

Abstract
We describe a 19F NMR method for detecting bromodomain–ligand interactions using fluorine-labeled aromatic amino acids due to the conservation of aromatic residues in the bromodomain binding site. We test the sensitivity, accuracy, and speed of this method with small molecule ligands (+)-JQ1, BI2536, Dinaciclib, TG101348, and acetaminophen using three bromodomains Brd4, BrdT, and BPTF. Simplified 19F NMR spectra allowed for simultaneous testing of multiple bromodomains to assess selectivity and identification of a new BPTF ligand. Fluorine labeling only modestly affected the Brd4 structure and function assessed by isothermal titration calorimetry, circular dichroism, and X-ray crystallography. The speed, ease of interpretation, and low concentration of protein needed for binding experiments affords a new method to discover and characterize both native and new ligands.
Bromodomains are epigenetic “reader” proteins that are the only known structural modules for recognizing the acetylated ε-nitrogen of lysine on histones and play essential roles in diverse diseases, particularly in cancer.1 Structural biology efforts in NMR2,3 and particularly X-ray crystallography have accelerated chemical probe development with more than 150 X-ray structures of 43 different bromodomains reported.4 The BET (bromodomain and extraterminal) subfamily of proteins, Brd2, -3, -4, and -T are the most intensely studied due to the discovery of tool compounds including pan-BET inhibitors such as (+)-JQ1, first used to validate Brd4 inhibition for treating NUT midline carcinoma.5,6 Selective targeting of BET bromodomains is one of the most significant challenges for this emerging therapeutic class.1 Discovery of new binding modes can yield insight into selective inhibitor designs. While BET research is developing rapidly, the majority of the remaining bromodomains are not as well-characterized due to a lack of both small molecule inhibitors and optimal screening methods.7 Here, we describe a new ligand discovery and characterization method for bromodomains using 19F NMR, and highlight the value of fluorine-labeled aromatic side-chains for selectivity and binding analyses.
Protein-observed NMR has proven to be a powerful ligand discovery method since the original reports from Abbott laboratories.8 In these experiments, a protein is labeled with NMR active nuclei, typically on the backbone amides. Small molecule induced changes in protein chemical shifts are used to characterize a binding event. This method can be used to elucidate the binding site, discriminate specific from nonspecific interactions, and quantify weak affinities of low molecular weight compounds (MW < 300) termed fragments, commonly used for NMR screening. Fragments can be missed in the current ligand discovery methods for bromodomains.9 This is particularly challenging in the absence of an initial ligand for competition studies, which is the case for many bromodomains.7 However, in amide detected, protein-observed NMR, the need for a high concentration of protein, detailed resonance assignments, and speed of experiment can be limiting.10
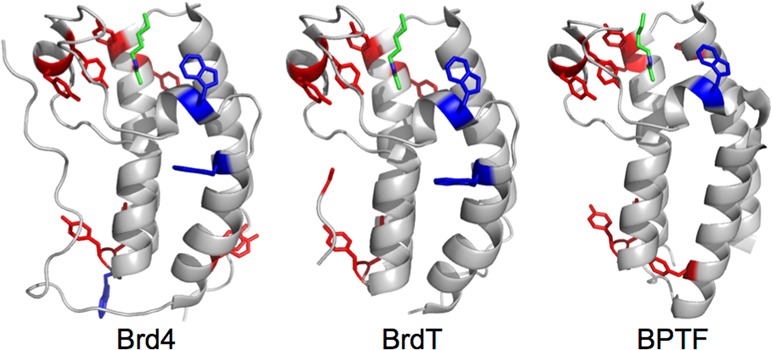
We described the use of protein-observed 19F NMR (PrOF NMR) for fragment screening using fluorinated aromatic amino acid side chains demonstrated by a 50 compound fragment pilot screen on a nonbromodomain containing protein.11 Here, we first test the PrOF NMR approach on the N-terminal bromodomain of Brd4 (Brd4(1)), using known ligands and include a small molecule whose binding affinity was too weak to characterize by prior methods. Sequence analyses of 61 bromodomains identified moderate to high conservation of multiple aromatic amino acids, many positioned near the histone binding site.4 For example, aromatic side chains in the histone binding site are found at positions 81, 97, and 139 of Brd4(1) and are conserved across bromodomains 31%, 97%, and 92% of the time, respectively. Therefore, for new selectivity studies, we apply this method to a second BET bromodomain BrdT, as well as a non-BET, BPTF, for which no small molecule probes are known. The hyper-responsiveness of the fluorine chemical shift to ligand binding and simplicity of the rapidly acquired NMR spectrum11,12 provides the speed and structural information necessary for developing potential inhibitors. These studies lead us to propose PrOF NMR as a structure-based tool for characterizing new and native ligands for the aromatic-rich bromodomains.
