

Abstract
The deposition of amyloid-beta (Aβ) aggregates in the brain is a major pathological hallmark of Alzheimer's disease (AD). Aβ is generated from the cleavage of C-terminal fragments of the amyloid precursor protein (APP-CTFs) by γ-secretase, an intramembrane-cleaving protease with multiple substrates, including the Notch receptors. Endogenous modulation of γ-secretase is pointed to be implicated in the sporadic, age-dependent form of AD. Moreover, specifically modulating Aβ production has become a priority for the safe treatment of AD because the inhibition of γ-secretase results in adverse effects that are related to impaired Notch cleavage. Here, we report the identification of the adipocyte differentiation protein APMAP as a novel endogenous suppressor of Aβ generation. We found that APMAP interacts physically with γ-secretase and its substrate APP. In cells, the partial depletion of APMAP drastically increased the levels of APP-CTFs, as well as uniquely affecting their stability, with the consequence being increased secretion of Aβ. In wild-type and APP/ presenilin 1 transgenic mice, partial adeno-associated virus-mediated APMAP knockdown in the hippocampus increased Aβ production by ∼20 and ∼55%, respectively. Together, our data demonstrate that APMAP is a negative regulator of Aβ production through its interaction with APP and γ-secretase. All observed APMAP phenotypes can be explained by an impaired degradation of APP-CTFs, likely caused by an altered substrate transport capacity to the lysosomal/autophagic system.
INTRODUCTION
In the neurodegenerative amyloid cascade of Alzheimer's disease (AD), quantitative and qualitative changes in the production of amyloid-beta (Aβ) peptides and their aggregation into senile plaques precede the formation of neurofibrillary tangles and initiate the pathological process causing the disease (1,2). Neurotoxic Aβ peptides are generated by successive cleavages of the amyloid precursor protein (APP) by β- and γ-secretases. APP is first processed by the β-secretase BACE1, leading to the formation of a 99-amino-acid long APP C-terminal fragment (APP-C99 or APP-CTFβ). The cleavage of APP-CTFβ by γ-secretase liberates Aβ peptides that are 38–43 amino acids in length, with Aβ42 being a major component of the senile plaques (3). Yet, APP is mainly cleaved within the Aβ region by the α-secretases ADAM10/17 to release APP-CTFα, the processing of which by γ-secretase results in N-terminally truncated and non-toxic Aβ peptides. γ-Secretase is a ubiquitously expressed membrane protease complex that processes a long list of type I transmembrane proteins, including APP, the Notch-1 receptor and several synaptic cell adhesion molecules (4–6). Active γ-secretase is comprised of five protein components: Nicastrin (NCT), Presenilin N- and C-terminal fragments (PS-NTF and PS-CTF), Aph1 and Pen-2 (7). Because they prevent the production of Aβ peptides, γ-secretase inhibitors (GSI) have been extensively investigated in the clinic for the treatment of AD. However, recent human GSI trials revealed significant side effects linked to impaired Notch cleavage (8). Therefore, specifically modulating γ-secretase and Aβ production, without interfering with other physiological processes, is an attractive strategy to safely treat AD (9). Because endogenous γ-secretase modulating factors are potential therapeutic targets, but also possibly implicated in the sporadic forms of AD, we aimed to identify new γ-secretase-interacting proteins (GSPs) that are capable of modulating Aβ production.
RESULTS
Identification of GSPs modulating Aβ production
Highly purified and active γ-secretase prepared from Chinese hamster ovary (CHO) cells overexpressing the four subunits of the protease complex (10) was resolved by blue-native gel electrophoresis (Fig. 1A), and the band corresponding to the complex was excised for protein content analysis by LC-MS/MS mass spectrometry. This approach led to the identification of seven GSPs encoded by the genes ATP1α1, RTN4, VAMP2, VAMP3, TSPAN6, APMAP and HSPA5 (Fig. 1A; peptides identified by LC-MS/MS are listed in Supplementary Material, Fig. S1). As expected from a γ-secretase overexpression system, the silver-stained SDS–PAGE of the protease preparation used for the native gel (Fig. 1A) confirmed the trace amounts of GSPs relative to the predominant γ-secretase subunits (Supplementary Material, Fig. S2). Because ATP1α1, VAMP2/3 and HSPA5 had previously been reported to interact physically with either γ-secretase or APP (11,12), we focused in our study on RTN4, APMAP and TSPAN6. RTN4 encodes for the myelin-associated multi-pass membrane protein reticulon-4 (Nogo), a neurite outgrowth inhibitor limiting plasticity in the healthy adult brain and neuronal regeneration during brain injury (reviewed in13). APMAP encodes for a 415-amino-acid long single-pass type-II glycosylated membrane protein that is implicated in the regulation of white adipose tissue differentiation (14,15). TSPAN6 belongs to the tetraspanin superfamily of multi-pass membrane proteins interacting with multiple immune-related molecules, including immune receptors, integrins, signaling molecules and functioning as important immune response modulators (16). The neurobiological functions of APMAP and TSPAN6 remain unknown.
Figure 1.
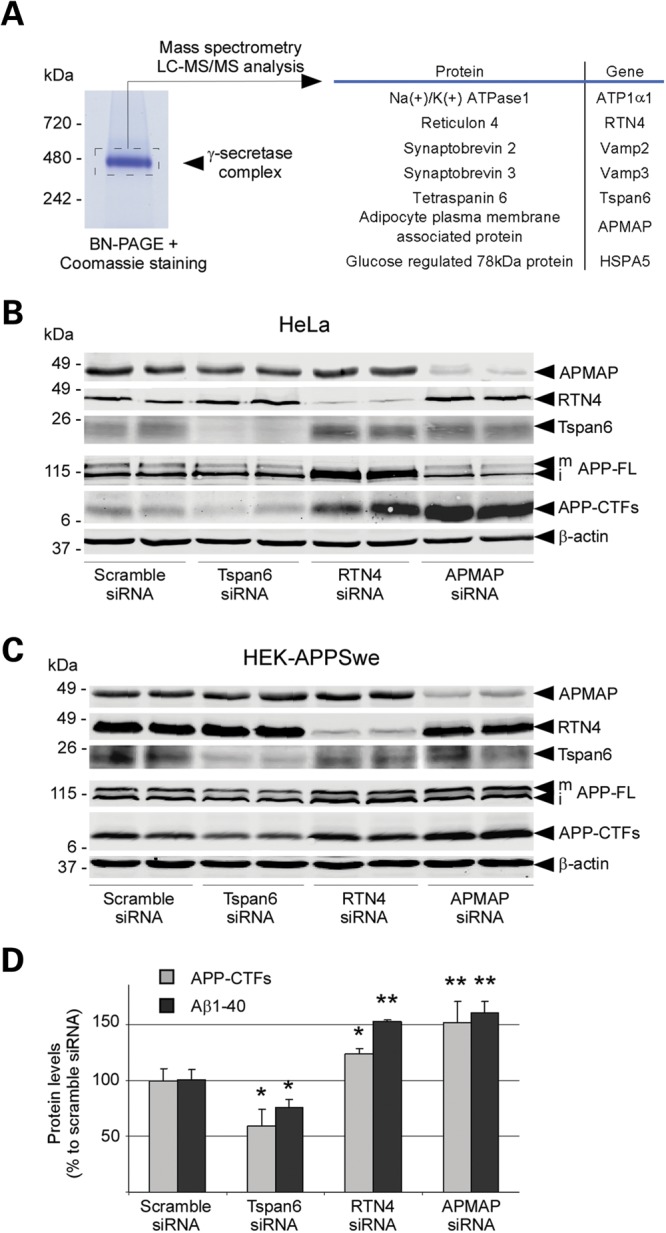
Identification of endogenous modulators of γ-secretase and Aβ production. (A) Analysis by LC-MS/MS of the protein components of the highly purified γ-secretase complex. (B and C) siRNA knockdown of TSPAN6, RTN4 and APMAP affects APP-CTFs in HeLa cells (B) and HEK cells overexpressing APP bearing the Swedish mutation that causes early-onset familial Alzheimer's disease (HEK-APPSwe; C). Biological duplicates are shown for each siRNA condition. (D) Correlation between APP-CTFs levels and Aβ40 production in HEK-APPSwe cells with reduced TSPAN6, RTN4 and APMAP expression. APP-CTFs levels were estimated by densitometric analysis, while Aβ40 levels were quantified by ELISA. Student's t-test was applied for statistical analysis; significance is shown as mean ± SD, *P < 0.05; **P < 0.01; Aβ40: n = 6/group; APP-CTFs: n = 4/Scramble, TSPAN6and APMAP groups; n = 3/RTN4 group. β-Actin served as a protein loading control. mAPP-FL and iAPP-FL: mature and immature APP full-length.
