

Abstract
MicroRNAs (miRNAs) are a class of small ∼22 nucleotide noncoding RNAs which regulate gene expression at the posttranscriptional level by either destabilizing and consequently degrading their targeted mRNAs or by repressing their translation. Emerging evidence has demonstrated that miRNAs are essential for normal mammalian development, homeostasis, and many other functions. In addition, deleterious changes in miRNA expression were associated with human diseases. Several muscle-specific miRNAs, including miR-1, miR-133, miR-206, and miR-208, have been shown to be important for normal myo-blast differentiation, proliferation, and muscle remodeling in response to stress. They have also been implicated in various cardiac and skeletal muscular diseases. miRNA-based gene therapies hold great potential for the treatment of cardiac and skeletal muscle diseases. Herein, we describe methods commonly applied to study the biological role of miRNAs, as well as techniques utilized to manipulate miRNA expression and to investigate their target regulation.
Keywords: MicroRNA, miRNA, Muscle, Gene expression, Posttranscriptional regulation, Muscle disease
1. Introduction
Formation, development, and physiology of skeletal muscle are of the utmost importance for the normal locomotion of an organism. Abnormal development, damage, or deterioration of skeletal muscle might result in muscle atrophy, paralysis, or even death. Skeletal muscle disorders are a group of diseases caused by different mechanisms, including defects in structural proteins, disorganization of the sarcomeres, and/or perturbed regulation of growth/maturation signaling pathways (1). These diseases can be classified as: 1) Neuromuscular, such as multiple sclerosis or 2) musculoskeletal, such as Duchenne Muscular Dystrophy, myotubular myopathy, and others. Elucidation of mechanisms that regulate muscle determination, differentiation, and proliferation is an important prerequisite to developing therapeutic strategies to correct or to circumvent skeletal muscle defects that accompany neuromuscular disease.
Recently, a large class of small ∼22 nucleotide (nt) noncoding RNAs have been discovered and are collectively referred to as microRNAs (miRNAs). To date, thousands of miRNA genes have been identified in multiple organisms from plants and nematodes, to fish and mammals (2). Similar to protein-encoding genes, the expression of a miRNA begins with the transcription of the miRNA gene by RNA polymerase II. Though many miRNAs are under the control of their own promoter, some miRNA genes are found in clusters sharing a single promoter (3); others are encoded within an intron and are coexpressed along with the host gene (4). After transcription within the nucleus, the large primary miRNA transcript is processed by the microprocessor complex (Drosha/DGCR8) into a hairpin intermediate commonly referred to as a pre-miRNA. However, a small subgroup of miRNAs found within short introns is known to bypass this step (5). Pre-miRNAs are then exported from the nucleus to the cytoplasm by the nuclear transporter exportin-5 (6); in the cytoplasm, they are further processed into miRNA duplexes by the cytosolic RNase III enzyme Dicer (7). Finally, the functional strand of the miRNA duplex is loaded into the RNA-induced silencing complex (RISC) to facilitate targeted mRNA degradation and/or translational repression (8).
Evolutionarily conserved miRNAs have been identified in multiple eukaryotes from the worm Caenorhabditis elegans, to the fruit fly Drosophila melanogaster, to the mouse Mus musculus, and to the human Homo sapiens. The C. elegans genome contains a single mammalian miR-1 ortholog (9), whereas in higher eukaryotes there are multiple genes encoding miR-1 (identical coding sequences of miR-1-1 and miR-1-2). miR-206, expressed specifically in skeletal muscles, is related to miR-1 and differs from miR-1 by only four nucleotides (10). Several mammalian miRNAs, including the miR-1/206 and miR-133 families, and miR-208a/b, are specifically expressed in cardiac and skeletal muscle (11–13). miR-1 is known to regulate skeletal muscle differentiation and proliferation in C2C12 myoblasts (12) as well as the neuromuscular junction in C. elegans (14). In addition, miR-1 expression is dependent upon the activity of the transcription factors Serum Response Factor (SRF) and MyoD, as evidenced in both D. melanogaster (15) and M. musculus (11). Together, these results strongly suggest that both the regulation and the function of miR-1 are conserved throughout eukaryotic evolution, and miR-1 plays an important role in several processes, such as cell proliferation, differentiation, migration, and apoptosis during both normal development and disease progression (16, 17).
Intriguingly, even though both miR-1 and miR-133 genes are clustered together and cotranscribed as a single primary transcript, they represent two distinct miRNAs, each with its own biological function (12). miR-1 overexpression has been shown to promote skeletal muscle myoblast differentiation in cultured C2C12 myoblasts (12). miR-1 also significantly impairs normal cardiac development (11, 12), inducing arrhythmias through negative regulation of Kcnj2 and Gja1 (18). Conversely, miR-133 induces cell proliferation and represses myogenic gene expression (12). miR-208 is specifically expressed in the myocardium and is required for stress-dependent cardiac growth and remodeling (13, 19). miR-206 is uniquely expressed in skeletal muscle cells (20); although its functions are not fully understood, they include a potential role in muscular hypertrophy, maintaining the ratio between αMHC and βMHC through regulating the activity of the retinoic acid receptor alpha (RXRα), a potential role in satellite cell specification through the regulation of Pax3, and a potential role in the switching of the fiber types by downregulating Utrophin, which could compensate for the loss of dystrophin in Duchenne muscular dystrophy syndrome (20). The expression of many miRNAs is altered under pathological conditions. Subsets of miRNAs are found to be both positively and negatively regulated in clinical human samples and animal models of cardiac and skeletal myopathies (21-24). In vivo overexpression of miR-195 in cardiomyocytes is sufficient to cause dilated cardiomyopathy and heart failure in the mouse (22). In addition, dystrophin-deficient mice were found to have significantly decreased expression of miR-133a and miR-206 (25). Together, these results indicate that proper expression of miRNAs is necessary for both normal development and function of skeletal and cardiac muscles.
