbc.272.44.27513. [DOI] [PubMed] [Google Scholar]
- Shibahara S, Mukai S, Nishihara T, Inoue H, Ohtsuka E, Morisawa H. Site-directed cleavage of RNA. Nucleic Acids Res. 1987;15:4403–4415. doi: 10.1093/nar/15.11.4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Liu C, Zhu J, Shu P, Yin B, Gong Y.et al. (2012MicroRNA-16 targets amyloid precursor protein to potentially modulate Alzheimer's-associated pathogenesis in SAMP8 mice Neurobiol Aging 33522–534. [DOI] [PubMed] [Google Scholar]
- Linsley PS, Schelter J, Burchard J, Kibukawa M, Martin MM, Bartz SR.et al. (2007Transcripts targeted by the microRNA-16 family cooperatively regulate cell cycle progression Mol Cell Biol 272240–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyhan AA, Vlassov AV, Johnston BH. RNA interference from multimeric shRNAs generated by rolling circle transcription. Oligonucleotides. 2006;16:353–363. doi: 10.1089/oli.2006.16.353. [DOI] [PubMed] [Google Scholar]
- Li Y, Kim HJ, Zheng C, Chow WH, Lim J, Keenan B.et al. (2008Primase-based whole genome amplification Nucleic Acids Res 36e79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern JA, Davis RH. Application of solution equilibrium analysis to in vitro RNA transcription. Biotechnol Prog. 1997;13:747–756. doi: 10.1021/bp970094p. [DOI] [PubMed] [Google Scholar]
- Abe N, Hiroshima M, Maruyama H, Nakashima Y, Nakano Y, Matsuda A.et al. (2013Rolling circle amplification in a prokaryotic translation system using small circular RNA Angew Chem Int Ed Engl 527004–7008. [DOI] [PubMed] [Google Scholar]
- Cheong HK, Hwang E, Lee C, Choi BS, Cheong C. Rapid preparation of RNA samples for NMR spectroscopy and X-ray crystallography. Nucleic Acids Res. 2004;32:e84. doi: 10.1093/nar/gnh081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidl CI, Lama L, Ryan K. Circularized synthetic oligodeoxynucleotides serve as promoterless RNA polymerase III templates for small RNA generation in human cells. Nucleic Acids Res. 2013;41:2552–2564. doi: 10.1093/nar/gks1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller S, Wolf J, Ivanov S. Current strategies for the synthesis of RNA. Curr Org Synth. 2004;1:293–307. [Google Scholar]
- Donzé O, Picard D. RNA interference in mammalian cells using siRNAs synthesized with T7 RNA polymerase. Nucleic Acids Res. 2002;30:e46. doi: 10.1093/nar/30.10.e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohail M, Doran G, Riedemann J, Macaulay V, Southern EM. A simple and cost-effective method for producing small interfering RNAs with high efficacy. Nucleic Acids Res. 2003;31:e38. doi: 10.1093/nar/gng038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler WC, Nahvi A, Roth A, Collins JA, Breaker RR. Control of gene expression by a natural metabolite-responsive ribozyme. Nature. 2004;428:281–286. doi: 10.1038/nature02362. [DOI] [PubMed] [Google Scholar]
- Martick M, Scott WG. Tertiary contacts distant from the active site prime a ribozyme for catalysis. Cell. 2006;126:309–320. doi: 10.1016/j.cell.2006.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray JB, Terwey DP, Maloney L, Karpeisky A, Usman N, Beigelman L.et al. (1998The structural basis of hammerhead ribozyme self-cleavage Cell 92665–673. [DOI] [PubMed] [Google Scholar]
- Sydow JF, Cramer P. RNA polymerase fidelity and transcriptional proofreading. Curr Opin Struct Biol. 2009;19:732–739. doi: 10.1016/j.sbi.2009.10.009. [DOI] [PubMed] [Google Scholar]
- Aiyar SE, Helmann JD. A mismatch bubble in double-stranded DNA suffices to direct precise transcription initiation by Escherichia coli RNA polymerase. J Biol Chem. 1994;269:13179–13184. [PubMed] [Google Scholar]
- Moretti JE, Müller UF. A ribozyme that triphosphorylates RNA 5′-hydroxyl groups. Nucleic Acids Res. 2014;42:4767–4778. doi: 10.1093/nar/gkt1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.