First, the expression of the RTN4, APMAP and TSPAN6 genes was reduced by small-interfering RNA (siRNA) in HeLa cells as well as in HEK cells overexpressing APP harboring the Swedish double mutation near the β-secretase cleavage site (mutation resulting in increased total Aβ levels; HEK-APPSwe). In HeLa and HEK-APPSwe cells, depletion by ∼60% of TSPAN6 lowered APP-CTFs levels by 38 and 40%, respectively (Fig. 1B and C). In contrast, depletion of RTN4 and APMAP (both by ∼75%) caused a strong accumulation of APP-CTFs, respectively, by 181 and 286% in HeLa cells and by 25 and 52% in HEK-APPSwe cells (Fig. 1B and C and Supplementary Material, Fig. S3). This siRNA knockdown of individual genes did not interfere with the maturation or protein levels of α-, β- or γ-secretases, with the exception of increased ADAM10 associated with reduced RTN4 (Supplementary Material, Fig. S4). Importantly, Aβ secreted from HEK-APPSwe cells depleted for the expression of TSPAN6, RTN4 and APMAP directly reflected the levels of intracellular APP-CTFs (Fig. 1D). Indeed, siRNAs targeting TSPAN6 reduced the levels of Aβ40 by 25 ± 7.5%, while siRNAs targeting RTN4, and APMAP raised Aβ40 levels by 52 ± 1.8 and 60 ± 10.1%, respectively (Fig. 1D). Yet, full-length APP (APP-FL) protein levels remained unchanged in the APMAP knockdown experiments (Fig. 1B and C and Supplementary Material, Fig. S3). In contrast, depletion of RTN4 in both HeLa and HEK-APPSwe cells, respectively, caused ∼170 and ∼140% increases in APP-FL levels (Fig. 1B and C).
Genetic inhibition of APMAP increases Aβ production
We further focused on the adipocyte plasma membrane-associated protein APMAP because (i) its knockdown displayed the biggest variations in APP-CTFs and Aβ40 (Fig. 1B–D), (ii) its knockdown was associated with a unique effect on the stability against proteolytic degradation of APP-CTFs (see below and Fig. 3), and (iii) its brain functions remain unknown. First, we confirmed by ELISA and a high-resolution Tris-Tricine urea gel that reduced APMAP expression in HEK-APPSwe cells increased the secretion of full-length total Aβ (Aβ1-x), Aβ1-40 and Aβ1-42 in a dose-dependent manner (Supplementary Material, Fig. S5), without affecting the levels or the maturation of the secretases (Supplementary Material, Fig. S6). Mass spectrometric analysis of secreted Aβ revealed no changes in the Aβ profile (Supplementary Material, Fig. S5), suggesting that APMAP does not affect the sequential and specific processing of APP by γ-secretase. Moreover, APMAP1 purified from E. coli or CHO cells did not interfere with the processing of APP-CTFβ in our cell-free γ-secretase activity assays, arguing against a direct effect of APMAP on the activity of the mature γ-secretase (Supplementary Material, Figs S7 and S8). Finally, purified APMAP1 did not prove to be a substrate for γ-secretase (Supplementary Material, Figs S7 and S8). The latter observation excludes an APP phenotype (accumulation of APP-CTFs) caused by substrate competition, after siRNA-dependent depletion of APMAP1.
Figure 3.

APMAP controls the levels and the stability of APP-CTFs. (A) siRNA knockdown of APMAP in HeLa cells quantitatively and qualitatively impacts APP-CTFs. Reduced APMAP expression correlates with increased APP-CTFs and the formation of three forms of APP-CTFα (CTFα1, α2 and α3), as revealed on a Tris-Tricine urea gel (left panel). This phenotype is further amplified in the presence of 1 µm GSI DAPT (right panel). Biological triplicates are shown. (B) MALDI-TOF mass spectrometric analysis of APP-CTFα1, α2 and α3 immunoprecipitated from DAPT-treated cells with siRNA-reduced APMAP expression. (C) Peptide sequences of N-terminal truncated APP-CTFα1 (C80), -CTFα2 (C74) and -CTFα3 (C71).
APMAP is expressed in neurons and interacts physically with γ-secretase and APP
Next, the physical interaction between APMAP1 and the γ-secretase complex and APP was confirmed in both HEK-APPSwe and CHO cells overexpressing APMAP1-Flag or an untagged control APMAP1 (Fig. 2 and Supplementary Material, Fig. S9). In these experiments, all subunits of the γ-secretase complex, as well as APP-FL and APP-CTFs, specifically and sequentially co-sedimented and co-precipitated with APMAP1-Flag (Fig. 2A). In sharp contrast, these proteins did not co-precipitate with the untagged control APMAP1 (Fig. 2A). Under our experimental conditions, other APP processing enzymes including BACE1 or ADAM10 did not bind to APMAP (Fig. 2A). Next, by combining immunohistochemistry and confocal imaging, we also found that APMAP1 is highly expressed in mouse primary cortical neurons where it adopts an axonal and somatodendritic distribution (Fig. 2B and Supplementary Material, Fig. S10). Our results further show that APMAP partially co-localizes with Nicastrin, a subunit of the γ-secretase complex, mainly in the neuronal cell body (Fig. 2B and Supplementary Material, Fig. S10). Consistent with the predicted topology of APMAP (type-II transmembrane protein), our data suggest that APMAP1 traffics via the secretory pathway to the plasma membrane and adopts a somatodendritic cellular distribution. We further found that APMAP co-localizes partially with APP-FL/APP-CTFs, both in cytosolic and neuritic cellular compartments (Fig. 2B and Supplementary Material, Fig. S10). Altogether, these data support the existence of neuronal high molecular weight complexes made of APMAP and γ-secretase, APMAP and APP-FL/CTFs, and possibly all three components.
Figure 2.

APMAP interacts physically and co-localizes with γ-secretase, APP-FL and APP-CTFs. (A) Velocity co-sedimentation and co-immunoprecipitation of APMAP with the γ-secretase complex, APP-FL and APP-CTFs. Total membrane protein extracts from HEK-APPSwe cells transiently overexpressing hAPMAP1 or hAPMAP1-Flag were sedimented on an 18–28% glycerol gradient containing 0.1% CHAPSO. Each fraction was collected and analyzed by western blot for APMAP1, APP-FL, APP-CTFs and mature and immature γ-secretase (top panels). Next, proteins interacting with hAPMAP1-Flag (Flag) were affinity-precipitated in the fractions labeled in red with M2 anti-Flag affinity resin (lower panel). Untagged APMAP (hAPMAP1, also labeled ‘-’ in the figure) served as a control for the specific co-precipitation. (B) Immunohistochemical co-localization of APMAP (green) with the γ-secretase subunit Nicastrin (red, upper panel) or APP (red, lower panel) in 14 days in vitro mouse primary cortical neurons. Scale bar: 10 µm. Both confocal images (left panels) and Z-stack projections (right panels) are shown with a microscope objective magnification of 40×. For comparison, un-merged images for APMAP, NCT, APP-CTFs and DAPI are shown in Supplementary Material, Fig. S10. mNCT and iNCT, mature and immature Nicastrin.