Strategies commonly used to investigate the biological function of a particular miRNA include both gain-of-function and loss-of-function approaches. Gain-of-function studies are usually performed in vitro, where cells can be transiently transfected with an expression construct encoding the pre-miRNA. Alternatively, synthetic miRNA duplexes and virus-based miRNA expression systems may also be employed. In vitro loss-of-function studies can be accomplished with either 2′-O-methyl miRNA antisense oligonucleotides or locked nucleic acid (LNA)-miRNA antisense oligonucleotides, which will block the function of an endogenous miRNA. The in vivo determination of a miRNA's function is best examined utilizing conventional transgenic and gene knockout strategies. Recently, a lentivirus targeting strategy that overexpresses short RNA fragments containing multiple miRNA target sequences has been shown to phenocopy a genetic miRNA knockout mouse (26). In addition, intravenous delivery of cholesterol-modified miRNA antisense oligonucleotides (antagomiRs) can inhibit miRNA function in vivo (27). These molecular approaches are invaluable in elucidating the biological function of miRNAs and may potentially lend themselves to future gene-based therapies.
A key to the understanding of the molecular mechanism of miRNA function is to identify miRNA targets. miRNAs are known to repress their targets primarily by targeting the 3′ UTRs of their target transcripts. However, identification of such targets has proved to be challenging in animals, primarily due to imperfect sequence match between miRNAs and their regulatory targets. Information technologies and bioinformatics databases are very useful tools to identify putative miRNA targets. In particular, computational algorithms for miRNA targets are publicly available through the world wide web (i.e., Pictar (28), miRanda (29), TargetScan (30), etc.). These algorithms allow investigators to search for possible miRNA target sites in the 3′ UTR of a candidate mRNA, or to predict possible regulatory targets of a specific miRNA. To date, it has been recognized that a single miRNA could target multiple mRNA transcripts (31, 32). On the other hand, the 3′ UTR of a gene might have multiple target sites for different miRNAs. Thus, miRNAs offer themselves as one additional layer in the posttranscriptional regulation of gene expression. Microarray technology is most commonly utilized for the basic purpose of comparing mRNA expression levels between two or more samples (i.e., dystrophic muscle vs. normal muscle) (21). The results are obtained in terms of expression folds either for upregulated or down-regulated genes. These up- or down-regulated genes are of most interest since they are the ones showing a dynamic expression. The results from microarray analyses are available through databases (i.e., NCBI database) from which one can extract the specifics for a gene. A further step in this technology is that of miRNA microarrays; these provide results for the up- or down-regulation of miRNAs in the compared samples. It is conceivable that a combination of conventional mRNA microarray and a miRNA microarray on the same sets of samples could be a powerful approach and will allow us to determine the correlation of miRNAs and their regulatory targets.
In this chapter, we will first describe how to document the expression of miRNAs by northern blot and qPCR analyses. We will then describe the method to define the regulatory targets of a miRNA using luciferase reporter assays. Finally, we will detail how to manipulate the expression level of muscle miRNAs in the C2C12 myoblast cell line and how to determine their biological function in muscle proliferation and differentiation.
2. Materials
2.1. Detecting the Expression of Muscle miRNAs by Northern Blotting and Quantitative RT-PCR Analyses
2.1.1. Northern Blot
Hoefer SE 400 vertical slab gel electrophoresis unit.
Hoefer TE77 Semidry transfer unit.
UV stratalinker 1800 (Stratagene).
Trizol Reagent (Invitrogen).
40% Acrylamide: AccuGel 29:1 (National Diagnostics).
10× TBE buffer: 0.9 M Tris base, 0.9 M boric acid, 0.02 M EDTA (pH 8.0), autoclave for 20 min.
Urea (molecular biology grade).
10% (w/v) Ammonium persulfate solution (APS). Aliquot and store at −20°C.
N,N,N,N′-Tetramethyl-ethylenediamine (TEMED).
Formamide.
Bromophenol blue solution: 10% (w/v) bromophenol blue.
Filter paper, sheet, grade 3, 460 × 570 MM (Whatman).
Zeta-Probe GT genomic tested blotting membranes (Bio-Rad).
T4 polynucleotide kinase (PNK).
Mini Quick Spin Oligo Columns (Roche).
Adenosine 5′-triphosphate [γ-32P], 3,000 Ci/mmol.
Anti-miRNA probe: the synthetic antisense oligonucleotide of the target miRNA.
Diethylpyrocarbonate (DEPC)-treated water: 1 mL DEPC in 1 L double-distilled H2O. Stir at room temperature for 1 h and autoclave.