We analyzed the crystal structure of Brd4(1) in complex with inhibitor (+)-JQ1 (Figure 1) to determine a fluorine-labeling strategy. The aromatic side chains of W81, Y97, and Y139 are within 3–5 Å of (+)-JQ1. Y98 and Y137 are within 10 Å of the thiophene ring of (+)-JQ1. Although W75 is not in close contact, it is located on the dynamic Z helix of Brd4(1) (Figures 1 and 2A). Fewer phenylalanine side chains were in close proximity.13 For these reasons, we decided to investigate the ligand detection ability of PrOF NMR using either 3-fluorotyrosine (3FY) or 5-fluorotryptophan (5FW)-labeled Brd4(1). High labeling efficiency with 3FY (>95%) and 5FW (78–90%) was achieved with good to high protein yields (10–60 mg/L).
Figure 1.

Aromatic amino acids are in close proximity to the bromodomain binding site. Ribbon diagram of Brd4(1) bound to (+)-JQ1 (PDB ID 3MXF). Tyrosine and tryptophan are indicated as sticks. Red and yellow indicate side-chains within 5 and 10 Å, respectively. (Right) (+)-JQ1 and fluorinated amino acids used in this study.
Figure 2.

Structure and function of 3FY-Brd4(1): (A) Cartoon representation of a 3FY-labeled (purple) Brd4(1) crystal structure complexed with (+)-JQ1 (green). Four of the seven 3FY residues adopt well-defined alternate conformations. Fluorine atoms are shown for both cases. PDB ID 4QZS. (B) Isothermal titration calorimetric analysis of binding of (+) JQ1 binding to 3FY-Brd4(1) at 25 °C. Kd = 89 ± 5 nM.
We used circular dichroism (CD), isothermal titration calorimetry (ITC), and X-ray crystallography to assess the structural and functional effects on fluorine incorporation into Brd4(1). Far UV CD spectra of the fluorinated proteins show similar levels of secondary structure but a slightly lower thermal stability (Tm = 50 °C vs 52 °C).13 We used ITC to assess how these effects may alter binding. (+)-JQ1 binds to Brd4(1) with a Kd of 49 nM at 15 °C.5 We detect a similar binding affinity of 75 ± 4 nM for unlabeled Brd4(1),13 89 ± 5 nM for 3FY-Brd4(1) (Figure 2B), and 78 ± 2 nM for 5FW-Brd4(1)13 at 25 °C. We solved the crystal structure of 3FY-Brd4(1) bound to (+)-JQ1 (Figure 2A). Aligning this structure onto the unlabeled protein complex with (+)-JQ1 yielded an RMSD of 0.089 Å. These results lead us to conclude that fluorine incorporation had only a modest effect on structure and function. Crystallization of 3FY-Brd4(1) is itself significant as less than 20 fluorinated protein crystal structures have been reported.
19F NMR spectra of 3FY and 5FW-labeled Brd4(1) display well-dispersed resonances indicative of folded proteins. From the 19F NMR spectrum of 5FW-Brd4(1), resonances for W75, W81, and W120 were clearly resolved (Figure 3A). The seven 3FY resonances in 3FY-Brd4(1) span over 12 ppm, suggesting a diverse environment of the aromatic side chains (Figure 3B). Two of the resonances at −136.6 ppm are partially overlapping. To our knowledge, this is the largest chemical shift range reported for a fluorinated protein but it remains to be tested if this range is similar in other bromodomains, which have similar arrangements of aromatic amino acids.
Figure 3.

PrOF NMR titration of (+)-JQ1 with Brd4(1). (A) Bottom to top: 5FW-Brd4(1) (25 μM), with 1 eq. (+)-JQ1, with 5FW-BPTF (25 μM), with 5FW-BPTF (25 μM) and 1 eq. (+)-JQ1, with unlabeled BrdT(1) (50 μM) and 1 eq. (+)-JQ1. (B) Bottom to top: 3FY-Brd4(1) (47 μM) titrated with 2 eq. (+)-JQ1.
