

Abstract
Diabetes is increasingly becoming a major cause of large-scale morbidity and mortality. Diabetes-induced oxidative stress alters numerous intracellular signaling pathways. Although testicular dysfunction is a major concern in diabetic men, the mechanistic alterations in the testes that lead to hypogonadism are not yet clear. Oxidative mitochondrial DNA damage, as indicated by 7,8-dihydro-8-oxo-2′-deoxyguanosine, and phosphorylation of p53 at ser315 residue (p-p53ser315) increased in a stage- and cell-specific manner in the testes of rats that were diabetic for 1 month (DM1). Prolongation of diabetes for 3 months (DM3) led to an increase in nuclear oxidative DNA damage in conjunction with a decrease in the expression of p-p53ser315. The nuclei of pachytene and preleptotene spermatocytes, steps 1, 11, and 12 spermatids, secondary spermatocytes and the Sertoli cells, and the meiotic figures showed an increase in the expression of p-p53ser315. An increase in the expression of a downstream target of p53 and protein 21cyclin-dependent kinase interacting protein 1/wild-type p53-activated factor 1 (p21CIP1/Waf1) in both diabetic groups did not show any time-dependent effects but occurred concurrent with an upregulation of p-p53ser315 in DM1 and a downregulation of the protein in DM3. In diabetic groups, the expression of p21CIP1/Waf1 was mainly cytoplasmic but also perinuclear in pachytene spermatocytes and round spermatids. The cytoplasmic localization of p21CIP1/Waf1 may be suggestive of an antiapoptotic role for the protein. The perinuclear localization is probably related to the cell cycle arrest meant for DNA damage repair. Diabetes upregulates p21CIP1/Waf1 signaling in testicular germ cells in association with alteration in p-p53ser315 expression, probably to counteract DNA damage-induced cell death.
Keywords: cell cycle control, diabetes, DNA damage repair, germ cell apoptosis, testicular dysfunction
Introduction
Diabetes, a group of metabolic disorders characterized by elevated blood glucose levels,1,2 induces oxidative stress in organ systems.3 The oxidative stress is responsible for the onset of several secondary effects4 of diabetes such as vascular diseases, kidney damage,5 blindness,6 and reproductive dysfunction.7 Both diabetic patients and animals commonly experience reproductive system deficiencies such as erectile and testicular dysfunctions. The diabetes-induced oxidative stress mainly orchestrates the onset of the reproductive dysfunction, although the underlying mechanisms of the latter are not yet entirely known.8–11 Diabetes induces structural changes in the testis and spermatozoa, germ cell apoptosis, impairment of sperm parameters, and hormonal changes, which results in subfertility or infertility.8,12–14 In men, diabetes imparts negligible adverse effects on conventional sperm tests, but on the other hand, the induced oxidative stress severely damages sperm DNA,15,16 which has an effect on the fertility outcome.9 Further, the increase in DNA damage, in sperm and reproductive tract epithelial cells of diabetic men and animal models correlates with high levels of advanced glycation end products and their receptors.17,18
A microarray analysis of spermatozoa collected from diabetic men showed the differential expression of 285 genes. Among them, 21 genes were mainly related to sperm structure and function, DNA replication and repair, transcription, and intracellular signaling.19 Diabetes induces apoptosis of germ cells in rats in parallel with increases in the expressions of transforming growth factor β1 and interleukin 1β and decreases in that of serine/threonine protein kinases.20 The diabetes-induced apoptosis is also associated with increases in the expressions of Bcl-2-associated X protein, apoptosis inducing factor, C/EBP homology protein, cleaved-caspase-12,21 oxidative stress, and protein 38-mitogen activated protein kinase (p38-MAPK).22 These reports indicate that diabetes-induced oxidative stress increases testicular germ cell apoptosis probably by inhibiting DNA damage repair mechanisms. The DNA damage repair machinery requires cell cycle arrest at G1/S and G2/M transitions to carry out the repair precisely, failing to which cells with damaged DNA undergo cell death.
The protein 21cyclin-dependent kinase interacting protein 1/wild-type p53-activated factor 1 (p53-p21CIP1/Waf1) signaling pathway is one of the main control mechanisms responsible for the cell cycle arrest meant for DNA damage repair. The tumor suppressor, p53, is necessary for the maintenance of normal cell cycle and to protect the genome from mutations.23 This protein, which contains 393 amino acids and 7 domains, is involved in DNA damage response and repair, cell cycle arrest, antitumorigenesis, and proapoptotic mechanisms.24,25 Upon DNA damage, ataxia telangiectasia mutated (ATM), ataxia telangiectasia mutated- related (ATR), and other upstream proteins stabilize p53, mainly by phosphorylation and also by acetylation and sumoylation. The stabilized p53 induces cell cycle arrest needed for DNA damage repair. If the repair does not take place due to the irreparable amount of the damage, then the protein mediates the onset of apoptosis.24 One of the main cell cycle arrest-related proteins that p53 transactivates is p21CIP1/Waf1, which mediates the cell cycle arrest at both G1/S and G2/M transitions.26 This protein induces the arrest through a mechanism that involves the inhibition of cyclin-dependent kinases (CDKs), but its dual roles in cell survival and cell death are largely dependent upon its intracellular localization.27
Although diabetes is known to induce DNA damage and apoptosis in the testis, its effects on cell cycle control mechanisms are not known. In view of this, the present study investigated oxidative DNA damage and p53-p21CIP1/Waf1 pathway in the testes of diabetic rats.
Materials and Methods
Animals
In the present study, male Sprague-Dawley rats (10-12 weeks old) were used. The animals were the offspring of a breeding colony maintained at The Animal Resources Center at the Kuwait University Faculty of Medicine. The animals were housed (12:12 dark–light cycle; room temperature, 22°C-24°C; and humidity, 50%-60%) in polypropylene cages (2-3 animals/cage) with sawdust bedding. The rats had free access to chow and tap water. All guidelines, stipulated by the University (Animal Ethics and Welfare Committee) as well as competent International Organizations for conducting animal experiments, were strictly followed.
Chemicals
The chemicals and reagents used in this study were of analytical grade as indicated in the following sections, wherever necessary.