1 M phosphate buffer: 71 g of anhydrous Na2HPO4, 4 mL of 85% H3PO4. Add DEPC-treated water to 1 L.
Hybridization buffer: 0.5 M phosphate buffer, 1 mM EDTA at pH 8.0, 7% (w/v) of sodium dodecyl sulfate (SDS), 1% (w/v) of bovine serum albumin (BSA), in DEPC-treated water.
20 × SSC: 3 M sodium chloride and 300 mM tri-sodium citrate dihydrate, pH 7.0.
Wash buffer: 1× SSC supplemented with 0.1% SDS.
Stripping buffer: 0.1× SSC supplemented with 0.1% SDS.
Storage phosphor screen (Amersham).
2.1.2. Quantitative RT-PCR
TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems).
TaqMan Universal PCR Master Mix, No AmpErase UNG (Applied Biosystems).
TaqMan MicroRNA Assays Kit (Applied Biosystems).
2.2. Studying the Regulation of Muscle miRNAs on Their Targets by Luciferase Reporter Assays
Mouse genomic DNA.
Primers for reporter construction: HDAC4-UTR-F, 5′-ATCGGAGCTCCAGCACTGGTGATAGACTTGG-3′; HDAC4-UTR-R, 5′-GTCTTATTGAACTTATTCTTAAGC TCGAGATCG-3′; HDAC4-Mut-F, 5′-GTTTCTTTCCT CAGATTTAAAATTCTTCACTGGTCACAGCCACG-3′; HDAC4-Mut-R, 5 ′-GTGACCAGTGAAGAATTTTAAATCT GAGGAAAGAAACACAACC-3′.
PfuTurbo DNA polymerase.
pGL3cM vector (modified by Chen JF and Wang DZ, the backbone is the pGL3-Control vector, Promega).
SacI restriction endonuclease.
XhoI restriction endonuclease.
T4 DNA ligase.
pRL-TK Vector for Renilla luciferase reporter (Promega).
NucleoBond plasmid Maxi kit (Macherey-Nagel).
HEK293T cells (ATCC).
CELLSTAR 12- and 24-well tissue culture plate.
Growth medium for HEK293T cells: Combine 1 L of 1× Dulbecco's modified Eagle medium (DMEM, high glucose with L-glutamine), 110 mL of fetal bovine serum (FBS), and 11 mL of 100× penicillin G–streptomycin (10,000 units penicillin; 10,000 μg streptomycin).
1× Trypsin–EDTA: 0.25% Trypsin, 1 mM EDTA/4Na.
Lipofectamine LTX and Plus Reagent (Invitrogen).
Opti-MEM I Reduced Serum Medium (Gibco).
miR-1 miRIDIAN miRNA mimic (Dharmacon).
10× Phosphate-buffered saline (PBS) solution: 80.6 mM sodium phosphate, 19.4 mM potassium phosphate, 27 mM KCl and 1.37 M NaCl at pH 7.4.
Dual-luciferase reporter assay system (Promega).
2.3. Overexpression and Knockdown of Muscle miRNAs in Cell Lines
Primers for miR-22 overexpression vector construction: miR22-F 5′-TAGCAGGTACCTTATTCAAGAACCCCTCA TTAG-3′, miR22-R 5′-GTATCTCTAGATTTCCCTCCCA TAAAGCCAT- 3′.
pcDNA3.1(+) vector (Invitrogen).
anti-miR-22 probe: antisense oligonucleotide to miR-22.
C2C12 cells (ATCC).
KpnI restriction endonuclease.
XbaI restriction endonuclease.
2′-O-methyl miR-133 antisense oligonucleotide (Dharmacon).
Growth medium for C2C12 cells: DMEM medium with 10% FBS and 1% penicillin G–streptomycin.
Differentiation medium for C2C12 cells: DMEM medium with 2% horse serum and 1% penicillin G–streptomycin.
Complete Protease Inhibitor Cocktail Tablets (Roche).
Cell lysis buffer: 40 mM Tris–HCl (pH7.5), 150 mM NaCl, 1% (v/v) Triton ×-100, Complete Protease Inhibitor Cocktail Tablet (1 tablet/50 mL).
Mini-PROTEAN 3 Electrophoresis Cell (Bio-Rad).
1× SDS-PAGE running buffer: 25 mM Tris, 200 mM glycine; 0.1% (w/v) SDS.
5× protein loading buffer: 10% (w/v) SDS, 10 mM beta-mer-capto-ethanol, 20% (v/v) glycerol, 0.2 M Tris-HCl (pH6.8), 0.05% (w/v) bromophenol blue.
PVDF membrane.
Transfer buffer: 25 mM Tris, 200 mM glycine, 20% (v/v) methanol.
Odyssey blocking buffer (LI-COR Biosciences).
Anti-SRF antibody from rabbit (Santa Cruz Biotechnology).
IRDye goat-anti-rabbit secondary antibody (LI-COR Biosciences).
3. Methods
3.1. Detecting the Expression of Muscle miRnAs by Northern Blot and Quantitative RT-PCR Analyses
Prepare total RNA from tissue or cultured cells with Trizol Reagent according to manufacturer's protocol (see Note 1).
