

Abstract
The activity of the allophanate hydrolase from Pseudomonas sp. strain ADP, AtzF, provides the final hydrolytic step for the mineralization of s-triazines, such as atrazine and cyanuric acid. Indeed, the action of AtzF provides metabolic access to two of the three nitrogens in each triazine ring. The X-ray structure of the N-terminal amidase domain of AtzF reveals that it is highly homologous to allophanate hydrolases involved in a different catabolic process in other organisms (i.e., the mineralization of urea). The smaller C-terminal domain does not appear to have a physiologically relevant catalytic function, as reported for the allophanate hydrolase of Kluyveromyces lactis, when purified enzyme was tested in vitro. However, the C-terminal domain does have a function in coordinating the quaternary structure of AtzF. Interestingly, we also show that AtzF forms a large, ca. 660-kDa, multienzyme complex with AtzD and AtzE that is capable of mineralizing cyanuric acid. The function of this complex may be to channel substrates from one active site to the next, effectively protecting unstable metabolites, such as allophanate, from solvent-mediated decarboxylation to a dead-end metabolic product.
INTRODUCTION
Atrazine (1-chloro-3-ethylamino-5-isopropylamino-2,4,6-triazine; Fig. 1) is one of the most heavily applied herbicides in the world and is registered for use in North and South America, Australia, Africa, Asia, and the Middle East. Atrazine is environmentally persistent (half-life, 4 to 57 weeks, depending on the location) and mobile, leading to the detection of atrazine in surface water, groundwater, and aquifers (1–3). Atrazine has been detected in the environment at concentrations of up to 4.6 μM in several countries (2, 3). It has been suggested that atrazine may be a carcinogen and an endocrine disrupter at such concentrations (4–6).
FIG 1.
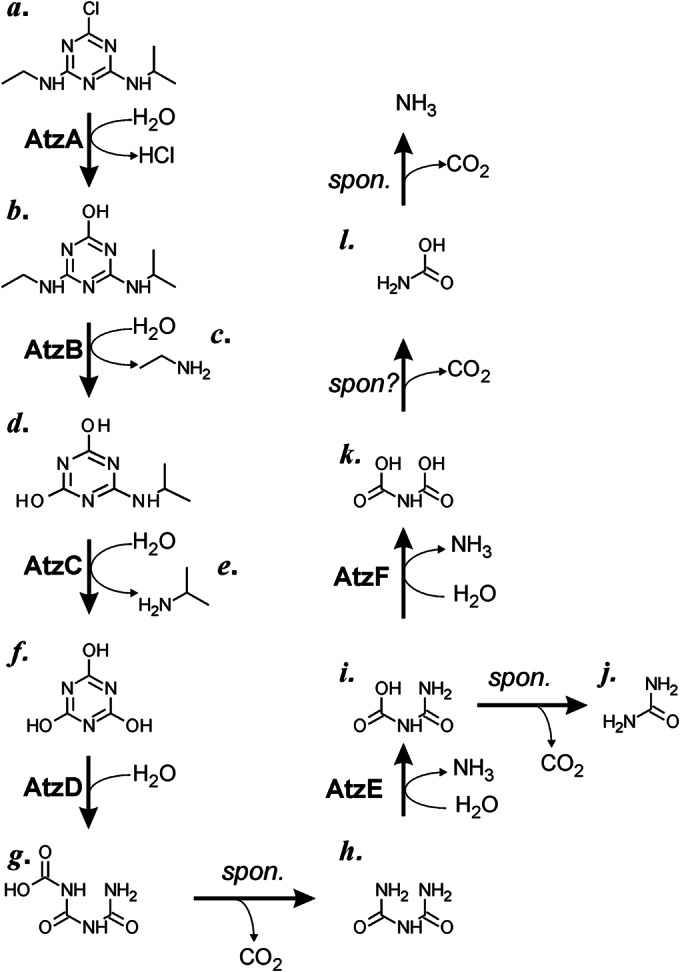
Mineralization of atrazine in Pseudomonas sp. strain ADP. Atrazine (a) is hydrolyzed by AtzA, releasing chloride. The product (b) is further hydrolyzed by AtzB, releasing ethylamine (c), and subsequent hydrolysis of the resulting product (d) releases isopropylamine (e), generating cyanuric acid (f). AtzD hydrolyzes cyanuric acid to produce the unstable 1-carboxybiuret (g), which decomposes to biuret (h). Biuret is deaminated by AtzE to yield allophanate (i), which can spontaneously decompose to urea (j) or can be deaminated by AtzF to produce the unstable N-carboxycarbamate (k), which spontaneously decomposes first to carbamate (l) and then to ammonia. spon., spontaneous.