Induction of Diabetes in Rats by Streptozotocin Injection
The animals were housed and acclimatized for a week in a separate room at The Animal Resources Center. Before the induction of diabetes, the animals were randomly divided into 3 groups (N = 6), and blood glucose level was measured (Accu-Check, Roche; measurement range, 10-600 mg/dL) to ensure that they were normoglycemic. Group 1 was given only citrate buffer (0.5 mL), which served as a control group. Each animal in groups 2 and 3 was given a single intraperitoneal injection of 55 mg/kg sreptozotocin mixed in 0.5 mL 0.1 mol/L citrate buffer (pH 4.5). The glucose level was measured after 2 days following the injection to confirm the persistent hyperglycemia in rats belonging to groups 2 and 3. Then onward, every week, the glucose level was measured to ascertain sustained hyperglycemia. The animals, which showed the glucose level >300 mg/dL, were considered diabetic and included in the study.12 The diabetic animals were killed at the end of first month (DM1) and third month (DM3) following the confirmation of hyperglycemia.
Tissue Collection and Paraffin Embedding
The animals were anesthetized and sacrificed by CO2 inhalation. Laparotomy was conducted by a plus-mark incision on the ventral abdominal wall and the reproductive organs were dissected out and placed in phosphate-buffered saline (PBS, pH 7.4). The testis was isolated from the epididymis, and the 2 ends of the organ were punctured with 24G needle and immersed in Bouin fluid for 18 hours. The organ was diced into small blocks and processed for paraffin embedding.28
Evaluation of Oxidative DNA Damage
The oxidative DNA damage was evaluated by immunohistochemical localization of 7,8-dihydro-8-oxo-2′-deoxyguanosine (8-oxo-dG) as described previously.29 Paraffin sections of 5-µm thick were dewaxed in xylene and taken to water through graded concentrations of ethanol. The sections were sequentially treated with 3% H2O2, 200 μL of proteinase K dissolved in PBS, RNase A, and 2 N HCl and 1 mol/L Tris–base, with PBS washes in between the treatments. The sections were covered with 10% goat serum followed by mouse monoclonal primary antibody for 8-oxo-dG (in PBS + 0.1% bovine serum albumin; 1:750 dilution; Cat#4354-mc-050; Trevigen Helgerman Ct., Gaithersburg). After 3 PBS washes, the sections were covered sequentially with the goat antimouse secondary antibody, horseradish peroxidase conjugate, and diaminobenzidine substrate. The nuclei of testicular cells were counterstained with Mayer hematoxylin, and the sections were dehydrated with graded concentrations of ethanol, cleared in xylene, and covered with coverslips. Appropriate positive and negative controls were maintained. The tissue sections were observed under light microscope and graded for the intensity of positive labeling. Qualitative assessment of the intensity of the expression of 8-oxo-dG was done by assigning grades, “−” for the lack of expression (no sample showed this result), “+” for the mild expression, and “++” for the moderate expression. Appropriate representative photomicrographs were captured and processed in Adobe Photoshop software.
Western Blotting for Phosphorylated p53 at ser315 Residue and p21CIP1/Waf1
The testis tissue was homogenized in the homogenization buffer (10 mmol/L Tris + 1.5 mmol/L EDTA + 10% v/v glycerol + 1.0 mmol/L dithiothreitol + 1 µg/mL leupeptin + 100 µg/mL + bacitracin + 2 µg/mL aprotinin + 1 µg/mL pepstain A, pH 7.4) and the protein concentration in the homogenate was quantified by the Bradford assay as described earlier.30 An appropriate amount of the homogenate containing 45 μg of proteins was adjusted to a volume of 20 μL by adding distilled H2O and mixed with an equal amount of loading buffer. The mixture was boiled for 3 minutes and centrifuged at 10 000g for 1 minute. The 4% to 12% gradient gels (sodium dodecyl sulfate [SDS] polyacrylamide gel electrophoresis; Invitrogen, Life Technologies) were set in Tris–hydroxyethyl piperazineethanesulfonic acid–SDS running buffer and the protein was loaded into each well. The proteins were electrophoresed (250 V), the gel cassette was removed, and the proteins were transferred to polyvinylidene difluoride membrane (Bio Rad Laboratories, Hercules; pH, 8.3) for 2 hours. The membrane was removed and blocked in 5% nonfat milk in 1 × Tris-buffered saline for an hour. The primary antibodies for phosphorylated-p53 at ser315 residue (p-p53ser315; rabbit polyclonal, 1:100; Abcam, Cambridge; cat# ab1647) and p21CIP1/Waf1 (1:100; mouse-monoclonal, Abcam; cat# ab80633) were filled in a plastic pouch along with the membranes and kept on a shaker overnight at 4°C. The structural protein, β-actin (mouse-monoclonal; Sigma Chemicals), was used as a loading control. The membranes were washed thrice with Tris-buffered saline and incubated for 1 to 2 hours with diluted secondary antibodies (mixed in blocking solution; 1:500) at room temperature. The membranes were removed and washed twice with Tris-buffered saline-Tween 20 (10 minutes each) and again thrice with Tris-buffered saline (10 minutes each). The membrane was covered with luminol reagent (Santa Cruz Biotechnology) and exposed to a photographic film for 2 minutes (Hyperfilm ECL, Invitrogen, Life Technologies). The film was developed and scanned for the density of the bands in a GS-800 Calibrated Densitometer (Quantity One 4.6.3, Bio-Rad Laboratories, Hercules). For phosphorylated p53 (p-p53), the line width was 2.9 and the sensitivity was 37.975; for p21CIP1/Waf1, the line width was 2.985 and sensitivity was 37.975). The band values of the proteins were divided by corresponding β-actin values.