APMAP controls the levels and stability of APP C-terminal fragments
Further supporting an association with APP, we found that partial depletion of APMAP in HeLa cells altered the profile of endogenous APP-CTFs. As observed on a high-resolution Tris-Tricine urea gel, APMAP siRNA combined with low concentrations of the GSI DAPT led to the generation of three distinct forms of APP-CTFα (CTFα1, α2 and α3), with two forms migrating with apparent sizes of ∼8.5 and 8.0 kDa, instead of 9.0 kDa (Fig. 3A). The mass spectrometric characterization of APP-CTFα1-3 (APP-C80, C74 and C71; Fig. 3B and C and Supplementary Material, Fig. S11) further suggested that endogenous APP-CTFs undergo either partial degradation by yet unknown proteases or further proteolytic processing [possibly by BACE2 (17)], supporting a possible APP stabilization function for APMAP.
APMAP modulates Aβ production in WT and Alzheimer's mice
Next, and to determine whether our findings are relevant in vivo, we induced the partial knockdown of APMAP in wild-type mice and transgenic APP/ presenilin 1 (PS1) Alzheimer's mice. Five week-old mice (males and females) were bilaterally injected in the dorsal hippocampus with serotype 9 adeno-associated viral vectors (AAV9) encoding shRNAs targeting APMAP (Fig. 4A and C and Supplementary Material, Figs S12 and S13). As recently demonstrated by Aschauer et al. (18), AAV9 vectors, when injected in the hippocampus, have a generally broad tropism for many different brain cell types, including oligodendrocytes, microglia, astrocytes and neurons (including inhibitory neurons). In our study, four weeks post-hippocampus injection, the global expression of APMAP was reduced by ∼50% in male WT mice (Fig. 4B) and by ∼35% in male APP/PS1 mice (Fig. 4D), whereas Aβ production was increased by ∼20% in male WT mice (Fig. 4B) and by ∼55% in male APP/PS1 mice (Fig. 4D). Interestingly, under our experimental conditions, no significant effect on Aβ production was found in female mice (Supplementary Material, Fig. S13), which may be linked to the weaker APMAP knockdown observed in these animals (APMAP knockdown by ∼35% in female WT mice and by ∼25% in female APP/PS1 mice). Finally, APMAP depletion in the brain of WT and AD mice does not seem to affect APP-CTF levels (Fig. 4B and D), in contrast to HeLa and HEK-APPSwe cells (Fig. 1B and C). This apparent discrepancy may result from the weaker APMAP knockdown in mice (by 35–50%), when compared with that observed in HeLa and HEK-APPSwe cells (by ∼75%). In support of this explanation, the dose-dependent reduction of APMAP in HEK-APPSwe cells revealed a critical step for APMAP depletion (∼50% depletion, 0.2 nm APMAP siRNA), above which small changes in APMAP expression are associated with big changes in APP-CTFs accumulation (Supplementary Material, Fig. S14).
Figure 4.

APMAP modulates Aβ production in vivo. Five-week-old wild-type (A and B) or APP/PS1 transgenic (C and D) male mice were injected in the dorsal hippocampus with AAV9 expressing APMAP shRNA or a scrambled control shRNA, together with a GFP reporter. Four weeks post-injection, AAV9 transduction is highly efficient and is mainly restricted to the hippocampus (A and C). Wild-type (B) or APP/PS1 (D) males displayed significant ∼50% and ∼35% decreases of APMAP expression (mean ± SD; ***P < 0.001; n = 6/group), associated with significant ∼20 and ∼55% increases in total Aβ levels in the hippocampus (*P < 0.05; **P < 0.01; n = 6/group). Student's t-test was applied for the statistical analysis. β-Actin served as a protein loading control. (A and C) Scale bar: 500 µm.
APMAP modulates APP-CTFs through the lysosomal pathway
Finally, we investigated whether the main APMAP phenotype, namely co-increases of both APP-CTFs and Aβ after APMAP knockdown, could be explained by an impaired degradation of APP-CTFs through the lysosomal–autophagic pathway. To do so, a pharmacological approach was used, by taking advantage of Chloroquine, a well-known inhibitor of the lysosomal–autophagic pathway. Chloroquine is a lysosomotropic agent that impedes the lysosomal pathway by inhibition of lysosomal enzymes and lysosomal protein degradation, and by preventing the fusion of endosomes and lysosomes (19). Chloroquine, because it raises the lysosomal pH, also inhibits autophagy (20). To test whether APMAP can regulate the levels of APP-CTFs through the lysosomal pathway, HEK-APPSwe and HeLa cells were incubated for 12 h with, respectively, 25 and 50 µm Chloroquine, in the presence of APMAP or after APMAP depletion. As shown in Fig. 5A and B, Chloroquine treatments in the presence of APMAP (scramble siRNA) were associated with a 2.1-fold increase of APP-CTFs in HEK-APPSwe cells and a 3.4-fold increase of the same proteins in HeLa cells, when compared with DMSO-treated controls. This observation clearly supports a lysosomal degradation of the APP-CTFs in these two cell types. Interestingly, APMAP depletion strongly reinforced the Chloroquine-dependent accumulation of APP-CTFs. The densitometric analysis of the western blots indeed revealed additional 2.2-fold and 1.6-fold increases of APP-CTFs in HEK-APPSwe and HeLa cells co-treated with Chloroquine and the APMAP siRNA, when compared with cells co-treated with Chloroquine and the scramble siRNA. The latter observation is consistent with a Chloroquine- and APMAP-dependent synergistic effect possibly caused by reduced lysosomal degradation and reduced lysosomal transport of APP-CTFs.
Figure 5.

APMAP regulates cellular APP-CTFs levels through the lysosomal–autophagic pathway. HEK-APPSwe (A) and HeLa (B) cells treated either with a scrambled control siRNA or with APMAP siRNA were incubated for 12 h with the lysosomal inhibitor Chloroquine (25 and 50 µM, respectively). Total membrane protein extracts were prepared and analyzed by western blot for APMAP1, APP-FL and APP-CTFs (left panels). Biological duplicates are shown and β-actin served as a protein loading control. Next, APP-CTFs bands were quantified by densitometry (right panels) and Student's t-test was applied for statistical analysis with n = 4/group. **P < 0.01; ***P < 0.001.
Altogether, our data suggest that APMAP modulates the levels of APP-CTFs through the lysosomal–autophagic system.
DISCUSSION
In this study, we analyzed the protein composition of highly purified and active γ-secretase complexes and identified seven proteins that interact physically with the enzymatic complex. Among these proteins, TSPAN6 was found to positively regulate the processing of APP and the production of Aβ, consistent with previous work on tetraspanins TSPAN12, TSPAN28 and TSPAN29 (12,21). Another of our proteins identified to be associated with the γ-secretase complex is RTN4, which has been shown previously to negatively regulate Aβ production by interacting with the β-secretase BACE1 (22). We confirmed this effect, as reduced RTN4 led to increased Aβ production (Fig. 1D) and suggest for the first time that the Aβ-suppressing function of RTN4 may also be based on a physical interaction with the γ-secretase complex.