Since atrazine was introduced in the 1950s, bacteria have evolved highly efficient catabolic pathways that allow the use of atrazine as a sole nitrogen and carbon source (7–10). These pathways have provided valuable insights into the evolutionary processes that drive the establishment of new enzyme function and new catabolic pathways (11–15). In addition, these pathways and cognate enzymes provide a potential biotechnological solution to atrazine contamination (i.e., bioremediation) (16–19).
The most intensively studied atrazine catabolism pathway was discovered in Pseudomonas sp. strain ADP in the mid-1990s and is comprised of six hydrolases: atrazine chlorohydrolase (AtzA; EC 3.8.1.8) (20, 21), N-ethylaminohydrolase (AtzB; EC 3.5.99.3) (22, 23), N-isopropylammelide isopropylaminohydrolase (AtzC; EC 3.5.99.4) (24, 25), cyanuric acid amidohydrolase (AtzD; EC 3.5.2.15) (15, 26, 27), biuret amidohydrolase (AtzE; EC 3.5.1.84) (28), and allophanate hydrolase (AtzF; EC 3.5.1.54) (29–31). These hydrolases sequentially dechlorinate (AtzA) and remove the two N-alkyl side groups (AtzB and AtzC) to produce cyanuric acid, which is then further hydrolyzed to biuret, allophanate, and ammonia via AtzD, AtzE, and AtzF, respectively (Fig. 1).
In other bacterial genera, such as Arthrobacter and Nocardioides, the function of AtzA is conducted by TrzN (32–36). Although TrzN is physiologically analogous to AtzA, it appears to have evolved independently, as the two enzymes share low sequence identity and have significantly different reaction mechanisms and substrate ranges.
Recently, there have been concerted efforts to understand the structural biology of the hydrolases of the atrazine catabolic pathway, with the structure of TrzN being obtained in 2010 (33) and the structure of AtzD (15), which possesses a previously unreported Toblerone protein fold, being reported in 2013. A structure for AtzC has also been deposited in the Protein Data Bank (PDB accession number 2QT3). Structures for AtzB, AtzE, and AtzF have yet to be reported, although structures for the ureolytic allophanate hydrolases (AHs) from Kluyveromyces lactis (AHKl) and Granulibacter bethesdensis (AHGb) were reported in 2013 (37, 38). Unlike the atrazine-degrading AHs, the ureolytic enzymes are found as multidomain enzymes along with a biotin- and ATP-dependent urea carboxylase, which is required to generate allophanate from urea (37, 39–42). The AH components of these complexes have considerable sequence conservation with each other and the triazine-related AHs (Fig. 2).
FIG 2.

Alignment of the allophanate hydrolases from Pseudomonas sp. strain ADP (AtzF), Kluyveromyces lactis (AHKl), and Granulibacter bethesdensis (AHGb). Single underline, amidase domain; double underline, C-terminal domain; diamonds, active-site residues of the amidase domain; triangle, proposed catalytic histidine residue in the C-terminal domain; arrow, position where AtzF was truncated to produce AtzF468; asterisks, identical residues; colons, highly similar residues; periods, similar.