Immunohistochemistry for p-p53ser315 and p21CIP1/Waf1
The immunohistochemistry for p-p53 and p21CIP1/Waf1 was conducted according to the procedure described earlier for 8-oxo-dG immunohistochemistry; however, the steps involving treatment with proteinase K, RNase A, HCl, and Tris–base were avoided. The primary antibodies for the proteins (same as in Western blotting) were diluted 1:50 in diluent (Dako antibody diluent, Dako Agilent Technologies; cat# S3022), and broad-spectrum secondary antibodies (Histostain plus broad spectrum, Invitrogen, Life Technologies; cat# 858943) were used. The sections were viewed under a light microscope and stage-dependent protein expression was evaluated in seminiferous tubules. The epithelial stages were classified into 4 groups, as some stages showed very similar expressions of the proteins. The 4 groups of the seminiferous epithelial stages were I to VI, VII to IX, X to XIII, and XIV. The intensity of the protein expressions was graded as “−” for the lack of protein expression, “+” for the mild expression, “++” for the moderate expression, “+++” for the intense expression, “++++” for the more-intense expression, and “+++++” for the highly intense expression. Representative photomicrographs were taken and processed in Adobe Photoshop software.
Statistical Analysis
The data were analyzed for statistical significance using SPSS software version 17. Each value was expressed as mean ± standard deviation and compared by Mann Whitney U test and Kruskal Wallis test. The P value <.05 was considered significant.
Results
Diabetes Induces Oxidative DNA Damage in Germ Cells
Although the objective of the present study was to investigate p-p53ser315 and p21CIP1/Waf1 signaling, first, the oxidative DNA damage was investigated by immunohistochemical labeling of 8-oxo-dG. In control rat testes, a very few cells showed the expression of the abnormal base, 8-oxo-dG (grade, +), suggesting some amount of oxidative DNA damage even in normal animals. In DM1 and DM3 groups, the intensity of 8-oxo-dG labeling was significantly increased (grade, ++). In DM1 group, the abnormal base was mainly localized in the cytoplasm of germ cells, but few nuclei also showed the expression of the oxidized base. In DM3 group, the localization was mainly in the nuclei of the germ cells, but few cells also showed cytoplasmic expression (Figure 1). The qualitative assessment of DNA damage in DM1 and DM3 groups (grade, ++), which was more than that in the control group, is in consensus with earlier studies indicating increased DNA damage in diabetic rat testis.16,31,32
Figure 1.

Effects of DM on expression of 8-oxo-dG in the testis (mouse-monoclonal antibody, clone 2E2 1:750; N = 6). In control (A) testis, a very few germ cells showed 8-oxo-dG labeling. The inset in A indicates a stage VII tubule indicating little or no DNA damage. In 1-month-long (DM1) diabetic rat testis (B), the labeling significantly increased mainly in cytoplasm, but also in nuclei of germ cells (D). In 3-month-long (DM3) diabetic rat testis, mainly nuclear DNA damage, but also cytoplasmic DNA damage, is observed (C and E); counterstained with Mayer hematoxylin; scale, 40 μm. DM indicates diabetes mellitus; 8-oxo-dG, 7,8-dihydro-8-oxo-2′-deoxyguanosine.
Diabetes Increases the Expression of p-p53 in DM1 but Decreases in DM3
Western blotting results showed the detection of p-p53ser315 at 63 kDa level (Figure 2A). The testes of normal control rats also showed the expression of p-p53. However, the expression significantly increased in DM1 group when compared to the control group (P < .05; Figure 2B). On the other hand, the expression of p-p53 showed a significant decrease in DM3 group when compared to the control and DM1 groups (P < .05; Figure 2B).
Figure 2.

The phosphorylated-p53ser315 in the testes of diabetic rats (N = 6). A, Representative Western blots indicating p-p53ser315 at 63 kDa level, and (B) densitometry data indicating altered protein activities in DM groups (*P < .05; mean + SD). p53ser315 indicates phosphorylated p53 at ser315 residue; DM, diabetes mellitus; SD, standard deviation.
The p-p53 Expression in Testicular Cells is Stage dependent
As diabetes quantitatively affected the p-p53ser315 levels in the testes, we further investigated the seminiferous epithelial stage-dependent expression of the protein. The immunohistochemistry showed nuclear localization in the germ cells (Figure 3). Table 1 shows qualitative analysis of p-p53ser315 labeling in different types of germ cells. In the testes of control rats, pachytene spermatocytes and step 1 round spermatids in stages I to VI tubules and preleptotene spermatocytes in stages VII to IX tubules showed mild to moderate protein expression. The zygotene and pachytene spermatocytes, step 12 spermatids, and the Sertoli cells in stages X to XIII tubules showed mild expression of the protein. In stage XIV tubules, no germ cells showed the expression of p-p53 (Table 1). In DM1 group, the pachytene spermatocytes showed moderate but step 1 round spermatids showed mild expression of the protein. In stages VII-IX tubules, preleptotene spermatocytes and the Sertoli cells showed intense and moderate p-p53 expression, respectively. In stages X to XIII tubules, the leptotene spermatocytes, step 11 spermatids, and the Sertoli cells showed intense protein expression, whereas the pachytene spermatocytes showed mild expression. In stage XIV tubules, the meiotic figures and secondary spermatocytes showed intense protein expression, whereas the pachytene spermatocytes showed mild expression (Figure 3; Table 1). In DM3 group, mild expression was observed in pachytene spermatocytes in stages I to VI tubules and leptotene and pachytene spermatocytes in stages X to XIII tubules; the other tubular stages did not show the protein expression (Figure 3; Table 1).
Figure 3.

Representative immunohistochemistry photomicrographs indicating nuclear localization of p-p53ser315 in germ and Sertoli cells (N = 6). The protein expression is indicated by brown diaminobenzidine labeling. Counterstained with Mayer hematoxylin, scale bar = 10 μm. A indicates spermatogonia of any type; L, leptotene spermatocytes; Me, meiotic figures (either of meiosis I or II); P, pachytene spermatocytes; Pl, preleptotene spermatocytes; S, Sertoli cells; Ss, secondary spermatocytes (rarely seen); Arabic numbers, spermatid steps; asterisks, retained spermatids due to delayed spermiation; p53ser315, phosphorylated p53 at ser315 residue.
Table 1.