Resonance assignments were made via a combination of site-directed mutagenesis and ligand binding experiments (explained below). For site-directed mutagenesis, a single tryptophan or tyrosine to phenylalanine mutant was expressed, and the disappearance of a single resonance was used to assign the side-chain in the parent 19F NMR spectrum.13 Complete resonance assignments enabled us to test the sensitivity of our 19F NMR method for characterizing binding footprints and quantifying the affinity of weak binding molecules for fragment-based screens.
Crystallographic evidence supports the role of W81 for determining the specificity of (+)-JQ1 for binding BET bromodomains in a region termed the WPF shelf (Figure 2A).4,5 W81 was first tested as a diagnostic residue for binding studies. Slow exchange binding was readily detected upon titrating (+)-JQ1 from an ethylene glycol stock solution, in which case the intensity of the W81 resonance at −126.3 ppm disappeared as a new downfield resonance at −124.0 ppm grew in (Figure 3A). The observed slow exchange of bound and free protein is consistent with submicromolar binding. Closer inspection of the spectrum revealed a small 0.14 ppm upfield shift of the W75 resonance located underneath W81 on the Z-helix of Brd4(1), but outside the binding site (Figures 3A, 4A). DMSO can also be used in these experiments but is a known bromodomain ligand so was avoided to only show (+)-JQ1 effects.14
Figure 4.
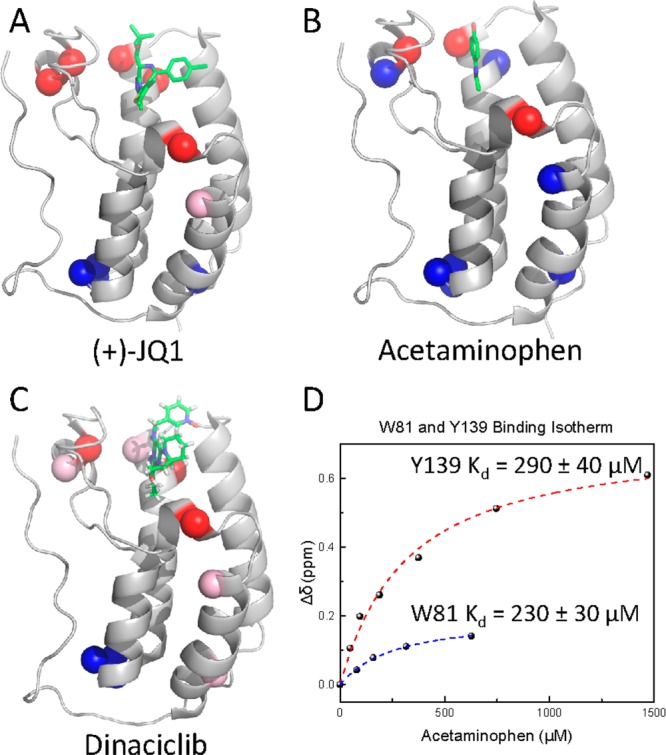
Binding footprint and affinity determination with Brd4(1) and various ligands. (A–C) Residues with fluorinated resonances not significantly perturbed are blue, resonances perturbed 0.05–0.1 ppm are light pink, and resonances perturbed greater than 0.1 ppm or broadened significantly in a dose dependent fashion are red. Acetaminophen was overlaid on Brd4(1) based on the crystal structure with Brd2(1) (PDB IDs 3MXF, 4O70, 4A9J). (D) Binding isotherms for acetaminophen titration with 3FY-and 5FW-Brd4(1).
Titrating (+)-JQ1 with 3FY-Brd4(1) provided further binding information. Y97, Y98, and Y139 are located within the binding site for (+)-JQ1. All three resonances shifted (slow exchange) and broadened upon titration (Figure 3B).13 The most upfield resonance was assigned to Y97, which forms a bridging hydrogen bond to (+)-JQ1 via a structurally conserved water molecule. Y137, on the outside of the binding site, moved downfield but remained sharp. The BC loop near Y137 is highly dynamic.5 Our observations are consistent with a protein conformational change with considerable flexibility at this site.