AH is a member of the amidase signature family, characterized by an ∼130-amino-acid-long region that is rich in serine and glycine residues and contains a characteristic Ser–cis-Ser–Lys catalytic triad (43, 44). Uniquely among this family of amidases, the AHs possess a conserved ca. 15-kDa extension at the C terminus of the amidase domain, of uncertain function, that is present in both eukaryotic and prokaryotic AHs (38). In a report from Fan et al., the authors proposed that the C-terminal domain of AHKl is involved in the hydrolysis of N-carboxycarbamate, the unstable product of AH-mediated allophanate deamination (Fig. 1) (37). Moreover, in silico substrate docking suggested that a C-terminal histidine residue (His492) plays an important catalytic role (37).
Herein, we present the X-ray structure of the N-terminal amidase domain of AtzF from Pseudomonas sp. strain ADP and compare it with the recently determined AH structures. Our in vitro biochemical data were unable to support a physiologically relevant catalytic function for the C-terminal domain of AtzF. However, we present evidence for a functional role for the ∼15-kDa C-terminal domain in coordinating the quaternary structure of the protein; AtzF forms a large multienzyme complex with AtzD and AtzE that mineralizes cyanuric acid. The function of this complex may be to protect unstable metabolites, such as allophanate, from solvent-mediated decarboxylation to a dead-end metabolic product.
MATERIALS AND METHODS
DNA manipulation.
The cloning of atzF and the truncated gene encoding the amidase (N-terminal) domain of AtzF (atzF467) is described elsewhere (45). A mutant of atzF that encoded the AtzF H488A variant was produced by the overlapping PCR method of Ho et al. (46), using atzF as the template DNA and primers atzF H488A fwd (5′-ACCAGCCCTTGAATGCTCAGCTCACGGAG-3′) and atzF H488A rev (5′-TCTCCGTGAGCTGAGCATTCAAGGGCTGG-3′). PCRs with Phusion DNA polymerase (New England BioLabs [NEB], Ipswich, MA, USA) were conducted under the following conditions: 30 s of denaturation (98°C) and 30 s of annealing (55°C), followed by a 2-min extension (72°C).
The atzF H488A amplicon was digested with NdeI and BamHI (NEB, Ipswich, MA, USA) and cloned into the pETCC2 expression vector (15). The resultant plasmid was used to transform electrocompetent Escherichia coli BL21 λ(DE3) cells (Invitrogen, CA) following the manufacturer's instructions.
Protein expression and purification.
Expression, postexpression harvest, and lysis of cells for the AtzF H488A variant were carried out under the same conditions used for AtzF and AtzF467 expression reported elsewhere (45). Purification of AtzF H488A from soluble cell extract was carried out by metal ion affinity chromatography using an Ni-nitrilotriacetic acid Superflow cartridge (Qiagen, MD). After loading the soluble fraction onto the column, a step gradient of 12.5 mM imidazole was applied for six column volumes, followed by a seven-column-volume wash with 50 mM imidazole, after which protein was eluted in seven column volumes of 250 mM imidazole.
Crystallization and structure solution.
Crystallization and data collection were done as previously described (45). Briefly, crystals grew from a reservoir containing 11 to 14% (wt/vol) polyethylene glycol 3350 and 2% Tacsimate reagent (pH 5) at 293 K and were used to collect X-ray data at the MX-2 beamline of the Australian Synchrotron using a wavelength of 0.9529 Å (13,011 eV). The structure was solved using molecular replacement (Phaser software) (47) of the structure with PDB accession number 2DQN (48), and a clear solution with six protomers in the asymmetric unit was found. The space group was found to be P21, and the resolution of the data extended to 2.5 Å (Table 1). The model was rebuilt by hand using Coot software (49) and refined using the Refmac program (50), with the noncrystallographic symmetry (NCS) restraints option being used during all stages of refinement. Four of the molecules (chains A to D) had a significantly better electron density than the other two molecules (chains E and F).
TABLE 1.