Importantly, we further identified the adipocyte differentiation protein APMAP (15) as an endogenous inhibitor of Aβ production that binds to both APP and γ-secretase, and controls the levels and the stability of APP-CTFs. Additionally, APMAP does not affect the processing of other γ-secretase substrates, including the Notch receptor, and the neuronal cell adhesion molecules Neurexin-3β and Neuroligin-1 (Supplementary Material, Fig. S15). Consistent with the Allen Brain and Human Protein Atlases (Supplementary Material, Fig. S16), we further showed that APMAP is expressed in the brain, and more specifically in neurons where it co-localizes at least partially with APP and γ-secretase (Fig. 2B). In mice, APMAP knockdown in both neuronal and glial cells (18; Fig. 4A and C) increased Aβ levels in the hippocampus 4 weeks after injection of the AAV9 with shRNAs targeting APMAP (Fig. 4). Yet, a decade of research on which forms and structures of Aβ impair neuronal functions has concluded that (i) small and diffusible assemblies of misfolded and self-aggregating Aβ peptides are probably the principal cytotoxic forms, but also that (ii) insoluble amyloid fibrils and the amyloid plaques cause neuritic alterations including a decreased efficiency of neurotransmission (reviewed in 23). Whether APMAP preferentially affects the production of soluble Aβ assemblies or the deposition of Aβ plaques remains unknown and will require specific and long-term investigations in AD transgenic mice.
At the molecular level, APP is cleaved into sAPP-α and CTF-α by α-secretase or cleaved into sAPP-β and CTF-β by β-secretase. Then, γ-secretase cleaves CTF-α into secreted 24–26 residues long P3 peptides and cytoplasmic APP-intracellular domain (AICD), and cleaves CTF-β into 38–43 residues long Aβ peptides and AICD. Under physiological conditions, APP processing by γ-secretase is associated with increased Aβ production and reduced CTFβ levels. In contrast, if a molecule inhibits γ-secretase, reduced Aβ production is associated with increased CTFβ levels. In our study, depletion of APMAP led to both increases of Aβ and CTFs (Figs 1 and 3). Moreover, depletion of APMAP did not affect the levels of sAPP-α or sAPP-β (Supplementary Material Figs 4 and 6), or the expression of APP-FL (Figs 1 and 3), suggesting that (i) α- and β-secretases are not targets for APMAP, and (ii) both cleavage and production of APP-FL are not affected by APMAP. All observed phenotypes including the co-increases of APP-CTFs and Aβ after APMAP knockdown could be explained by an impaired degradation of APP-CTFs (depicted in Fig. 6). This hypothesis is supported by the observation that APMAP depletion caused an increased accumulation of APP-CTFs in cells treated with Chloroquine, a well-known inhibitor of the lysosomal–autophagic pathway (Fig. 5).
Figure 6.

Molecular hypothesis for the regulation of APP-CTFs/Aβ levels by APMAP. In this hypothesis, APMAP is implicated in the transport of APP-CTFs to the lysosomal–autophagic system, where it undergoes substrate degradation. Consistent with the observed cellular phenotypes (Figs 1 and 3), depletion of APMAP would impair this transport capacity and consequently trigger increased APP-CTFs levels, which in turn cause increased Aβ production. Impaired degradation would further make APP-CTFs substrates accessible to yet unidentified proteases, explaining the N-terminal truncated species observed in cells with depleted APMAP (Fig. 3).
In the molecular hypothesis proposed in Figure 6, APMAP, by interacting physically with APP-CTFs (Fig. 2), would promote the transport of these substrates from the endosomes to the lysosomes, with the consequence being their degradation, thus reducing both APP-CTFs and Aβ levels. Indeed, it has been previously reported that APP-CTFs can be delivered to the lysosomes by two pathways: the endosomal–lysosomal pathway (heterophagy) and the authopagic pathway, causing the degradation of the Aβ precursor proteins (24–26). Similar to the phenotypes observed in our study with reduced APMAP, genetic or pharmacological inhibition of the lysosomal–autophagic system led to the significant intracellular accumulation of APP-CTFs and increased Aβ secretion (27,28). Thus, APMAP depletion would lead to impaired degradation, with the consequence being both increases of APP-CTFs and Aβ. Moreover, impaired APP-CTFs degradation would make these γ-secretase substrates accessible to yet unidentified proteases and trigger their proteolysis (Fig. 3). Alternatively, and still by interacting physically with APP-CTFs, APMAP could also transport these substrates to the γ-secretase complex, explaining the possible physical interaction between APMAP, APP-CTFs and the enzyme (Fig. 2). Since abnormalities in the neuronal lysosomal–autophagic system have been associated with the pathology of AD (reviewed in 29), understanding the precise molecular mechanisms by which APMAP can regulate APP-CTFs degradation deserves further investigation.
Yet, APP-CTFs do not seem to only be degraded through the lysosomal–autophagic system. Indeed, Bustamante et al. (30) have recently demonstrated in human H4 neuroglioma cells that APP-C99 is actively degraded in the endoplasmic reticulum in an ubiquitin and proteasome-dependent manner. Whether APMAP can play an active role in the proteasome-dependent degradation of APP-CTFs (including a crosstalk between the lysosomal and proteasomal systems) also requires further investigation.
APMAP also controls adipogenesis and consequently the adipocytic release of potential γ-secretase modulators, including leptin (31,32) and cholesterol (33–35). Moreover, the expression of APMAP is regulated by the peroxisome proliferation-activated receptor PPARγ (14,15), which has also been shown to decrease Aβ production (36). In return, the processing of Notch by γ-secretase inhibits PPARγ and adipocyte differentiation (37). Thus, reducing Aβ production by interfering with the PPARγ/APMAP/γ-secretase regulatory loop could potentially represent a therapeutic approach to slow down the pathology of AD. Moreover, APMAP, by participating in the gut/adipose tissue/brain crosstalk and regulating cerebral Aβ levels, could possibly provide new molecular insight into the statistical link between AD and obesity or diabetes.
MATERIALS AND METHODS
γ-Secretase purification, tryptic digestion and mass spectrometry
The γ-secretase complex was purified as previously described (10) and was run on a NativePAGE Novex® Bis–Tris 4–16% gel for BN-PAGE analysis (Invitrogen, Carlsbad, CA, USA). Next, the complex was visualized by Coomassie staining, excised and cut into small pieces. Proteins were reduced, alkylated and subjected to in-gel digestion with trypsin. Briefly, gel pieces were destained, desiccated by incubating twice in 200 µl of 50 mm ammonium bicarbonate and 50% ethanol for 20 min and dried with a vacuum concentrator. The samples were then incubated overnight at 37°C with trypsin (12.5 ng/µl). For liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) analysis after extraction from gel slices, peptides were resuspended in 2% Acetonitril/0.1% Formic Acid and separated by reverse-phase chromatography on a Dionex Ultimate 3000 RSLC nanoUPLC system connected in-line with an Orbitrap Elite (Thermo Fischer Scientific, Waltham, MA, USA). The instrument was operated in an information-dependent mode where peptide masses were selected for collision-induced dissociation to generate tandem mass spectra. A database search was performed using Mascot 2.3 (Matrix Science, Boston, MA, USA) and SEQUEST in Proteome Discoverer v.1.3 against a human database (UniProt release 2013_01; 87613 sequences) and Mammalian database (UniProt release 2011_07; 91104 sequences). All searches were performed with trypsin cleavage specificity, with up to three missed cleavages allowed, an ion mass tolerance of 10 ppm for the precursor and 0.5 Da for the fragments. Carbamidomethylation was set as a fixed modification, whereas oxidation (M), acetylation (protein N-term) and phosphorylation (STY) were considered variable modifications. Data were further processed and inspected using Scaffold 3 software (Proteome Software).