Qualitative Analysis and Grading of p-p53ser315 Immunohistochemistry.a
| Groups | Stages | Spermatogonia | Primary Spermatocytes | MF/SS | Round Spermatids | Elongating Spermatids | Sertoli Cells |
|---|---|---|---|---|---|---|---|
| Control | I-VI | Spermatogonia − | Pachytene +/++ | NA | Steps 1-6 + | Steps 15-18 − | Sertoli cells − |
| VII-IX | Spermatogonia − | Pachytene − Preleptotene ++ Leptotene − | NA | Steps 7-9 − | Step 19 − | Sertoli cells − | |
| X-XIII | Spermatogonia − | Leptotene − Zygotene + Pachytene + | NA | NA | Steps 10-13 + | Sertoli cells −/+ | |
| XIV | Spermatogonia − | Pachytene − | MF − SS − | NA | Step 14 − | Sertoli cells − | |
| DM3 | I-VI | Spermatogonia − | Pachytene ++ | NA | Steps 1-6 + | Steps 15-18 − | Sertoli cells − |
| VII-IX | Spermatogonia − | Pachytene − Preleptotene +++ Leptotene − | NA | Steps 7-9 − | Step 19 − | Sertoli cells ++ | |
| X-XIII | Spermatogonia − | Leptotene +++ Zygotene − Pachytene + | NA | NA | Steps 10-13 +++ | Sertoli cells +++ | |
| XIV | Spermatogonia − | Pachytene + | MF +++ SS +++ | NA | Step 14 − | Sertoli cells − | |
| DM3 | I-VI | Spermatogonia − | Pachytene + | NA | Steps 1-6 − | Steps 15-18 − | Sertoli cells − |
| VII-IX | Spermatogonia − | Pachytene − Preleptotene − Leptotene − | NA | Steps 7-9 − | Step 19 − | Sertoli cells − | |
| X-XIII | Spermatogonia − | Leptotene + Zygotene − Pachytene + | NA | NA | Steps 10-13 − | Sertoli cells − | |
| XIV | Spermatogonia − | Pachytene − | MF − SS − | NA | Step 14 − | Sertoli cells − |
Abbreviations: I-XIV, seminiferous epithelial stages; DM1, 1-month-long diabetic group; DM3, 3-month-long diabetic group; MF, meiotic figures; NA, not applicable as the particular feature is not seen in that tubular stage; SS, secondary spermatocytes; steps 1-19, different steps of spermiogenesis; p-p53ser315, phosphorylated p53 at ser315 residue.
a The grading of immunohistochemical labeling of p-p53ser315 was done in a blinded manner (N = 6). The phosphorylated protein was localized in nuclei of testicular cells in all groups. Negative or equivocal labeling was graded as “−,” mild labeling as “+,” moderate labeling as “++,” and intense labeling as “+++.”
Diabetes Upregulates the Expression of p21CIP1/Waf1 in Testicular Cells
As the p-p53ser315 expression was upregulated in DM1 and downregulated in DM3, we further investigated the expression of one of its main downstream targets, the cell cycle arrest inducer-p21CIP1/Waf1. Western blotting for this protein showed a single band at 19 kDa level (Figure 4A). In contrast to p-p53ser315 expression, the levels of p21CIP1/Waf1 were significantly upregulated in both diabetic groups (P < .05; Figure 4B). The protein expression did not show any time-dependent effects, although a marginal nonsignificant increase was observed in DM3 when compared to DM1 (Figure 4B).
Figure 4.
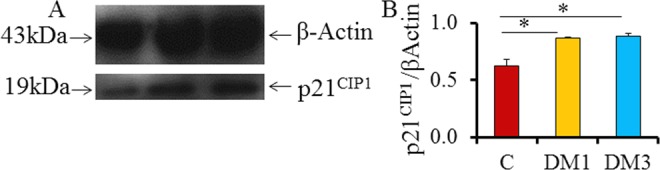
The p21CIP1/Waf1 expression in the testes of control and diabetic rats (N = 6). A, Representative Western blots indicating p21CIP1/Waf1 at 19 kDa level, and (B) densitometry for the protein indicates an increase in protein activities in DM groups (*P < .05; mean + SD). p21CIP1/Waf1 indicates protein 21cyclin-dependent kinase interacting protein 1/wild-type p53-activated factor 1; SD, standard deviation.
The p21CIP1/Waf1 is Mainly Expressed in Cytoplasm of Testicular Cells in Diabetic Rats
The protein was mainly localized in the cytoplasm and also in perinuclear regions of germ cells in a stage-dependent manner (Figure 5). Table 2 indicates the qualitative analysis of the protein expression. In control rat testes, the step 1 round spermatids in stages I to VI tubules and step 7 round spermatids in stages VII to IX tubules showed mild expression of the protein. In stages X to XIII tubules, the pachytene spermatocytes showed mild but step 10 spermatids showed intense expression of the protein. In stage XIV tubules, some meiotic figures showed mild labeling for the protein (Figure 5; Table 2). In DM1 group, in all but the stage XIV tubules some pachytene spermatocytes showed mild expression of the protein. In stages I to VI tubules, the step 1 round spermatids, and in stages VII to IX tubules, the step 7 round spermatids, showed intense expression of the protein. The step 10 spermatids in stages X to XIII tubules showed moderate expression of the protein. In stage XIV tubules, the meiotic figures and secondary spermatocytes showed intense nuclear expression of the protein (Figure 5; Table 2). The pachytene spermatocytes and step 1 round spermatids in stages I to VI tubules of rats in DM3 group showed an intense expression of the protein. Further, the pachytene spermatocytes and step 7 round spermatids in stages VII to IX tubules showed more- intense labeling for the protein. Similarly, the stages X to XIII tubules also showed more intense nuclear, perinuclear, and cytoplasmic expression in pachytene spermatocytes, and highly intense perinuclear and cytoplasmic expression in step 10 spermatids. In stage XIV tubules, the meiotic figures and the cytoplasm of dividing cells and step 14 spermatids showed intense expression of the protein (Figure 5; Table 2).
Figure 5.

The immunohistochemistry photomicrographs showing cytoplasmic localization of p21CIP1/Waf1 in germ cells (brown diaminobenzidine labeling; N = 6). Counterstained with Mayer hematoxylin, scale bar = 10 μm. A indicates spermatogonia of any type; L, leptotene spermatocytes; Me, meiotic figures (either of meiosis I or II); P, pachytene spermatocytes; Pl, preleptotene spermatocytes; S, Sertoli cells; and Ss, secondary spermatocytes (rarely seen); Arabic numbers, spermiogenesis steps. (The color version of this figure is available at rs.sagepub.com.)