Data collection and refinement statistics
| Parametera | Value for structure with PDB accession no. 4CP8b |
|---|---|
| Data collection statistics | |
| Space group | P21 |
| Cell dimensions | |
| a, b, c (Å) | 82.45, 179.23, 112.61 |
| α, β, γ (°) | 90, 106.63, 90 |
| Resolution (Å) | 2.50 (2.64–2.50) |
| Rmerge | 0.150 (0.810) |
| I/σI | 12.6 (2.8) |
| Completeness (%) | 100 (100) |
| Redundancy | 7.7 (7.7) |
| Refinement statistics | |
| Resolution (Å) | 40.0–2.50 |
| No. of reflections | 102, 548 |
| Rwork/Rfree (%) | 22.4/25.9 |
| No. of: | |
| Atoms | 20, 462 |
| Proteins | 20, 132 |
| Ligands/ions | 28 |
| Water molecules | 312 |
| B-factors (Å2) for: | |
| Proteins | 44.2 |
| Ligands/ions | 30.3 |
| Water | 24.2 |
| RMSD | |
| Bond lengths (Å) | 0.010 |
| Bond angles (°) | 1.367 |
Rmerge = Σ|I − 〈I〉/ΣI × 100, where I is the intensity of a reflection and 〈I〉 is the mean intensity.
Values in parentheses are for the highest-resolution shell.
The crystals were grown in the Tacsimate reagent (Hampton Research), which is predominantly malonic acid, and there was an unambiguous density for malonic acid in the four protomers with a good electron density. Despite the poor density for two of the molecules in the asymmetric unit, the model refined to Rwork and Rfree values of 22.4% and 25.9%, respectively (Table 1). The Ramachandran plot (from Coot software) shows that 94.3% of the residues were in the most favorable region, 4.3% were in the allowed region, and were 1.5% in the outlier region.
SAXS.
AtzF and AtzF467 were dialyzed overnight into 50 mM Tris buffer, pH 7.5, 100 mM NaCl. The same buffer used as the buffer standard during data collection was used. A dilution series of AtzF (from 6.8 to 0.2 mg/ml) and AtzF467 (from 13.1 to 0.4 mg/ml) was prepared, and scattering data were collected for 1 s using a Pilatus 1 M photon-counting detector (Dektris) with a sample-to-detector distance of 1.6 m. Ten replicate images were collected for each sample and averaged, with outlier rejection, to control for radiation damage. Data were measured in a Q (scattering vector) range from 0.01 to 0.5 Å−1, and at the highest protein concentration, the scattering remained above the noise threshold to the edge of the detector.
AtzF (75 μl, 6 mg/ml) and AtzF467 (75 μl, 6 mg/ml) were injected onto a size-exclusion column (Wyatt silica bead column; pore size, 300 Å) that had been preequilibrated with the PO4-NaCl buffer. The column was developed at 0.2 ml/min, and a single peak eluted. The small-angle X-ray scattering (SAXS) showed no change over the peak (unpublished observations).
The radius of gyration (Rg) and total forward scatter [I(0)] for each concentration in the dilution series was calculated using the autorg routine in the PRIMUS system (51). Fits to atomic models were performed using the CRYSOL program (52), and dummy atom models were calculated using the DAMMIF program (53) and superimposed on the high-resolution models using the SUPCOMB program (54). Molecular weight was calculated from I(0) (55), together with protein concentration measurements and the scattering length density and partial specific volume calculated from the protein sequences, using the web application MULCh (56).
Substrate preparation and kinetic assays.
The potassium salt of allophanate was prepared by hydrolyzing ethyl allophanate (Sigma-Aldrich, St. Louis, MO) with a 5 M excess of 1 M potassium hydroxide at 40°C for 3 h (42). Potassium allophanate was precipitated overnight in an ice-cold mixture of 5 volumes of ethanol and 1 volume of diethyl ether. The precipitate was separated by filtration and subsequently dried and stored in a desiccator.