Prepare 15% denaturing gel for electrophoresis separation of miRNAs. Carefully wash, dry, and assemble the Hoefer SE 400 vertical slab gel electrophoresis unit. Prepare denaturing gel containing 18.75 mL of 40% acrylamide, 2.5 mL of 10× TBE buffer, 12.5 mL of DEPC-treated water, and 20 g of urea. Mixture may need to be gently heated in 37°C water bath in order for urea to completely dissolve. To polymerize, add 400 μL of 10% APS; 40 μL of TEMED, mix well, and quickly pour. Allow the gel to polymerize for 1 h.
Prerun denaturing gel for 30 min at 200 V. Use 0.5× TBE for running buffer.
Prepare RNA samples for electrophoresis. Mix the RNA sample (40 μg) 1:1 (v/v) with formamide, and incubate at 65°C for 10 min. Chill RNA on ice for 3 min and add 2 μL of bromophenol blue solution. Mix well.
Load the sample(s) into the well(s) and run the gel at 250 V (see Note 2). Use 0.5× TBE for running buffer. Voltage can be stopped when the loading dye reaches the bottom of the plate.
Transfer the RNA from the gel to a membrane with Hoefer TE77 Semidry transfer unit. Soak the membrane and six pieces of filter paper in 0.5× TBE. Set up the transfer in the order from top (−) to bottom (+) as: three pieces of filter paper, gel, membrane, three pieces of filter paper (see Note 3). Transfer with constant current (0.8 mA/cm2 of gel area) for 1 h.
After transfer, wash the membrane with 0.5× TBE and perform UV crosslink using the auto crosslink option.
Prepare isotope-labeled probe for hybridization. Mix 5 μL of adenosine 5′-triphosphate [γ-32P], 5 μL of 1 μM anti-miRNA probe, 2 μL of 10× PNK buffer, 1 μL of T4 polynucleotide kinase, and 7 μL of double-distilled water and incubate at 37°C for 1 h.
Purify the [γ-32P]-labeled probe using a mini Quick Spin Oligo Column according to manufacturer's protocol (see Note 4).
Prehybridize the membrane for 1 h at 37°C with 5–10 mL of hybridization buffer.
Add the labeled anti-miRNA probe into the hybridization buffer and incubate overnight at 37°C.
Remove the hybridization buffer and wash the membrane 3 times with wash buffer (10 min per wash).
Expose the membrane to the storage phosphor screen for 4–24 h. The length of exposure depends upon strength of signal and will vary with different miRNA probes (see Note 5).
Scan the screen with Typhoon phosphor-imager (see Figs. 1a, 3b).
If you want to probe the membrane with a different miRNA probe or if you want to store the membrane for long term, add the membrane to heated stripping buffer (>95°C). Incubate for approximately 10 min while rocking.
After stripping, rinse membrane with fresh stripping buffer, and allow drying. The membrane may be reprobed immediately following the steps outlined above. The membrane can also be stored at −20°C for future use.
To measure the expression of muscle miRNAs by quantitative RT-PCR, dilute total RNA sample to 2 ng/μL and perform reverse transcription. Mix the following reagents, including 0.15 μL 100 mM dNTPs, 1 μL MultiScribe reverse transcriptase, 1.5 μL 10× reverse transcription buffer, 0.2 μL RNase inhibitor, 4.15 μL nuclease-free water, 5 μL diluted RNA sample, and 3 μL RT primer (see Note 6), into a 0.2 mL polypropylene PCR tube on ice. Briefly centrifuge the mixture and incubate the tube on ice for 5 min. Perform reverse transcription in thermal cycler with the following program: 16°C for 30 min→42°C for 30 min→85°C for 5 min→hold at 4°C. The cDNA can be used immediately or store in -20°C for further use.
Perform quantitative PCR by mixing the following reagents: 1 μL 20× TaqMan MicroRNA Assay (a mixture contains both PCR primer pair and TaqMan probe for the specific miRNA), 1.5 μL product from RT reaction, 10 μL TaqMan 2× universal PCR master mix (no AmpErase UNG), and 7.5 μL nuclease-free water, into a 0.2 mL quantitative PCR tube on ice. Briefly centrifuge the mixture and perform quantitative PCR in realtime PCR system with the default program for TaqMan quantitative PCR (see Fig. 1b).
Fig. 1.
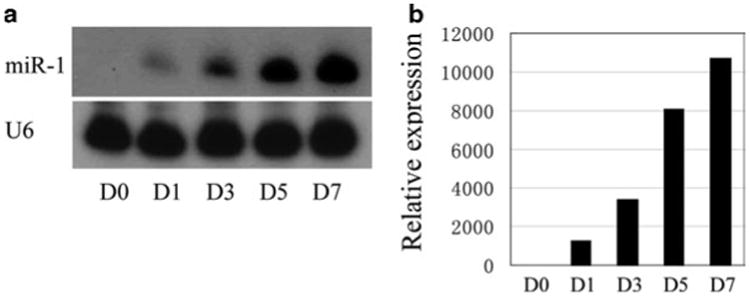
Determination of miR-1 expression during C2C12 myoblast cell differentiation by (a) northern blotting analyses and (b) qPCR assays. Total RNAs isolated from C2C12 myo-blasts which are switched into differentiation conditions at indicated time points (day-0 (D0) to day-7 (D7)) were used for northern blot and qPCR according to the protocols described in this chapter. U6 snRNA serves as a loading control.
Fig. 3.