Table 2.
Qualitative Analysis and Grading of p21CIP1/Waf1 Immunohistochemistry.a
| Groups | Stages | Spermatogonia | Primary spermatocytes | MF/SS | Round spermatids | Elongating spermatids | Sertoli cells |
|---|---|---|---|---|---|---|---|
| Control | I-VI | Spermatogonia − | Pachytene − | NA | Steps 1-6 + | Steps 15-18 − | Sertoli cells − |
| VII-IX | Spermatogonia − | Pachytene − Preleptotene − Leptotene − | NA | Steps 7-9 + | Step 19 − | Sertoli cells − | |
| X-XIII | Spermatogonia − | Leptotene – Zygoteine – Pachytene + | NA | NA | Steps 10-13 +++ | Sertoli cells − | |
| XIV | Spermatogonia − | Pachytene − | MF −/+ SS −/+ | NA | Step 14 − | Sertoli cells − | |
| DM1 | I-VI | Spermatogonia − | Pachytene −/+ | NA | Steps 1-6 +++ | Steps 15-18 − | Sertoli cells − |
| VII-IX | Spermatogonia − | Pachytene −/+ Preleptotene −/+ Leptotene −/+ | NA | Steps 7-9 +++ | Steps 19 − | Sertoli cells − | |
| X-XIII | Spermatogonia − | Leptotene −/+ Zygotene −/+ Pachytene −/+ | NA | NA | Steps 10-13 ++ | Sertoli cells − | |
| XIV | Spermatogonia − | Pachytene − | MF +++ SS +++ | NA | Step 14 − | Sertoli cells − | |
| DM3 | I-VI | Spermatogonia − | Pachytene +++ | NA | Steps 1-6 +++ | Steps 14-18 − | Sertoli cells − |
| VII-IX | Spermatogonia − | Pachytene ++++ Preleptotene ++++ Leptotene ++++ | Na | Steps 7-9 ++++ | Step 19 − | Sertoli cells − | |
| X-XIII | Spermatogonia − | Leptotene ++++ Zygotene ++++ Pachytene ++++ | NA | NA | Steps 10-13 +++++ | Sertoli cells − | |
| XIV | Spermatogonia − | Pachytene − | MF +++ SS +++ | NA | Step 14 +++ | Sertoli cells − |
Abbreviations: I-XIV, seminiferous epithelial stages; DM1, 1-month-long diabetic group; DM3, 3-month-long diabetic group; MF, meiotic figures; NA, not applicable as the particular feature is not seen in that tubular stage; p21CIP1/Waf1, protein 21cyclin-dependent kinase interacting protein 1/wild-type p53-activated factor 1; SS, secondary spermatocytes; steps 1-19, different steps of spermiogenesis.
a The grading of immunohistochemical labeling of the protein was done in a blinded manner (N = 6). The protein was localized predominantly in cytoplasm, but also in perinuclear regions of testicular cells. Negative or equivocal labeling was graded as “−,” mild labeling as “+,” moderate labeling as “++,” intense labeling as “+++,” more intense labeling as “++++,” and highly intense labeling as “+++++.”
Discussion
The main findings of this study are diabetes-induced oxidative DNA damage and alterations in the expressions of p-p53 and p21CIP1/Waf1 in the testis. Diabetes induces seminiferous tubular atrophy, oxidative stress, and mitochondrial dysfunction.33,34 It also causes germ cell apoptosis,35 decrease in sperm count and motility,20 and modulations in numerous intracellular signaling pathways that are responsible for maintaining normal cell functions.14,21,22 Diabetes also induces DNA damage36 and the formation of advanced glycation end products and their receptors.18,37 These adverse effects of diabetes may have significance as regards to the fertility outcome.9 In our study, we showed an increase in mitochondrial oxidative DNA damage in DM1 and predominantly nuclear oxidative DNA damage in DM3. The decrease in mitochondrial oxidative DNA damage in DM3. might be suggestive of its repair, and the persistent nuclear oxidative DNA damage in this group might be due to the lack of repair, although this assumption has to be investigated further. The oxidized base, 8-oxo-dG, is a reliable marker for DNA damage, which actually is formed due to the formation of free radicals in cells. As diabetes is known to induce oxidative stress in the testis,13 the base oxidation seen in our study can be attributed to it, and our results are in agreement with that of a recent study.31 However, our results provide a new insight, that is, during early period of diabetes, mitochondrial DNA is affected by the free radicals, which logically should, as the DNA is located near the site of free radical formation in mitochondria. At a later stage, the free radicals attack nuclear DNA, which appears not to be repaired or to suffer impaired repair process.
Our results indicate that the phosphorylation of p53 at ser315 was induced during early period (in DM1) but not during a prolonged period (in DM3) of diabetes. The increase in the p-p53ser315 expression and its nuclear localization suggest that the DNA damage was responsible for the activation of the protein.38 However, the decrease in the p-p53 expression seen in DM3 is difficult to interpret, as it is not in congruity with either the increase in DNA damage or p21CIP1/Waf1 expression. Upon DNA damage or receiving other forms of stress, p53 is phosphorylated at one or more than one residues (7 serines and 2 threonines on the N-terminal, and 2 serines on the C-terminal), including the one at ser315.24 The phosphorylation at ser315 occurs following DNA damage.39 Our finding that the increase in DNA damage along with the concomitant increase in p-p53ser315 in DM1 indicates that the protein is stabilized in response to diabetes-induced DNA damage. However, it is not clear why p-p53ser315 levels were significantly decreased in DM3 when compared to both control and groups, despite the increased oxidative DNA damage. It may be that due to prolonged period of hyperglycemia, the dephosphorylation of the protein at its ser315 residue took place, as seen in humans by a protein called hCdc14,40 or the new synthesis of the protein itself was probably inhibited. The other possibility for the decrease in p-p53ser315 in our study might reside in Aurora kinase A signaling. This kinase phosphorylates p53 at ser315 residue, which is meant for Mdm2-mediated degradation of the protein in the proteosome.41 Hence, the decrease in p-p53ser315 expression in DM3 group may have some relationship with the increase in Aurora kinase A signaling in the diabetic rat testes. However, these assumptions have to be investigated further. Moreover, at present, it is also unknown whether or not hyperglycemia causes posttranslational modifications such as acetylation, phosphorylation, and sumoylation at other residues in p53.