Cell lines, cell cultures and treatments with siRNA or GSI
Human embryonic kidney cells (HEK 293T), human cervical carcinoma cells (HeLa) and CHO cells were routinely grown in DMEM with 10% fetal bovine serum and penicillin/streptomycin in a humidified 5% CO2 atmosphere. The CHO cells stably expressing Flag-tagged APMAP and the HEK cells stably expressing APP with the Swedish mutation [HEK-APPSwe (38)] were maintained in DMEM with 10% FBS supplemented with 150 µg/ml Geneticin G418 (Gibco, Thermo Fischer Scientific). HEK cells stably expressing the γ-secretase substrate NotchΔE [HEK-N7 (39)] were cultured in DMEM with 10% FBS supplemented with 250 µg/ml Zeocin (Gibco). For siRNA knockdown experiments, cells were treated for 3 days with allstar negative control siRNA (Qiagen, Germantown, MD, USA), APMAP (5′-TTCACCGATTCTAGCAGCAAA-3′, Qiagen), RTN4 (5′-CAGGGCATATCTGGAATCTGA-3′, Qiagen) or Tspan6 (5′-CTCTCGTGCCATAACAAATAA-3′, Qiagen) siRNA duplexes complexed with Lipofectamine RNAiMax from Invitrogen. For Aβ peptides secreted in the conditioned medium of HEK-APPSwe cells, Aβ1-X (IBL), Aβ1-40 (Invitrogen) and Aβ1-42 (Invitrogen) were quantitatively measured by ELISA, according to the protocol provided by the manufacturer. For inhibition of cellular γ-secretase activity, cells were incubated for 4 h with 10 µm of the γ-secretase-specific inhibitor N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester (DAPT; Sigma-Aldrich, GmbH, Buchs, Switzerland). For inhibition of the lysosomal pathway, HEK-APPSwe and HeLa cells were incubated for 12 h with 25 and 50 µm of Chloroquine (Sigma-Aldrich), respectively.
Western blotting and antibodies
Whole cell extracts were prepared in 50 mm HEPES buffer containing 1% NP40 and complete protease inhibitor cocktail (Roche Applied Sciences, Basel, Switzerland) and were separated by electrophoresis on homemade 12% acrylamide Tris-glycine SDS–PAGE or NuPAGE Novex® 4–12% Bis–Tris gels (Invitrogen) for SDS–PAGE analysis of APMAP purification and γ-secretase activity. High separation of APP-CTFs or Aβ peptides was resolved using 15% acrylamide and 6 m urea Tris-Tricine SDS–PAGE as described previously (40) or by a Tris-bicine-urea gel (41), respectively. Proteins were transferred onto nitrocellulose membranes and probed with antibodies against APP-FL and APP-CTFs (CT15, Sigma-Aldrich, Saint Louis, MS, USA), as well as sAPPα (6E10; Covance, Berkeley, CA, USA) and total sAPP (22C11; Millipore, Billerica, MA, USA), NCT (N1660 or NCT164, BD Bio-Sciences, Bedford, MA, USA), PS1-NTF (MAB1563, Chemicon International, Temecula, CA, USA), PS1-CTF (MAB5232, Chemicon International), Aph1 (O2C2, Covance), Pen-2 (UD1, a gift from Helena Karlström, KI Stockholm, Sweden), ADAM10 (Prosci, Poway, CA, USA), BACE1 (EE17, Sigma-Aldrich), APMAP (4F6, AbCam, Cambridge, UK), RTN4 (Ab2, Sigma-Aldrich), Tspan6 (AbCam), Notch-ICD (Val1744, Cell Signaling, Boston, MA, USA) or β-actin (Sigma-Aldrich). Anti-mouse, rabbit and rat IgGs conjugated to Alexa 680 were purchased from Invitrogen, and the Odyssey infrared imaging system (LICOR, Lincoln, NE, USA) was used to detect the fluorescent signal.
Glycerol gradient velocity sedimentation and co-immunoprecipitation
Membranes were prepared by osmotic shock from HEK-APPSwe cells transiently expressing either Flag-tagged or untagged human APMAP1, as previously described (4). Next, membrane proteins solubilized in 1% CHAPSO were loaded onto an 18–28% glycerol gradient containing 11 × 950 µl fractions prepared in 0.1% CHAPSO. After centrifugation for 15 h at 220 000g, 950 µl fractions were collected and analyzed by western blotting. For co-immunoprecipitation experiments, all fractions collected after glycerol velocity sedimentation were diluted twice with 0.2% digitonin in Tris-buffered saline (TBS) and incubated with 50 µl of M2 resin (Sigma-Aldrich) at 4°C overnight. The beads were then washed three times for 15 min in TBS with 0.1% digitonin and collected in 50 µl of diluted (1:2 in water) Laemmli sample buffer.
Immunostaining
Mouse primary cortical neurons were prepared from embryonic day 17 Of1 fetal mouse brains. Cortices were digested in medium containing papain (20 U/ml, Sigma-Aldrich) and dissociated by mechanical trituration. Cells were plated in the neurobasal medium (Invitrogen) supplemented with B27 (Invitrogen) and 2 mm Glutamax (Life Technologies, Thermo Fischer Scientific) on poly-l-ornithine (Sigma-Aldrich)-coated plates at 37°C in a humidified 5% CO2 atmosphere. At in vitro day 14, neurons were fixed with 4% paraformaldehyde and permeabilized in phosphate buffered saline (PBS) containing 0.1% Triton X-100 for 10 min. The cells were then incubated in citrate buffer (10 mm sodium citrate, 0.05% Tween-20, pH 8.0) for 30 min at 95–100°C. After cooling at room temperature, non-specific antibody binding was prevented by incubation in 0.25% cold water fish gelatin (Sigma-Aldrich) diluted in PBS for 45 min. Following the blocking step, the cells were incubated with antibodies against APMAP (GTX46051, Gentex, Irvine, CA, USA), APP-CTFs (4G8, AbCam Cambridge, UK) or Nicastrin (NCT164, BD Bio-Sciences, Franklin Lakes, NJ, USA). Finally, the fixed cells were incubated with anti-rabbit Alexa 488 and anti-mouse Alexa 568 (Invitrogen) secondary antibodies. Image acquisition was performed with a Zeiss LSM700 UP2 microscope using a 40× objective.
Immunoprecipitation-mass spectrometry (IP-MS) analysis of Aβ and APP-CTFs
Aβ secreted from HEK-APPSwe cells treated with scrambled or APMAP siRNAs were immunoprecipitated overnight using the monoclonal anti-Aβ antibody 4G8 (AbCam) and protein G-coupled agarose (Roche Applied Sciences). Aβ was eluted using a 1:20:20 (vol:vol:vol) mix of 0.1% (vol/vol) trifluoroacetic with acid:acetonitrile:H20 mixed 1:1 (vol:vol) with saturated CHCA (α-cyano-4-hydroxycinnamic acid) and analyzed by MALDI-TOF mass spectrometry in the reflectron mode on an ABI 4800 MALDI-TOF/TOF mass spectrometer (Applied Biosystems, Thermo Fischer Scientific). Intracellular APP-CTFs from whole protein extracts prepared from cells treated with APMAP siRNA and/or DAPT were immunoprecipitated with an antibody targeting the CTF portion of APP (CTF15; Sigma-Aldrich GmbH, Buchs, Switzerland). Next, APP-CTFs were washed two times in 1% NP40-HEPES and two more times in deionized water, and then eluted in 0.1% (vol/vol) trifluoroacetic acid with acetonitrile:H20 mixed 1:1 (vol:vol), dried with a vacuum pump and finally resolubilized in the same elution buffer saturated with sinapinic acid (3–4-hydroxy-3,5-dimethoxyphenyl prop-2-enoic acid). All spectra were acquired in the linear mode with an ABI 4800 MALDI-TOF/TOF mass spectrometer. Molecular masses were accurately measured and searched against known amino acid sequences of human APP.