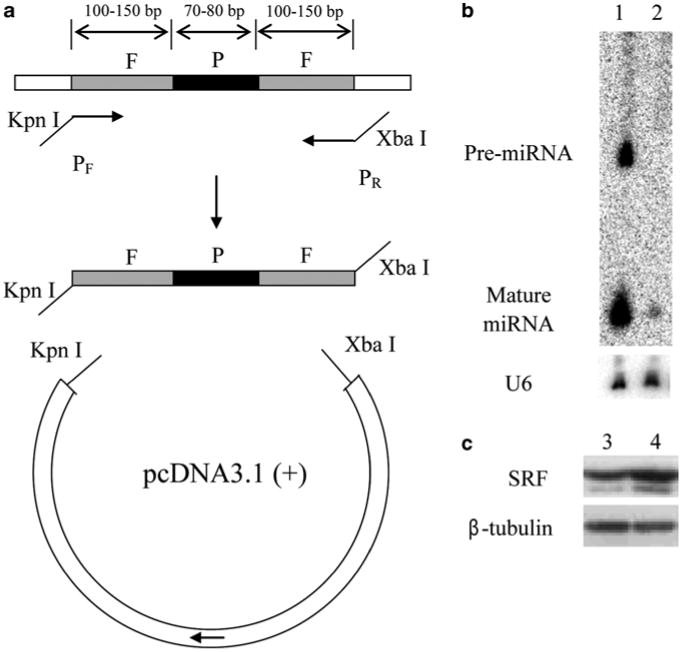
Overexpression and knockdown of miRNAs in C2C12 myoblast cells. (a) The strategy for the construction of miR-22 overexpression vector. P miRNA hairpin precursor; F flanking sequences. (b) Verification of miR-22 overexpression in HEK293T cells by northern blot analyses. Lane 1, cells transfected with the miR-22 overexpression vector; Lane 2, cells transfected with a control vector. U6 snRNA serves as a loading control. (c) Western blot showing SRF protein expression repressed by miR-133. Lane 3, C2C12 cells transfected with control 2′-O-methyl oligonucleotide; Lane 4, C2C12 cells transfected with 2′-O-methyl miR-133 antisense oligonucleotide. β-tubulin serves as a loading control.
3.2. Studying the Regulation of Muscle miRNAs on Their Targets by Luciferase Reporter Assays
Here we show an example using the luciferase reporter vectors which contains either the wild-type or the mutant HDAC4 3′ UTR.
Generate the ∼400 bp HDAC4 gene 3′ UTR DNA fragment containing the seed sequence for miR-1 by PCR reaction using mouse genomic DNA as the template and the HDAC4-UTR-F and HDAC4-UTR-R primers. The SacI and XhoI sites are introduced at the 5′ and 3′-ends, respectively, by the PCR primers. The UTR PCR products are cloned into the SacI/XhoI sites of the pGL3cM vector (see Note 7). The resulting Luc-WT-UTR reporter contains the wild-type 3′ UTR of the HDAC4 gene (see Fig. 2a, b).
Generate the Luc-Mut-UTR reporter by using the plasmidgenerated in step 1 as a template and introducing mutationswith HDAC4-Mut-F and HDAC4-Mut-R primers (see Fig. 2b).
Prepare high-quality Luc-WT-UTR reporter, Luc-Mut-UTR reporter, and pRL-TK plasmid for reporter assays with NucleoBond Plasmid Maxi Kit. These plasmids will be used for HEK293T cell transfection.
At 1 day before transfection, plate HEK293T cells in a 24-well plate at 5 × 104 cells per well in 500 μL of growth medium (see Note 8). This will yield 50–80% confluence at the day of transfection.
To generate the transfection complex for one well, add 25 ng reporter (either Luc-WT-UTR or Luc-Mut-UTR), 25 ng pRL-TK plasmid, and 0.5 μL of 10 μM miRIDIAN miRNA mimic to 100 μL of Opti-MEM I Reduced Serum Medium and mix gently. To this mixture, add 0.5 μL of PLUS Reagent, mix gently, and incubate for 5–10 min at room temperature. Finally, add 1.25 μL of Lipofectamine LTX Reagent, mix gently, and incubate for 30 min at room temperature.
Add the transfection complex (∼100 μL) to the well. Mix gently by rocking the plate back and forth.
At 24 h after transfection, remove cell culture medium and wash cells twice with 1× PBS.
Lyse cells with 100 μL of 1× passive lysis buffer by gentle shaking at room temperature for 15 min.
Mix 20 μL of cell lysate with 50 μL of firefly luciferase substrate and measure the firefly luciferase activity with a scintillation counter.
Add 50 μL Stop&Glo reagent into the mixture from step 9 and measure the Renilla luciferase activity with a scintillation counter.
Normalize firefly luciferase activity with Renilla luciferase activity and plot results (see Fig. 2c).
Fig. 2.