In this study, we also found an increase in the expression of p21CIP1/Waf1 in a stage-dependent manner in the testis, and the expression was predominantly cytoplasmic. The phosphorylation of p21CIP1/Waf1 by protein kinase B (AKT) and extracellular-signal-regulated kinase results in cytoplasmic translocation of the stabilized protein, and by this mechanism the protein imparts its antiapoptotic function.27,42 The nuclear localization of the protein happens in response to DNA damage, which results in the cell cycle arrest at G1/S and G2/S transitions.26,43 Hence, the perinuclear expression of the protein seen in our study was probably due to DNA damage. The p21CIP1/Waf1 protein manages the cell cycle arrest by many modulations of numerous intracellular events, but mainly by inhibiting cyclin/CDK interactions, downregulating proliferating cell nuclear antigen functions and retinoblastoma protein activities.44 The p21CIP1/Waf1 protein is the key down-stream target of p53 pathway meant to induce the cell cycle arrest.38 The protein is stabilized by the phorphorylation at various serine/threonine residues by up-stream proteins such as c-Jun N-terminal kinases, p38-MAPKs, and AKT.27,45
The expression of p21CIP1/Waf1 in control rat testicular cells implies some degree of DNA damage and, therefore, the antiapoptotic function of the protein, as the testes of normal animals also show a small number of apoptotic cells.30 In the diabetic rats, the expression of the protein in pachytene spermatocytes (in stages X-XIII in control, and in all except in XIV stages, in DM1 and DM3) was increased, especially in DM3, which suggested mainly antiapoptotic (cytoplasmic expression) and also the cell cycle arrest (perinuclear expression) related roles for the protein. In DM3, however, the pachytene spermatocytes, round spermatids (steps 1, 7, and 10), and in step 14 elongating spermatids showed much higher intensity of expression of the protein in both cytoplasm and perinuclear regions than that did in DM1. Moreover, the meiotic figures and secondary spermatocytes (these cells are rarely visible) showed both nuclear and cytoplasmic expressions of the protein indicating their susceptibility for the oxidative DNA damage stimulated apoptosis. These results indicate that the oxidative DNA damage was more in DM3 as indicated by the increased 8-oxo-dG expression than that in DM1. The augmented cytoplasmic expression may be due to an increased need for antiapoptotic stimuli in testicular cells to prevent the latter from entering into apoptosis due to DNA damage. However, diabetes is known to induce apoptosis of germ cells in the testes,22,31,35 but our findings indicate an antiapoptotic role for p21CIP1/Waf1 due to its predominant cytoplasmic localization. These disparate findings might be due to 2 possible mechanisms: first, there may be some other stronger proapoptotic signals than the antiapoptotic influence of p21CIP1/Waf1, which overpowers the latter; and, second, the studies that reported the apoptosis have drawn their conclusions largely based on terminal deoxynucleotidyl transferase dUTP nick end labeling assay results. The latter assay labels DNA double-strand breaks presenting 3′OH ends, which are commonly seen in both apoptosis and necrosis46,47; therefore, the cell death observed in the testes of diabetic animals need not be apoptosis.
Although the colocalization of the proteins was not done, both the proteins showed their expression in primary spermatocytes but not in round or elongating spermatids (Tables 1 and 2). Moreover, the intensity of their expressions also did not match with each other. These observations indicate that the expression of p21CIP1/Waf1 in diploid primary spermatocytes has some correlation with p-p53ser315 expression in the same cells, especially in DM1. The haploid spermatids do not show any relationship between p21CIP1/Waf1 and p-p53ser315 expressions indicating the possible existence of a p-p53ser315-independent mechanism in these cells to upregulate p21CIP1/Waf1. The p21CIP1/Waf1 protein can be upregulated in a p53-independent manner,48 which may also be the reason for the lack of p-p53ser315 expression in DM3 group, but upregulated p21CIP1/Waf1expression in the same group. In addition, the decrease in p-p53ser315 in DM3, in contrast to the increase in p21CIP1/Waf1, may also be due to negative regulation of p53 by p21CIP1/Waf1.49 In other words, the highly intense p21CIP1/Waf1expression in round spermatids led to downregulation of p-p53ser315. In conclusion, our results indicate that hyperglycemia-induced DNA damage upregulates the p21CIP1/Waf1 expression mainly in primary spermatocytes and round spermatids. This expression is seen in parallel with the nuclear expression of p-p53ser315 after the short period of hyperglycemia. The increase in p21CIP1/Waf1 in the testes after the long period of hyperglycemia is seen concurrently with the decrease in p-p53ser315 expression. Moreover, it appears that p21CIP1/Waf1 has dual roles in cell cycle arrest and antiapoptotic stimulus in the testicular germ cells of diabetic rats.
Acknowledgments
We gratefully acknowledge the technical assistance of Ms S. Verghese, Ms J. Prashanth, Dr S. Jacob, Dr S. Mohan, and Ms Lizamma.
Footnotes
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Kuwait University Grant # YM10/13 and General Facility Grant# SRUL 02/13.