Fragment screening is a promising method for early stage ligand discovery and is well-suited for NMR analyses due to the ability to characterize protein ligand interactions at the high concentrations needed to detect small molecule binding.10 Acetaminophen represented an ideal test case as it was shown to bind BET bromodomains via X-ray crystallography, but its affinity for its target was reported as too weak for characterizing Brd4(1) binding by fluorescence anisotropy or TR-FRET.9 The small size of this molecule (MW = 160 g/mol) is representative of many compounds in fragment libraries. Using 3FY-Brd4(1), we detect binding at ligand concentrations as low as 47 μM with chemical shift changes >0.6 ppm when fully bound (Figure 4B,D). Based on the crystal structure with Brd29 and site directed mutant analysis,13 we assigned this shifted resonance to Y139. Y97, which is expected to make a water-mediated hydrogen bond, also broadens. Titration of acetaminophen yielded a Kd of 290 ± 40 μM. This results in a high ligand efficiency (0.44 kcal/mol/non-hydrogen atom) for acetaminophen as a bromodomain inhibitor scaffold. Titration with 5FW-Brd4(1) yielded a similar Kd based on the chemical shift perturbation of W81 (230 ± 30 μM, Figure 4D). The two Kd’s are within error, arguing against a specific fluorine perturbing binding. Due to the speed of data collection (chemical shift information can be acquired in <5 min),13 low protein concentration (40–50 μM), and conserved aromatic contacts, we anticipate this method will be well-suited for bromodomain fragment screening. This is especially useful where there is a lack of suitable ligands for competition-based experiments or affinities are weak (Kd > 100 μM).13
BET bromodomains are also inhibited by several classes of kinase inhibitors affording molecules with dual modes of actions and a new source of ligand diversity for screening against bromodomains.15 We chose three inhibitors covering a wide range of IC50 values, BI2536 (0.025 μM), TG101348 (0.29 μM), and Dinaciclib (19 μM), to compare their binding footprints to X-ray structures with Brd4. During Dinaciclib titration, the resonance for W81 broadened significantly. Perturbation of the fluorine resonance for W75 was in fast exchange. We used this perturbation to estimate a Kd of 70 μM ± 20.13 BI2536 and TG101348 have nanomolar binding affinity. In both cases, W81 was highly perturbed (Δδ = 0.4–1 ppm). With BI2536, a new resonance in slow exchange grows in upfield and is consistent with ring current effects based on analysis of the cocrystal structure and the reported nanomolar affinity.13 Similar to (+)-JQ1 binding, the resonance for W75 showed a small perturbation. W75 shows the most pronounced effect for TG101348 binding (Δδ > 0.25 ppm).13 Inspection of crystal structure overlays reveals very minor perturbations to the residues and neighboring side-chains highlighting the sensitivity of 19F NMR for identifying small conformational effects. 3FY-Brd4(1) was used to further characterize the kinase inhibitor binding modes for Dinaciclib (Figure 4C), BI2536,13 and TG101348.13 The results were consistent with X-ray data and induced conformational effects along the Z-helix.
On the basis of the Brd4(1) results, we tested the generality of using PrOF NMR with a second BET bromodomain, BrdT, of interest as a male contraception target,16 and a non-BET bromodomain, BPTF, for which no small molecule screens nor small molecule X-ray complexes have been reported. Both proteins contain a WPF shelf; therefore, we labeled these proteins with 5FW. BI2536 and (+)-JQ1 bind to BrdT. We detect a perturbation of the fluorinated protein resonances in slow exchange, consistent with their reported nanomolar affinity. (+)-JQ1 is selective for BETs, and thus in the presence of 5FW-BPTF, we do not detect binding. Surprisingly, PrOF NMR reveals that BI2536 binds to 5FW-BPTF,13 which went undetected in existing selectivity screens using BROMOscanSM and thermal shift assays,15,17 highlighting the sensitivity of 19F NMR. Deconstruction of the BI2536 ligand may lead to a useful starting point for BPTF chemical probe development to study its role in cancer.18
Finally, screening in the presence of other BET proteins can increase the stringency and binding information for finding selective BET inhibitors. Binding induced shifts from pan-BET inhibitor (+)-JQ1 were reversed by 46% in the presence of excess unlabeled BrdT(1) (Figure 3A). The 19F NMR spectrum with BI2536 is similarly perturbed.13 The non-BET, BPTF bromodomain has one tryptophan in the WPF shelf. As a new experiment, due to the significant chemical shift dispersion and simplified 19F NMR spectra, we decided to simultaneously test two fluorinated proteins. Consistent with single protein binding studies, the addition of (+)-JQ1 to 5FW-Brd4(1) and 5FW-BPTF shows selectivity for Brd4(1) (Figure 3A), whereas BI2536 exhibits binding to both bromodomains.13