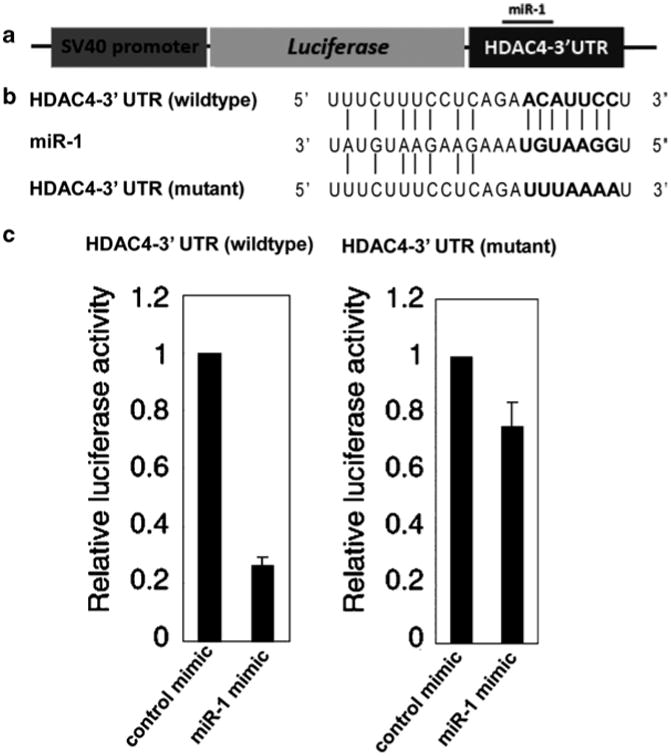
Luciferase reporter assays of miR-1 and HDAC4-3′ UTR. (a) Graphic representation of the modified luciferase vector containing the 3′ UTR of the HDAC4 gene. (b) Sequence comparison among HDAC4 wild-type 3′ UTR, HDAC4 mutant 3′ UTR sequence, and miR-1. (c) miR-1 significantly represses the luciferase reporter activity of HDAC4 wild-type 3′ UTR, but not that of mutant 3′ UTR.
3.3. Overexpression and Knockdown of Muscle miRNAs in Cell Lines
In this section, we describe steps to overexpress miR-22 in HEK293T cells and to knockdown miR-133 in C2C12 myoblasts.
For the overexpression study, use PCR to generate a ∼35 bp DNA fragment containing the intact hairpin for the miR-22 precursor plus the flanking sequences on both ends (see Note 9). Use mouse genomic DNA as the template. The KpnI and XbaI sites are introduced at the 5′ and 3′-ends, respectively, by the PCR primers. Clone the PCR product into the KpnI/XbaI sites in the pcDNA3.1(+) vector (see Note 10). The resulting construct is termed the miR-22 overexpression vector (see Fig. 3a).
Prepare high-quality plasmid for transfection with NucleoBond Plasmid Maxi Kit.
Transfect the miR-22 overexpression vector into HEK293T cells following the steps outlined in Subheading 3.2. Use 6-well plates for the transfection and adjust the amount of transfection reagents accordingly.
Extract total RNA from cells 48 h after transfection using the Trizol reagent according to manufacturer's protocol.
Evaluate the overexpression of miR-22 by northern blot according to the protocol described in Subheading 3.1 (see Fig. 3b). Similarly, the miR-22 overexpression vector can also be evaluated in other cells such as C2C12 myoblasts.
For miR-133 knockdown study, plate C2C12 myoblasts in a 6-well plate at 2 × 105 cells per well in 2 mL of growth medium 1 day before transfection. This will yield 50–80% confluence at the day of transfection.
Transfect C2C12 myoblasts with 200 nM 2′-O-methyl miR-133 antisense oligonucleotides (see Note 11). Adjust the amount of transfection reagents accordingly.
Change growth medium 4–6 h after transfection and continue to culture the cells for an additional 24 h.
24 h after transfection, replace growth medium with differentiation medium and culture the cells for an additional 12 h.
Confirm miR-133 knockdown by northern blotting analysis according to the protocol described in Subheading 3.1.
Prepare cell lysate with cell lysis buffer (100 μL per well) and examine the up-regulation of SRF, a target regulated by miR-133 (12), by western blot analysis.
Prepare SDS–PAGE gel for electrophoresis separation of proteins. Carefully wash, dry, and assemble the Bio-Rad Mini-PROTEAN 3 Electrophoresis Cell. Prepare 9% running gel containing 2.4 mL of 30% acrylamide, 2 mL of 1.5 M Tris-HCl (pH8.8), 3.5 mL of double-distilled water, and 80 μL of 10% SDS. To polymerize, add 40 μL of 10% APS, 5.5 μL of TEMED, mix well, and quickly pour. Allow the gel polymerize for 30 min. Then, prepare stacking gel containing 0.53 mL of 30% acrylamide, 0.5 mL of 1 M Tris–HCl (pH 6.8), 2.97 mL of double-distilled water, and 40 μL of 10% SDS. To polymerize, add 20 μL of 10% APS, 4 μL of TEMED, mix well, and quickly pour on the top of the polymerized running gel. Allow the gel polymerize for 30 min.
Prepare protein samples for SDS–PAGE electrophoresis. Mix the cell lysis sample (50 μg) with 5× protein loading buffer, and incubate at 95°C for 5 min.
Load the sample(s) into the well(s) and run the gel at 100 V. Use 1× running buffer. Voltage can be stopped when the loading dye reaches the bottom of the plate.
Transfer the protein from the gel to the PVDF membrane with 1× transfer buffer. Wet the PVDF membrane with methanol. Soak the PVDF membrane, gel, and six pieces of filter papers in 1× transfer buffer for 10 min. Set up the transfer in the order from cathode (−) to anode (+) as: three pieces of filter paper, gel, membrane, three pieces of filter paper (see Note 12). Transfer at either 100 V for 3 h or 30 V overnight at 4°C.