Purification of APMAP in E. coli and CHO
For the purification of recombinant human APMAP1-His6 in E. coli, a pET21b vector encoding human APMAP1-His6 cDNA was expressed in BL21(DE3) cells and induced with 1 mm isopropyl-β-d-thiogalactopyranoside for 16 h at 24°C at a 600 nm optical density of 0.8. For the purification of human APMAP1-His6, harvested bacteria were lysed with 10 mm Tris pH 7.4, 150 mm NaCl, 1% CHAPSO and complete protease inhibitor cocktail (Roche) and passed three times through a high-pressure homogenizer (Emulsiflex-C5; Avestin, Inc., Mannheim, Germany) at a pressure greater than 1000 psi. The obtained lysate was incubated for 1 h on ice and spun down at 5150g for 20 min at 4°C, and the resulting supernatant was incubated for 30 min with Ni-NTA agarose beads (Invitrogen) at room temperature. Bound proteins were eluted five times in 10 mm Tris pH 7.4, 150 mm NaCl and 1% CHAPSO containing 250 mm imidazole (pH 7.4) and analyzed by Coomassie-stained SDS–PAGE.
For the purification of mammalian APMAP-Flag, a total of 2.64 × 109 CHO cells stably transfected with human APMAP1-Flag were resuspended in MES buffer [50 mm MES pH 6.0, 150 mm NaCl, 5 mm MgCl2, 5 mm CaCl2 and protease inhibitor cocktail (Roche)]. The cells were lysed by three passages in a high-pressure homogenizer at a pressure greater than 1000 psi and centrifuged at 3000g for 20 min. The supernatant was further centrifuged at 100 000g for 1 h to pellet microsomes. The pellet was resuspended by pipetting up and down 30 times in bicarbonate buffer (0.1 m NaHCO3, pH 11.3) and incubated for 20 min at 4°C to remove non-integral proteins. The washed microsomes were pelleted by centrifugation at 100 000g for 1 h. The supernatant was discarded, and the membranes were solubilized in ice-cold 1% CHAPSO-HEPES lysis buffer [50 mm HEPES, 150 mm NaCl, 5 mm MgCl2, 5 mm CaCl2, 1% CHAPSO and protease inhibitor cocktail (Roche)] for 1 h at 4°C. The lysate was centrifuged at 16 000g, saved and diluted twice in HEPES buffer [50 mm HEPES, 150 mm NaCl, 5 mm MgCl2, 5 mm CaCl2 and protease inhibitor cocktail (Roche)]. The lysate was further diluted six times in 0.1% digitonin-TBS buffer (50 mm Tris–HCl pH 7.4, 150 mm NaCl) and bound to M2 anti-Flag affinity resin (Sigma-Aldrich) overnight. Following three washes in 0.1% digitonin-TBS buffer, the bound proteins were eluted in 1.5 ml of 0.1% digitonin-TBS buffer containing 0.2 mg/ml Flag peptides and used for the in vitro γ-secretase assays. In both protocols, the purity of human APMAP1-His6 and APMAP1-Flag was confirmed by SDS–PAGE using Coomassie staining.
γ-Secretase activity assays
γ-Secretase assays using recombinant human APMAP1 and APP-C100-Flag were performed as previously reported (42–44). Briefly, γ-secretase purified from SS20 cells was solubilized in 0.2% (wt/vol) CHAPSO, 50 mm HEPES (pH 7.0), 150 mm NaCl, 5 mm MgCl2 and 5 mm CaCl2 and incubated at 37°C for 4 h with 1 µm substrate, 0.1% (wt/vol) phosphatidylcholine and 0.025% (wt/vol) phosphatidylethanolamine. The resulting products, AICD-Flag and Aβ, were detected with Flag-specific M2 antibodies (Sigma-Aldrich) and Aβ-specific 6E10 (Covance), respectively.
Mice
Male and female WT and APP/PS1 transgenic mice (obtained from Prof. M. Jucker, HIH, Tübingen, Germany) were used. APP/PS1 animals co-express the KM670/671NL Swedish mutation of human APP and the L166P mutation of human PS1 under the control of the Thy-1 promoter and show age-dependent accumulation of parenchymal Aβ plaques (45). Both WT and APP/PS1 mice were generated on a C57BL/6J background. We conducted all animal experiments in accordance with local and federal directives for the care and use of laboratory animals.
Production and titration of AAV2/9 vectors
The production and titration of AAV vector particles pseudotyped with a serotype 9 capsid has been described previously (46). Briefly, the vectors were obtained by transient co-transfection of 293-AAV cells (Agilent, Santa Clara, CA, USA) with the shuttle plasmid and a pDP9rs.gck helper plasmid, which encodes the REP and CAP adeno-associated viral genes, as well as genes that are essential to the adenoviral helper function (plasmid kindly provided by Dr J. Kleinschmidt, Deutsches Krebsforschungszentrum, Heidelberg, Germany). Viral vector particles were purified from cell lysates by centrifugation on an iodixanol gradient (OptiPrep, Axis-Shield, Oslo, Norway) followed by anion exchange affinity chromatography on a HiTrap Q-FF column (GE Healthcare Bio-Sciences AB, Uppsala, Sweden). The suspension medium was replaced with Dulbecco's PBS (DPBS, Gibco), and the viral particles were concentrated to a final volume of ∼100 µl using Centricon Plus-20 filtration devices (regenerated cellulose, 100 000 MWCO, Millipore). Viral titers were determined by measuring the number of viral genomes present in a given volume of the vector suspension using real-time PCR. We used a primer and probe set that anneals to the β-globin intron (fw: 5′-CGTGCCAAGAGTGACGTAAG-3′; rv: 5′-TGGTGCAAAGAGGCATGATA-3′; TaqMan probe: 5′-FAM-TTGCCCTGAAAGAAAGAGATTAGGGAA-BHQ-1-3′). The reactions were run in a Corbett Rotor-Gene RG-3000 cycler (Qiagen Inc., Valencia, CA, USA) using the following program: one cycle of 20 min at 95°C for capsid denaturation followed by 40 cycles of 3 s at 95°C and 10 s at 60°C.
Injection of mice and Aβ measurements
The equivalent of 2.53 × 1010 and 1.10 × 1010 viral particles were injected into 5-week-old WT and APP/PS1 mice, respectively. Bilateral injection was performed at the following stereotaxic coordinates: (in mm, anterior, lateral, ventral to bregma) −2.1, 1.5, 1.7. After 4 weeks, the mice were euthanatized, and the right hemisphere was homogenized in a buffer (1 ml buffer/100 mg brain) containing 50 mm NaCl, 0.2% DEA and complete protease inhibitor cocktail (Roche). After centrifugation at 200 000g for 35 min at 4°C, soluble Aβ peptide analysis in WT and APP/PS1 brain lysates was performed using anti-human/rat AβX-40 (Wako Chemicals, Richmond, VA, USA) and anti-human Aβ1-X (IBL International GmbH, Hamburg, Germany), respectively, according to the manufacturer's instructions. Total proteins were subsequently extracted in 1% NP40 and complete protease inhibitor cocktail (Roche) for western blot analysis.
Statistical analyses
All data are presented as the mean ± SD. Student's t-test (two-tailed) was applied for statistical analysis, and statistical significance is shown as P < 0.05 (one asterisks), P < 0.01 (two asterisks) or P < 0.001 (three asterisks).