Using a validated therapeutic epigenetic protein, we evaluated fluorinated bromodomains as sensitive tools for detecting ligand binding modes via 19F NMR. By labeling two different aromatic amino acids, we characterized variations in binding modes of (+)-JQ1 and new ligands repurposed from the kinase field. We further showed that our method can detect protein binding with weak fragments, DMSO and acetaminophen. This is encouraging due to the success of developing fragments into lead molecules in drug discovery campaigns. On the basis of the high number of aromatic amino acids across 61 bromodomains and enrichment of aromatics at protein–protein interaction sites in general,19 we anticipate that our method should be generalizable for bromodomains, many lacking specific tool compounds to study their biology such as BPTF. We further demonstrate that fluorine incorporation was only modestly perturbing for Brd4(1), by solid and solution state methods. Future studies will use PrOF NMR in a full fragment screen. This side-chain labeling technique has been employed for studying small- to medium-sized proteins (10 to 60 kDa),12,20 as well as G-protein coupled receptors.21,22 Here, we apply it to bromodomains both in isolation and in mixtures. Due to the speed of PrOF NMR, ease of interpretation, and availability of 19F-tuned NMR probes in academic and industrial settings, this approach should be of broad appeal for early stage ligand discovery.
Methods
Brd4(1)(42–168), BrdT(1)(29–134), and BPTF (2793–2911) Protein Expression
The pNIC28-BSA4 plasmids containing the Brd4(1) and BPTF genes were a kind gift from the laboratory of Stefan Knapp. For protein expression, either the E. coli Rosetta (DE3) strain (Novagen) was first transformed with the respective expression plasmid or the BL21(DE3) strain was cotransformed along with the pRARE (Novagen) plasmid and plated onto agar plates containing kanamycin (100 mg/L) and chloramphenicol (35 mg/L). Following overnight incubation at 37 °C, a single colony was selected from the agar plate and inoculated in 50 mL of LB media containing kanamycin (100 mg/L) and chloramphenicol (35 mg/L). The primary culture was grown overnight at 25 °C while shaking at 250 rpm. For secondary culture growth, 1 L of LB media containing kanamycin (100 mg/L) was inoculated with the primary culture and cultured at 37 °C while shaking at 250 rpm. When the O.D. of culture at 600 nm reached 0.6, the shaker temperature was reduced to 20 °C. After 30 min, the expression was induced with 1 mM IPTG overnight for 12–16 h. Cells were harvested by centrifugation.
Expression of 3FY and 5FW Labeled Brd4(1), 5FW-BrdT(1), and 5FW-BPTF
3FY and 5FW labeled Brd4(1) were expressed based on established methods11,23 using E. coli DL39(DE3) + pRARE and E. coli Bl21(DE3) + pRARE strains, respectively. To express the labeled protein, the secondary culture in LB media was grown until an O.D. at 600 nm of 0.6 was reached followed by harvesting and washing the cells with PBS. Washed cells were resuspended in defined media of Muchmore et al.24 containing either 3FY (70 mg/L) in place of tyrosine or 5-fluoroindole (60 mg/L) in place of tryptophan. The resuspended E. coli were incubated at 37 °C while shaking for 1 h followed by the cooling to 20 °C and media temperature equilibration for 30 min. Protein expression was induced with 1 mM IPTG overnight (14–16 h) at 20 °C. The cells were harvested and stored at −20 °C. 5FW-BrdT(1) and 5FW-BPTF were expressed by the same protocol. Cell pellets were thawed at RT followed by the addition of lysis buffer (50 mM Tris pH 7.4, 150 mM NaCl and 10% v/v glycerol) containing protease inhibitor PMSF (5 mM) and purified according to methods described in the Supporting Information using Ni-affinity chromatography. Yields following purification are 60 mg/L Brd4(1), 60 mg/L 5FW-Brd4(1) (78–90% 5FW incorporation) and 10 mg/L 3FY-Brd4(1) (>95% 3FY incorporation), 26 mg/L 5FW-BPTF (94%, incorporation) and 22 mg/L BrdT, and 11 mg/L 5FW-BrdT(1) (91%, incorporation). Purity of proteins was assessed by SDS-PAGE. Fluorinated amino acid incorporation efficiency in proteins was measured by mass spectrometry as described in the Supporting Information. Concentration was determined via absorbance at 280 nm.11
1D 19F NMR Parameters
19F NMR spectra were acquired at 470 MHz on a Bruker 500 spectrometer with a 5 mm Prodigy TCI Cryoprobe without proton decoupling. Samples containing 40–50 μM bromodomains were labeled in 50 mM TRIS, 100 mM NaCl, 1 mM CHAPS, 2 mM DTT, and 5% D2O, pH 7.4 for binding assays unless otherwise stated. Spectra were referenced to trifluoroacetate (−76.55 ppm). Measurement parameters included a relaxation delay time of 0.7 s for 5FW-Brd4(1) and a 90° flip angle and a relaxation delay of 0.2 s for 3FY-Brd4(1) containing a 30° flip angle. An acquisition time of 0.05 s was used for all experiments. A sweepwidth of 10 ppm was used for 5FW-Brd4(1) spectra and 18 ppm for 3FY-Brd4(1). A 20 Hz line-broadening was applied after 500–3000 transients unless otherwise stated.