After transfer, incubate the membrane with Odyssey blocking buffer for 1 h at room temperature.
Dilute SRF first antibody with 1:500 dilution into Odyssey blocking buffer and incubate the membrane with diluted first antibody overnight.
Wash the membrane with 1× PBS 3 times at room temperature (15 min per wash).
Dilute IRDye goat-anti-rabbit secondary antibody with 1:7,500 dilution into Odyssey blocking buffer and incubate the membrane with diluted second antibody for 1 h at room temperature.
Wash the membrane with 1× PBS 3 times at room temperature (15 min per wash).
Scan the membrane with the Odyssey infrared imaging system (see Fig. 3c).
Acknowledgments
We thank members of the Wang laboratory for discussion and support. Research in the Wang lab was supported by the March of Dimes Birth Defect Foundation, National Institutes of Health and Muscular Dystrophy Association. ZP Huang is a postdoctoral fellow and DZ Wang is an Established Investigator of the American Heart Association.
Footnotes
RNase(s) rapidly degrade RNA and are abundant in the environment. When extracting total RNA from samples, RNase-free tubes, DEPC-treated water, and solutions made with DEPC-treated water are highly recommended. RNA samples can be preserved in pellet for more than 1 year if stored in 100% ethanol at −80°C.
Prior to loading the RNA sample into the denaturing gel, wash the well by flushing with 0.5 × TBE running buffer. Excess urea in the well will prevent the RNA sample from sinking to the bottom of the well.
Exclude air bubbles when assembling the “sandwich” for RNA transfer.
It is important to have enough protection when conducting the isotope-related experiments. Always wear personal protective equipment when handling radioisotopes.
Besides the phosphor-imager system, northern blot can also be imaged with X-ray autography. In general, the membrane needs to be exposed to film for 1 day to 1 week.
Up to five different miRNA RT primers can be added in one reverse transcription reaction. In this case, the total volume of all the RT primers should be 3 μL for one reaction. It is noticed that some combinations of miRNA RT primers may not work well. Test the combination before performing experiments.
To generate the pGL3cM vector, the multiple cloning site (MCS) is removed from pGL3-control vector by KpnI/BglII digestion and filled in by Klenow. The 53 bp oligonucleotide containing the MCS is then introduced into the XbaI site.
At least 12 wells are needed for one experiment to examine four combinations of transfection reagents including Luc-WT-UTR reporter and miR-1 miRIDIAN mimic, Luc-WT-UTR reporter and control miRIDIAN mimic, Luc-Mut-UTR reporter and miR-1 miRIDIAN mimic, and Luc-Mut-UTR reporter and control mimic. Each combination of transfection reagents is performed in triplicate.
Different cloning strategies can be applied to generate a miRNA overexpression vector. In this protocol, our strategy is to clone the fragment containing the whole hairpin (miRNA precursor) and a 100–150 bp flanking sequence on both the 5′ and 3′ ends of the miRNA sequence. Alternatively, the full-length noncoding transcript can be cloned into the expression vector. However, this is only applicable for miRNAs generated from a nonprotein-coding gene.
Besides pcDNA3.1(+), other expression vectors can be used to construct a miRNA overexpression plasmid. Virus-based expression vectors have already been reported for miRNA overexpression (33).
In this protocol, a 2′-O-methyl miRNA antisense oligonucleotide is used to knockdown the endogenous miRNA. Alternatively, LNA antisense oligonucleotides can be used to obtain similar effects.
Exclude air bubbles when assembling the “sandwich” for protein transfer.
References
- 1.Wagner KR. Genetic diseases of muscle. Neurol Clin. 2002;20:645–678. doi: 10.1016/s0733-8619(02)00002-6. [DOI] [PubMed] [Google Scholar]
- 2.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 3.He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe SW, Hannon GJ, Hammond SM. A microRNA polycistron as a potential human oncogene. Nature. 2005;435:828–833. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rodriguez A, Griffiths-Jones S, Ashurst JL, Bradley A. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004;14:1902–1910. doi: 10.1101/gr.2722704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ruby JG, Jan CH, Bartel DP. Intronic microRNA precursors that bypass Drosha processing. Nature. 2007;448:83–86. doi: 10.1038/nature05983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yi R, Qin Y, Macara IG, Cullen BR. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003;17:3011–3016. doi: 10.1101/gad.1158803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hutvágner G, McLachlan J, Pasquinelli AE, Bálint E, Tuschl T, Zamore PD. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science. 2001;293:834–838. doi: 10.1126/science.1062961. [DOI] [PubMed] [Google Scholar]
- 8.Schwarz DS, Hutvágner G, Du T, Xu Z, Aronin N, Zamore PD. Asymmetry in the assembly of the RNAi enzyme complex. Cell. 2003;115:199–208. doi: 10.1016/s0092-8674(03)00759-1. [DOI] [PubMed] [Google Scholar]
- 9.Lee RC, Ambros V. An extensive class of small RNAs in Caenorhabditis elegans. Science. 2001;294:862–864. doi: 10.1126/science.1065329. [DOI] [PubMed] [Google Scholar]