SUPPLEMENTARY MATERIAL
AUTHORS' CONTRIBUTIONS
S.M., A.M., J.R.A., M.D. and J.P. performed the experiments; B.L.S. and P.C.F. designed the research; P.C.F. supervised the project; S.M. and P.C.F. wrote the manuscript. All authors edited the manuscript.
FUNDING
This work was supported by the Swiss National Science Foundation (to P.C.F. and S.M., grant 31003A_152677/1) and the Strauss foundation (to P.C.F. and M.D.). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript. Funding to pay the Open Access publication charges for this article was provided by EPFL.
Supplementary Material
ACKNOWLEDGMENTS
The authors also thank M. Moniatte and R. Hamelin (Proteomics Core Facility, School of Life Sciences, Ecole Polytechnique Fédérale de Lausanne, Lausanne, Switzerland) for technical assistance with mass spectrometry. The authors further thank L. Aeschbach and P. Colin for technical help, and V. Padrun and F. Pidoux for technical assistance with AAV production.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Hardy J.A., Higgins G.A. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 2.Kawabata S., Higgins G.A., Gordon J.W. Amyloid plaques, neurofibrillary tangles and neuronal loss in brains of transgenic mice overexpressing a C-terminal fragment of human amyloid precursor protein. Nature. 1991;354:476–478. doi: 10.1038/354476a0. [DOI] [PubMed] [Google Scholar]
- 3.Iwatsubo T., Odaka A., Suzuki N., Mizusawa H., Nukina N., Ihara Y. Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: evidence that an initially deposited species is A beta 42(43) Neuron. 1994;13:45–53. doi: 10.1016/0896-6273(94)90458-8. [DOI] [PubMed] [Google Scholar]
- 4.Bot N., Schweizer C., Ben Halima S., Fraering P.C. Processing of the synaptic cell adhesion molecule neurexin-3beta by Alzheimer disease alpha- and gamma-secretases. J. Biol. Chem. 2011;286:2762–2773. doi: 10.1074/jbc.M110.142521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Strooper B., Annaert W., Cupers P., Saftig P., Craessaerts K., Mumm J.S., Schroeter E.H., Schrijvers V., Wolfe M.S., Ray W.J., et al. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature. 1999;398:518–522. doi: 10.1038/19083. [DOI] [PubMed] [Google Scholar]
- 6.Haapasalo A., Kovacs D.M. The many substrates of presenilin/gamma-secretase. J. Alzheimers Dis. 2011;25:3–28. doi: 10.3233/JAD-2011-101065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fraering P.C. Structural and functional determinants of gamma-secretase, an intramembrane protease implicated in Alzheimer's disease. Curr. Genomics. 2007;8:531–549. doi: 10.2174/138920207783769521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Imbimbo B.P. Therapeutic potential of gamma-secretase inhibitors and modulators. Curr. Top. Med. Chem. 2008;8:54–61. doi: 10.2174/156802608783334015. [DOI] [PubMed] [Google Scholar]
- 9.Dimitrov M., Alattia J.R., Lemmin T., Lehal R., Fligier A., Houacine J., Hussain I., Radtke F., Dal Peraro M., Beher D., et al. Alzheimer's disease mutations in APP but not gamma-secretase modulators affect epsilon-cleavage-dependent AICD production. Nat. Commun. 2013;4:2246. doi: 10.1038/ncomms3246. [DOI] [PubMed] [Google Scholar]
- 10.Alattia J.R., Matasci M., Dimitrov M., Aeschbach L., Balasubramanian S., Hacker D.L., Wurm F.M., Fraering P.C. Highly efficient production of the Alzheimer's gamma-secretase integral membrane protease complex by a multi-gene stable integration approach. Biotechnol. Bioeng. 2013;110:1995–2005. doi: 10.1002/bit.24851. [DOI] [PubMed] [Google Scholar]
- 11.Yang Y., Turner R.S., Gaut J.R. The chaperone BiP/GRP78 binds to amyloid precursor protein and decreases Abeta40 and Abeta42 secretion. J. Biol. Chem. 1998;273:25552–25555. doi: 10.1074/jbc.273.40.25552. [DOI] [PubMed] [Google Scholar]
- 12.Wakabayashi T., Craessaerts K., Bammens L., Bentahir M., Borgions F., Herdewijn P., Staes A., Timmerman E., Vandekerckhove J., Rubinstein E., et al. Analysis of the gamma-secretase interactome and validation of its association with tetraspanin-enriched microdomains. Nat. Cell Biol. 2009;11:1340–1346. doi: 10.1038/ncb1978. [DOI] [PubMed] [Google Scholar]
- 13.Teng F.Y., Tang B.L. Cell autonomous function of Nogo and reticulons: the emerging story at the endoplasmic reticulum. J. Cell Physiol. 2008;216:303–308. doi: 10.1002/jcp.21434. [DOI] [PubMed] [Google Scholar]
- 14.Albrektsen T., Richter H.E., Clausen J.T., Fleckner J. Identification of a novel integral plasma membrane protein induced during adipocyte differentiation. Biochem. J. 2001;359:393–402. doi: 10.1042/0264-6021:3590393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bogner-Strauss J.G., Prokesch A., Sanchez-Cabo F., Rieder D., Hackl H., Duszka K., Krogsdam A., Di Camillo B., Walenta E., Klatzer A., et al. Reconstruction of gene association network reveals a transmembrane protein required for adipogenesis and targeted by PPARgamma. Cell Mol. Life Sci. 2010;67:4049–4064. doi: 10.1007/s00018-010-0424-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hemler M.E. Tetraspanin functions and associated microdomains. Nat. Rev. Mol. Cell Biol. 2005;6:801–811. doi: 10.1038/nrm1736. [DOI] [PubMed] [Google Scholar]
- 17.Sun X., He G., Song W. BACE2, as a novel APP theta-secretase, is not responsible for the pathogenesis of Alzheimer's disease in Down syndrome. FASEB J. 2006;20:1369–1376. doi: 10.1096/fj.05-5632com. [DOI] [PubMed] [Google Scholar]
- 18.Aschauer D.F., Kreuz S., Rumpel S. Analysis of transduction efficiency, tropism and axonal transport of AAV serotypes 1, 2, 5, 6, 8 and 9 in the mouse brain. PLoS ONE. 2013;8:e76310. doi: 10.1371/journal.pone.0076310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hart P.D., Young M.R., Jordan M.M., Perkins W.J., Geisow M.J. Chemical inhibitors of phagosome-lysosome fusion in cultured macrophages also inhibit saltatory lysosomal movements. A combined microscopic and computer study. J. Exp. Med. 1983;158:477–492. doi: 10.1084/jem.158.2.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poole B., Ohkuma S. Effect of weak bases on the intralysosomal pH in mouse peritoneal macrophages. J. Cell Biol. 1981;90:665–669. doi: 10.1083/jcb.90.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu D., Sharma C., Hemler M.E. Tetraspanin12 regulates ADAM10-dependent cleavage of amyloid precursor protein. FASEB J. 2009;23:3674–3681. doi: 10.1096/fj.09-133462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murayama K.S., Kametani F., Saito S., Kume H., Akiyama H., Araki W. Reticulons RTN3 and RTN4-B/C interact with BACE1 and inhibit its ability to produce amyloid beta-protein. Eur. J. Neurosci. 2006;24:1237–1244. doi: 10.1111/j.1460-9568.2006.05005.x. [DOI] [PubMed] [Google Scholar]