References
- 1. Zimmet P, Alberti KGMM, Shaw J. Global and societal implications of the diabetes epidemic. Nature. 2001;414 (6865):782–787. [DOI] [PubMed] [Google Scholar]
- 2. Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nature Rev. 2011;11 (2):98–107. [DOI] [PubMed] [Google Scholar]
- 3. Evans JL, Ira ED, Maddux GBA, Grodsky GM. Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of type 2 diabetes. Endocrine Rev. 2002;23 (5):599–622. [DOI] [PubMed] [Google Scholar]
- 4. Golbidi S, Badran M, Laher I. Antioxidant and anti-inflammatory effects of exercise in diabetic patients. Exp Diabetes Res. 2012;2012:941868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Marshall SM, Flyvbjerg A. Diabetic nephropathy. In: Holt R, Cockram C, Flyvbjerg A, Goldstein B, eds. Textbook of Diabetes, 4th Edition Hoboken, NJ: Blckwell Publishing; 2010:599–614. [Google Scholar]
- 6. Sun JK, Keenan HA, Cavallerano JD, et al. Protection from retinopathy and other complications in patients with type 1 diabetes of extreme duration: the Joslin 50-year medalist study. Diabetes Care. 2011;34 (4):968–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Saboor Aftab SA, Kumar S, Barber TM. The role of obesity and type 2 diabetes mellitus in the development of male obesity-associated secondary hypogonadism. Clin Endocrinol (Oxf). 2013;78 (3):330–337. [DOI] [PubMed] [Google Scholar]
- 8. Shrilatha B, Muralidhara Early oxidative stress in testis and epididymal sperm in streptozotocin-induced diabetic mice: its progression and genotoxic consequences. Reprod Toxicol. 2007;23 (4):578–587. [DOI] [PubMed] [Google Scholar]
- 9. Rama Raju GA, Jaya Prakash G, Murali Krishna K, Madan K, Siva Narayana T, Ravi Krishna CH. Noninsulin-dependent diabetes mellitus: effects on sperm morphological and functional characteristics, nuclear DNA integrity and outcome of assisted reproductive technique. Andrologia. 2012;44 (suppl 1):490–498. [DOI] [PubMed] [Google Scholar]
- 10. Condorelli RA, Calogero AE, Vicari E, et al. Vascular regenerative therapies for the treatment of erectile dysfunction: current approaches. Andrology. 2013;1 (4):533–540. [DOI] [PubMed] [Google Scholar]
- 11. Narayana K, Yousif MH, El-Hashim AZ, Makki B, Akhtar S, Benter IF. Role of angiotensin II and angiotensin-(1-7) in diabetes-induced oxidative DNA damage in the corpus cavernosum. Fertil Steril. 2013;100 (1):226–233. [DOI] [PubMed] [Google Scholar]
- 12. Chandrashekar KN, Muralidhara Evidence of oxidative stress and mitochondrial dysfunctions in the testis of prepubertal diabetic rats. Int J Impot Res. 2009;21 (3):198–206. [DOI] [PubMed] [Google Scholar]
- 13. Kanter M, Aktas C, Erboga M. Curcumin attenuates testicular damage, apoptotic germ cell death, and oxidative stress in streptozotocin-induced diabetic rats. Mol Nutr Food Res. 2013;57 (9):1578–1585. [DOI] [PubMed] [Google Scholar]
- 14. Zhao Y, Zhao H, Zhai X, et al. Effects of Zn deficiency, antioxidants, and low-dose radiation on diabetic oxidative damage and cell death in the testis. Toxicol Mech Methods. 2013;23 (1):42–47. [DOI] [PubMed] [Google Scholar]
- 15. Agbaje IM, Rogers DA, McVicar CM, et al. Insulin dependant diabetes mellitus: implications for male reproductive function. Hum Reprod. 2007;22 (7):1871–1877. [DOI] [PubMed] [Google Scholar]
- 16. Kushwaha S, Jena GB. Enalapril reduces germ cell toxicity in streptozotocin-induced diabetic rat: investigation on possible mechanisms. Naunyn Schmiedebergs Arch Pharmacol. 2012;385 (2):111–124. [DOI] [PubMed] [Google Scholar]
- 17. Mallidis C, Agbaje IM, Rogers DA, et al. Advanced glycation end products accumulate in the reproductive tract of men with diabetes. Int J Androl. 2009;32 (4):295–305. [DOI] [PubMed] [Google Scholar]
- 18. O'Neill J, Czerwiec A, Agbaje I, et al. Differences in mouse models of diabetes mellitus in studies of male reproduction. Int J Androl. 2010;33 (5):709–716. [DOI] [PubMed] [Google Scholar]
- 19. Mallidis C, Agbaje I, O'Neill J, McClure N. The influence of type 1 diabetes mellitus on spermatogenic gene expression. Fertil Steril. 2009;92 (6):2085–2087. [DOI] [PubMed] [Google Scholar]
- 20. Roy S, Metya SK, Rahaman N, Sannigrahi S, Ahmed F. Ferulic acid in the treatment of post-diabetes testicular damage: relevance to the down regulation of apoptosis correlates with antioxidant status via modulation of TGF-β1, IL-1β and Akt signalling. Cell Biochem Funct. 2014;32 (1):115–124. doi: 10.1002/cbf.2983. [DOI] [PubMed] [Google Scholar]
- 21. Jiang X, Zhang C, Xin Y, et al. Protective effect of FGF21 on type 1 diabetes-induced testicular apoptotic cell death probably via both mitochondrial- and endoplasmic reticulum stress-dependent pathways in the mouse model. Toxicol Lett. 2013;219 (1):65–76. [DOI] [PubMed] [Google Scholar]
- 22. Zhao Y, Tan Y, Dai J, et al. Exacerbation of diabetes-induced testicular apoptosis by zinc deficiency is most likely associated with oxidative stress, p38 MAPK activation, and p53 activation in mice. Toxicol Lett. 2011;200 (1-2):100–106. [DOI] [PubMed] [Google Scholar]
- 23. May P, May E. Twenty years of p53 research: structural and functional aspects of the p53 protein. Oncogene. 1999;18 (53):7621–7636. [DOI] [PubMed] [Google Scholar]
- 24. Appella E, Anderson CW. Post-translational modifications and activation of p53 by genotoxic streses. Eur J Biochem. 2001;268 (10):2764–2772. [DOI] [PubMed] [Google Scholar]