- 10.Williams AH, Liu N, van Rooij E, Olson EN. MicroRNA control of muscle development and disease. Curr Opin Cell Biol. 2009;21:461–469. doi: 10.1016/j.ceb.2009.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao Y, Samal E, Srivastava D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardio-genesis. Nature. 2005;436:214–220. doi: 10.1038/nature03817. [DOI] [PubMed] [Google Scholar]
- 12.Chen JF, Mandel EM, Thomson JM, Wu Q, Callis TE, Hammond SM, Conlon FL, Wang DZ. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat Genet. 2006;38:228–233. doi: 10.1038/ng1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science. 2007;316:575–579. doi: 10.1126/science.1139089. [DOI] [PubMed] [Google Scholar]
- 14.Simon DJ, Madison JM, Conery AL, Thompson-Peer KL, Soskis M, Ruvkun GB, Kaplan JM, Kim JK. The microRNA miR-1 regulates a MEF-2-dependent retrograde signal at neuromuscular junctions. Cell. 2008;133:903–915. doi: 10.1016/j.cell.2008.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kwon C, Han Z, Olson EN, Srivastava D. MicroRNA1 influences cardiac differentiation in Drosophila and regulates Notch signaling. Proc Natl Acad Sci USA. 2005;102:18986–18991. doi: 10.1073/pnas.0509535102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Callis TE, Wang DZ. Taking microRNAs to heart. Trends Mol Med. 2008;14:254–260. doi: 10.1016/j.molmed.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 17.Chen JF, Callis TE, Wang DZ. microRNAs and muscle disorders. J Cell Sci. 2009;122:13–20. doi: 10.1242/jcs.041723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang B, Lin H, Xiao J, Lu Y, Luo X, Li B, Zhang Y, Xu C, Bai Y, Wang H, Chen G, Wang Z. The muscle-specific microRNA miR-1 regulates cardiac arrhyth-mogenic potential by targeting GJA1 and KCNJ2. Nat Med. 2007;13:486–91. doi: 10.1038/nm1569. [DOI] [PubMed] [Google Scholar]
- 19.Callis TE, Pandya K, Seok HY, Tang RH, Tatsuguchi M, Huang ZP, Chen JF, Deng Z, Gunn B, Shumate J, Willis MS, Selzman CH, Wang DZ. MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. J Clin Invest. 2009;119:2772–86. doi: 10.1172/JCI36154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McCarthy JJ. MicroRNA-206: the skeletal muscle-specific myomiR. Biochim Biophys Acta. 2008;1779:682–691. doi: 10.1016/j.bbagrm.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eisenberg I, Eran A, Nishino I, Moggio M, Lamperti C, Amato AA, Lidov HG, Kang PB, North KN, Mitrani-Rosenbaum S, Flanigan KM, Neely LA, Whitney D, Beggs AH, Kohane IS, Kunkel LM. Distinctive patterns of microRNA expression in primary muscular disorders. Proc Natl Acad Sci USA. 2007;104:17016–17021. doi: 10.1073/pnas.0708115104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Rooij E, Sutherland LB, Liu N, Williams AH, McAnally J, Gerard RD, Richardson JA, Olson EN. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci USA. 2006;103:18255–18260. doi: 10.1073/pnas.0608791103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Rooij E, Sutherland LB, Thatcher JE, DiMaio JM, Naseem RH, Marshall WS, Hill JA, Olson EN. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci USA. 2008;105:13027–13032. doi: 10.1073/pnas.0805038105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tatsuguchi M, Seok HY, Callis TE, Thomson JM, Chen JF, Newman M, Rojas M, Hammond SM, Wang DZ. Expression of microRNAs is dynamically regulated during cardiomyocyte hypertrophy. J Mol Cell Cardiol. 2007;42:1137–1141. doi: 10.1016/j.yjmcc.2007.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCarthy JJ, Esser KA, Andrade FH. MicroRNA-206 is overexpressed in the diaphragm but not the hindlimb muscle of mdx mouse. Am J Physiol Cell Physiol. 2007;293:C451–457. doi: 10.1152/ajpcell.00077.2007. [DOI] [PubMed] [Google Scholar]
- 26.Gentner B, Schira G, Giustacchini A, Amendola M, Brown BD, Ponzoni M, Naldini L. Stable knockdown of microRNA in vivo by lentiviral vectors. Nat Methods. 2009;6:63–66. doi: 10.1038/nmeth.1277. [DOI] [PubMed] [Google Scholar]
- 27.Krützfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438:685–689. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- 28.Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M, Rajewsky N. Combinatorial microRNA target predictions. Nat Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 29.John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS. Human MicroRNA targets. PLoS Biol. 2004;2:e363. doi: 10.1371/journal.pbio.0020363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by ade-nosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 31.Selbach M, Schwanhäusser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008;455:58–63. doi: 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
- 32.Baek D, Villén J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of microRNAs on protein output. Nature. 2008;455:64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stegmeier F, Hu G, Rickles RJ, Hannon GJ, Elledge SJ. A lentiviral microRNA-based system for single-copy polymerase II-regulated RNA interference in mammalian cells. Proc Natl Acad Sci USA. 2005;102:13212–13217. doi: 10.1073/pnas.0506306102. [DOI] [PMC free article] [PubMed] [Google Scholar]