Ligand Binding Studies
Small molecules were titrated into the protein solution from concentrated stock solutions of ethylene glycol (10 mM for all small molecules except acetaminophen, which was 50 mM). Final ethylene glycol concentrations were kept below 4% ethylene glycol. For small molecule titrations, 500 scans were acquired with 5FW-Brd4(1) and 3000 3FY-Brd4(1) to ensure good S/N resolution for improved fitting of the data. However, good chemical shift estimates can be readily acquired at 200–400 scans in under 5 min for initial screening as described in the Supporting Information. Stock solutions of kinase inhibitors were prepared from preweighed 5 mg samples which were used to estimate final concentrations. Kd values were obtained using a one-site-binding equation accounting for ligand depletion. Reported errors are from the nonlinear regression fit of the data.
Structure Determination
Protein crystallization was performed with the mosquito LCP (TTP Labtech) crystallization robot at 18 °C using the sitting drop vapor diffusion method. Crystals of 3FY-Brd4(1) were grown in the presence of 1 mM (+)-JQ1 and 10% (v/v) DMSO from 0.2 M NH4C2H3O2, 0.1 M HEPES (pH 7.5), and 25% (w/v) PEG 3350; harvested in cryoprotectant (reservoir containing 25% (v/v) ethylene glycol and 0.5 mM ligand); and flash frozen in a stream of nitrogen gas. X-ray diffraction data were collected at −180 °C using station 22-ID, SER-CAT, Advanced Photon Source, Argonne National Laboratories.
Acknowledgments
We would like to kindly thank J.E. Bradner for the generous donation of (+)-JQ1, and Dr. Margaret Biddle for helpful discussion in preparation for this study. This project was funded in part by the NICHD (1U01HD076542 and HHSN275201300017C, E.S.) and NSF-CAREER Award CHE-1352091 (W.C.P). ITC instrumentation was purchased by the University Minnesota Medical Fund and an NIH shared instrumentation grant NS10-OD017982.
Supporting Information Available
Additional experimental procedures, tables, figures and characterization data can be accessed free of charge via the Internet at http://www.pubs.acs.org.