- 25. Sanli T, Steinberg GR, Singh G, Tsakiridis T. AMP-activated protein kinase (AMPK) beyond metabolism: a novel genomic stress sensor participating in the DNA damage response pathway. Cancer Biol Ther. 2014;15 (2):156–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Abbas T, Dutta A. P21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009;9 (6):400–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cmielová J, Rezáčová M. p21Cip1/Waf1 protein and its function based on a subcellular localization. J Cell Biochem. 2011;112 (12):3502–3506. [DOI] [PubMed] [Google Scholar]
- 28. Narayana K, Verghese S, Jacob S. L-Ascorbic acid partially protects two cycles of cisplatin chemotherapy-induced testis damage and oligo-astheno-teratospermia in a mouse model. Exp Toxicol Pathol. 2009;61 (6):553–563. [DOI] [PubMed] [Google Scholar]
- 29. Narayana K. Cisplatin induces duplex 3' overhangs and 5' blunt ends in epididymal epithelium in a Bax-dependent manner without any protection from L-ascorbic acid. Eur J Pharmacol. 2010;641 (2-3):238–245. [DOI] [PubMed] [Google Scholar]
- 30. Narayana K, Al-Bader M, Mousa A, Khan KM. Molecular effects of chemotherapeutic drugs and their modulation by antioxidants in the testis. Eur J Pharmacol. 2012;674 (2-3):207–216. [DOI] [PubMed] [Google Scholar]
- 31. Tsounapi P, Saito M, Dimitriadis F, et al. Antioxidant treatment with edaravone or taurine ameliorates diabetes-induced testicular dysfunction in the rat. Mol Cell Biochem. 2012;369 (1-2):195–204. [DOI] [PubMed] [Google Scholar]
- 32. Broedbaek K, Weimann A, Stovgaard ES, Poulsen HE. Urinary 8-oxo-7,8-dihydro-2'-deoxyguanosine as a biomarker in type 2 diabetes. Free Radic Biol Med. 2011;51 (8):1473–1479. [DOI] [PubMed] [Google Scholar]
- 33. Kyathanahalli C, Bangalore S, Hanumanthappa K, Muralidhara Experimental diabetes-induced testicular damage in prepubertal rats. J Diabetes. 2013;6 (1):48–59. [DOI] [PubMed] [Google Scholar]
- 34. Rato L, Duarte AI, Tomás GD, et al. Pre-diabetes alters testicular PGC1-α/SIRT3 axis modulating mitochondrial bioenergetics and oxidative stress. Biochim Biophys Acta. 2014;1837 (3):335–344. [DOI] [PubMed] [Google Scholar]
- 35. Koh PO. Streptozotocin-induced diabetes increases the interaction of Bad/Bcl-XL and decreases the binding of pBad/14–3–3 in rat testis. Life Sci. 2007;81 (13):1079–1084. [DOI] [PubMed] [Google Scholar]
- 36. Vendramini V, Cedenho AP, Miraglia SM, Spaine DM. Reproductive function of the male obese Zucker Rats: alteration in sperm production and sperm DNA damage. Reprod Sci. 2014;21 (2):221–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mallidis C, Agbaje IM, Rogers DA, et al. Advanced glycation end products accumulate in the reproductive tract of men with diabetes. Int J Androl. 2008;32 (4):295–305. [DOI] [PubMed] [Google Scholar]
- 38. Mirzayans R, Andrais B, Scott A, Murray D. New insights into p53 signaling and cancer cell response to DNA damage: implications for cancer therapy. J Biomed Biotechnol, 2012;2012:170325 doi: 10.1155/2012/170325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Waterman MJ, Stavridi ES, Waterman JL, Halazonetis TD. ATM- dependent activation of p53 involves dephosphorylation and association with 14-3-3 proteins. Nat Genet. 1998;19 (2):175–178. [DOI] [PubMed] [Google Scholar]
- 40. Li L, Ljungman M, Dixon JE. The human Cdc14 phosphatases interact with and dephosphorylate the tumor suppressor protein p53. J Biol Chem. 2000;275 (4):2410–2414. [DOI] [PubMed] [Google Scholar]
- 41. Katayama H, Sasai K, Kawai H, et al. Phosphorylation by aurora kinase A induces Mdm2-mediated destabilization and inhibition of p53. Nat Genet. 2004;36 (1):55–62. [DOI] [PubMed] [Google Scholar]
- 42. Heo JI, Oh SJ, Kho YJ, et al. ERK mediates anti-apoptotic effect through phosphorylation and cytoplasmic localization of p21Waf1/Cip1/Sdi in response to DNA damage in normal human embryonic fibroblast (HEF) cells. Mol Biol Rep. 2011;38 (4):2785–2791. [DOI] [PubMed] [Google Scholar]
- 43. Cazzalini O, Scovassi AI, Savio M, Stivala LA, Prosperi E. Multiple roles of the cell cycle inhibitor p21(CDKN1A) in the DNA damage response. Mutat Res. 2010;704 (1-3):12–20. [DOI] [PubMed] [Google Scholar]
- 44. Rousseau D, Cannella D, Boulaire J, Fitzgerald P, Fotedar A, Fotedar R. Growth inhibition by CDK-cyclin and PCNA binding domains of p21 occurs by distinct mechanisms and is regulated by ubiquitin-proteasome pathway. Oncogene. 1999;18 (30):4313–4325. [DOI] [PubMed] [Google Scholar]
- 45. Hwang CY, Lee C, Kwon KS. Extracellular signal-regulated kinase 2-dependent phosphorylation induces cytoplasmic localization and degradation of p21Cip1. Mol Cell Biol. 2009;29 (12):3379–3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Didenko VV, Ngo H, Baskin DS. Early necrotic DNA degradation: presence of blunt-ended DNA breaks, 3' and 5' overhangs in apoptosis, but only 5' overhangs in early necrosis. Am J Pathol. 2003;162 (5):1571–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Narayana K, Raghupathy R. DNA damage in lead-exposed hepatocytes: coexistence of apoptosis and necrosis? Drug Chem Toxicol. 2012;35 (2):208–217. [DOI] [PubMed] [Google Scholar]
- 48. Macleod KF, Sherry N, Hannon G, et al. p53-dependent and independent expression of p21 during cell growth, differentiation, and DNA damage. Genes Dev. 1995;9 (8):935–944. [DOI] [PubMed] [Google Scholar]
- 49. Javelaud D, Besancon F. Inactivation of p21WAF1 sensitizes cells to apoptosis via an increase of both p14ARF and p53 levels and an alteration of the Bax/Bcl-2 ratio. J Biol Chem. 2002;277 (40):37949–37954. [DOI] [PubMed] [Google Scholar]