- 23.Mucke L., Selkoe D.J. Neurotoxicity of amyloid beta-protein: synaptic and network dysfunction. Cold Spring Harb. Perspect. Med. 2012;2:a006338. doi: 10.1101/cshperspect.a006338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen F., Yang D.S., Petanceska S., Yang A., Tandon A., Yu G., Rozmahel R., Ghiso J., Nishimura M., Zhang D.M., et al. Carboxyl-terminal fragments of Alzheimer beta-amyloid precursor protein accumulate in restricted and unpredicted intracellular compartments in presenilin 1-deficient cells. J. Biol. Chem. 2000;275:36794–36802. doi: 10.1074/jbc.M006986200. [DOI] [PubMed] [Google Scholar]
- 25.Nixon R.A. Autophagy, amyloidogenesis and Alzheimer disease. J. Cell Sci. 2007;120:4081–4091. doi: 10.1242/jcs.019265. [DOI] [PubMed] [Google Scholar]
- 26.Tian Y., Chang J.C., Greengard P., Flajolet M. The convergence of endosomal and autophagosomal pathways: implications for APP-CTF degradation. Autophagy. 2014;10:694–696. doi: 10.4161/auto.27802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tian Y., Bustos V., Flajolet M., Greengard P. A small-molecule enhancer of autophagy decreases levels of Abeta and APP-CTF via Atg5-dependent autophagy pathway. FASEB J. 2011;25:1934–1942. doi: 10.1096/fj.10-175158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tian Y., Chang J.C., Fan E.Y., Flajolet M., Greengard P. Adaptor complex AP2/PICALM, through interaction with LC3, targets Alzheimer's APP-CTF for terminal degradation via autophagy. Proc. Natl Acad. Sci. USA. 2013;110:17071–17076. doi: 10.1073/pnas.1315110110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salminen A., Kaarniranta K., Kauppinen A., Ojala J., Haapasalo A., Soininen H., Hiltunen M. Impaired autophagy and APP processing in Alzheimer's disease: the potential role of Beclin 1 interactome. Prog. Neurobiol. 2013;106–107:33–54. doi: 10.1016/j.pneurobio.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 30.Bustamante H.A., Rivera-Dictter A., Cavieres V.A., Munoz V.C., Gonzalez A., Lin Y., Mardones G.A., Burgos P.V. Turnover of C99 is controlled by a crosstalk between ERAD and ubiquitin-independent lysosomal degradation in human neuroglioma cells. PLoS ONE. 2013;8:e83096. doi: 10.1371/journal.pone.0083096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tezapsidis N., Johnston J.M., Smith M.A., Ashford J.W., Casadesus G., Robakis N.K., Wolozin B., Perry G., Zhu X., Greco S.J., et al. Leptin: a novel therapeutic strategy for Alzheimer's disease. J. Alzheimers Dis. 2009;16:731–740. doi: 10.3233/JAD-2009-1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fewlass D.C., Noboa K., Pi-Sunyer F.X., Johnston J.M., Yan S.D., Tezapsidis N. Obesity-related leptin regulates Alzheimer's Abeta. FASEB J. 2004;18:1870–1878. doi: 10.1096/fj.04-2572com. [DOI] [PubMed] [Google Scholar]
- 33.Yu B.L., Zhao S.P., Hu J.R. Cholesterol imbalance in adipocytes: a possible mechanism of adipocytes dysfunction in obesity. Obes. Rev. 2010;11:560–567. doi: 10.1111/j.1467-789X.2009.00699.x. [DOI] [PubMed] [Google Scholar]
- 34.Thirumangalakudi L., Prakasam A., Zhang R., Bimonte-Nelson H., Sambamurti K., Kindy M.S., Bhat N.R. High cholesterol-induced neuroinflammation and amyloid precursor protein processing correlate with loss of working memory in mice. J. Neurochem. 2008;106:475–485. doi: 10.1111/j.1471-4159.2008.05415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Di Paolo G., Kim T.W. Linking lipids to Alzheimer's disease: cholesterol and beyond. Nat. Rev. Neurosci. 2011;12:284–296. doi: 10.1038/nrn3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Escribano L., Simon A.M., Gimeno E., Cuadrado-Tejedor M., Lopez de Maturana R., Garcia-Osta A., Ricobaraza A., Perez-Mediavilla A., Del Rio J., Frechilla D. Rosiglitazone rescues memory impairment in Alzheimer's transgenic mice: mechanisms involving a reduced amyloid and tau pathology. Neuropsychopharmacology. 2010;35:1593–1604. doi: 10.1038/npp.2010.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang Y., Yang X., Wu Y., Jing W., Cai X., Tang W., Liu L., Liu Y., Grottkau B.E., Lin Y. Gamma-secretase inhibitor induces adipogenesis of adipose-derived stem cells by regulation of Notch and PPAR-gamma. Cell Prolif. 2010;43:147–156. doi: 10.1111/j.1365-2184.2009.00661.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haass C., Capell A., Citron M., Teplow D.B., Selkoe D.J. The vacuolar H(+)-ATPase inhibitor bafilomycin A1 differentially affects proteolytic processing of mutant and wild-type beta-amyloid precursor protein. J. Biol. Chem. 1995;270:6186–6192. doi: 10.1074/jbc.270.11.6186. [DOI] [PubMed] [Google Scholar]
- 39.Liao Y.F., Wang B.J., Cheng H.T., Kuo L.H., Wolfe M.S. Tumor necrosis factor-alpha, interleukin-1beta, and interferon-gamma stimulate gamma-secretase-mediated cleavage of amyloid precursor protein through a JNK-dependent MAPK pathway. J. Biol. Chem. 2004;279:49523–49532. doi: 10.1074/jbc.M402034200. [DOI] [PubMed] [Google Scholar]
- 40.Schagger H. Tricine-SDS-PAGE. Nat. Protoc. 2006;1:16–22. doi: 10.1038/nprot.2006.4. [DOI] [PubMed] [Google Scholar]
- 41.Wiltfang J., Smirnov A., Schnierstein B., Kelemen G., Matthies U., Klafki H.W., Staufenbiel M., Huther G., Ruther E., Kornhuber J. Improved electrophoretic separation and immunoblotting of beta-amyloid (A beta) peptides 1–40, 1–42, and 1–43. Electrophoresis. 1997;18:527–532. doi: 10.1002/elps.1150180332. [DOI] [PubMed] [Google Scholar]
- 42.Cacquevel M., Aeschbach L., Osenkowski P., Li D., Ye W., Wolfe M.S., Li H., Selkoe D.J., Fraering P.C. Rapid purification of active gamma-secretase, an intramembrane protease implicated in Alzheimer's disease. J. Neurochem. 2008;104:210–220. doi: 10.1111/j.1471-4159.2007.05041.x. [DOI] [PubMed] [Google Scholar]
- 43.Wu F., Schweizer C., Rudinskiy N., Taylor D.M., Kazantsev A., Luthi-Carter R., Fraering P.C. Novel gamma-secretase inhibitors uncover a common nucleotide-binding site in JAK3, SIRT2, and PS1. FASEB J. 2010;24:2464–2474. doi: 10.1096/fj.09-148031. [DOI] [PubMed] [Google Scholar]
- 44.Alattia J.R., Kuraishi T., Dimitrov M., Chang I., Lemaitre B., Fraering P.C. Mercury is a direct and potent gamma-secretase inhibitor affecting Notch processing and development in Drosophila. FASEB J. 0000;25:2287–2295. doi: 10.1096/fj.10-174078. [DOI] [PubMed] [Google Scholar]
- 45.Radde R., Bolmont T., Kaeser S.A., Coomaraswamy J., Lindau D., Stoltze L., Calhoun M.E., Jaggi F., Wolburg H., Gengler S., et al. Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 2006;7:940–946. doi: 10.1038/sj.embor.7400784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Low K., Aebischer P., Schneider B.L. Direct and retrograde transduction of nigral neurons with AAV6, 8, and 9 and intraneuronal persistence of viral particles. Hum. Gene Ther. 2012;24:613–629. doi: 10.1089/hum.2012.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.