Author Contributions
§ These authors contributed equally
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Belkina A. C.; Denis G. V. (2012) Nat. Rev. Cancer 12, 465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borah J. C.; Mujtaba S.; Karakikes I.; Zeng L.; Muller M.; Patel J.; Moshkina N.; Morohashi K.; Zhang W. J.; Gerona-Navarro G.; Hajjar R. J.; Zhou M. M. (2011) Chem. Biol. 18, 531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L.; Li J. M.; Muller M.; Yan S.; Mujtaba S.; Pan C. F.; Wang Z. Y.; Zhou M. M. (2005) J. Am. Chem. Soc. 127, 2376. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P.; Picaud S.; Mangos M.; Keates T.; Lambert J. P.; Barsyte-Lovejoy D.; Felletar I.; Volkmer R.; Muller S.; Pawson T.; Gingras A. C.; Arrowsmith C. H.; Knapp S. (2012) Cell 149, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P.; Qi J.; Picaud S.; Shen Y.; Smith W. B.; Fedorov O.; Morse E. M.; Keates T.; Hickman T. T.; Felletar I.; Philpott M.; Munro S.; McKeown M. R.; Wang Y. C.; Christie A. L.; West N.; Cameron M. J.; Schwartz B.; Heightman T. D.; La Thangue N.; French C. A.; Wiest O.; Kung A. L.; Knapp S.; Bradner J. E. (2010) Nature 468, 1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicodeme E.; Jeffrey K. L.; Schaefer U.; Beinke S.; Dewell S.; Chung C. W.; Chandwani R.; Marazzi I.; Wilson P.; Coste H.; White J.; Kirilovsky J.; Rice C. M.; Lora J. M.; Prinjha R. K.; Lee K.; Tarakhovsky A. (2010) Nature 468, 1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung C. W.; Witherington J. (2011) J. Biomol. Screening 16, 1170. [DOI] [PubMed] [Google Scholar]
- Shuker S. B.; Hajduk P. J.; Meadows R. P.; Fesik S. W. (1996) Science 274, 1531. [DOI] [PubMed] [Google Scholar]
- Chung C. W.; Dean A. W.; Woolven J. M.; Bamborough P. (2012) J. Med. Chem. 55, 576. [DOI] [PubMed] [Google Scholar]
- Stockman B. J.; Dalvit C. (2002) Prog. Nucl. Magn. Reson. Spectrosc. 41, 187. [Google Scholar]
- Pomerantz W. C.; Wang N. K.; Lipinski A. K.; Wang E. W.; Cierpicki T.; Mapp A. K. (2012) ACS Chem. Biol. 7, 1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitevski-LeBlanc J. L.; Prosser R. S. (2012) Prog. Nucl. Magn. Reson. Spectrosc. 62, 1. [DOI] [PubMed] [Google Scholar]
- Please see Supporting Information.
- DMSO can compete for binding to the histone recognition site. (Mol. Biosyst. 2011, 7, 2899–2908) which can be readily detected in the 19F NMR spectrum of 3FY and 5FW-Brd4(1). Although ligand binding experiments can also be performed in DMSO (see Supporting Information),unless stated, all binding experiments presented here have been carried out with ethylene glycol as a titrating solvent which does not perturb the NMR spectrum below 3%.
- Ciceri P.; Muller S.; O’Mahony A.; Fedorov O.; Filippakopoulos P.; Hunt J. P.; Lasater E. A.; Pallares G.; Picaud S.; Wells C.; Martin S.; Wodicka L. M.; Shah N. P.; Treiber D. K.; Knapp S. (2014) Nat. Chem. Biol. 10, 305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzuk M. M.; McKeown M. R.; Filippakopoulos P.; Li Q. L.; Ma L.; Agno J. E.; Lemieux M. E.; Picaud S.; Yu R. N.; Qi J.; Knapp S.; Bradner J. E. (2012) Cell 150, 673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ember S. W. J.; Zhu J.-Y.; Olesen S. H.; Martin M. P.; Becker A.; Berndt N.; Georg G. I.; Schönbrunn E. (2014) ACS Chem. Biol. 9, 1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buganim Y.; Goldstein I.; Lipson D.; Milyavsky M.; Polak-Charcon S.; Mardoukh C.; Solomon H.; Kalo E.; Madar S.; Brosh R.; Perelman M.; Navon R.; Goldfinger N.; Barshack I.; Yakhini Z.; Rotter V. (2010) PLoS One 5, e9657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogan A. A.; Thorn K. S. (1998) J. Mol. Biol. 280, 1. [DOI] [PubMed] [Google Scholar]
- Rule G. S.; Pratt E. A.; Simplaceanu V.; Ho C. (1987) Biochemistry 26, 549. [DOI] [PubMed] [Google Scholar]
- Kim T. H.; Chung K. Y.; Manglik A.; Hansen A. L.; Dror R. O.; Mildorf T. J.; Shaw D. E.; Kobilka B. K.; Prosser R. S. (2013) J. Am. Chem. Soc. 135, 9465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J. J.; Horst R.; Katritch V.; Stevens R. C.; Wuthrich K. (2012) Science 335, 1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley P. B.; Kyne C.; Monteith W. B. (2012) Chem. Commun. 48, 10681. [DOI] [PubMed] [Google Scholar]
- Muchmore D. C.; McIntosh L. P.; Russell C. B.; Anderson D. E.; Dahlquist F. W. (1989) Methods in Enzymol. 177, 44. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.