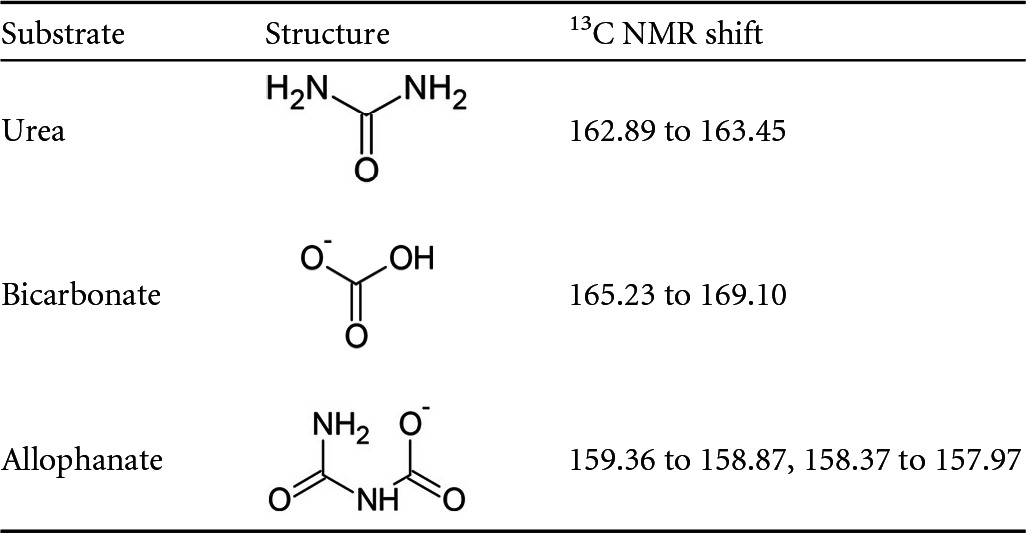
The synthesis of allophanate was confirmed by 13C nuclear magnetic resonance (NMR) (see Fig. S1 in the supplemental material) using a Varian Inova-500 NMR spectrometer. The substrate was dissolved in 1 M KOH prepared in D2O, and 2,048 scans were performed in total. The purity of the potassium allophanate produced was assessed using a glutamate dehydrogenase (GDH)-coupled assay. Complete AtzF-mediated hydrolysis of allophanate was performed, and the GDH-based ammonia assay was used to determine the purity of the allophanate prepared. The GDH-dependent decrease in the absorbance at 340 nm was measured using a SpectraMax M2 spectrophotometer (Molecular Devices, CA) to follow the consumption of NADH during the reductive amination of α-ketoglutaric acid to form l-glutamate. The hydrolysis of allophanate by AtzF was assumed to have reached completion, as a high concentration of AtzF (∼1 μM) was added, the reaction components of the GDH-linked ammonia assay were added well in excess of the allophanate concentration, and the decrease in NADH absorbance plateaued before all of the NADH was consumed. Allophanate prepared by the method described above was found to be 86% pure (Table 2).
TABLE 2.
13C NMR shifts of allophanate and possible impurities
To study the pH dependence of AtzF activity, kinetic assays were performed in the pH range of 7 to 9.5 using allophanate as a substrate, and kcat/Km values were plotted against the pH (see Fig. S3 in the supplemental material). The kinetic assays were carried out in buffer comprised of 100 mM HEPES and 100 mM CHES (N-cyclohexyl-2-aminoethanesulfonic acid) adjusted to the desired pH. The combined buffer system was used to keep the conditions similar throughout the wide pH range used in these assays. Kinetic assays were performed for AtzF, AtzF467, and AtzF H488A using allophanate as the substrate at pH values ranging from 7.0 to 9.0 and temperatures of 28°C and 4°C. To account for the temperature-dependent decrease in AtzF activity, the reaction mixtures in assays conducted at 28°C contained 27 nM enzyme, and the reaction mixtures in assays carried out at 4°C contained 53 nM enzyme. In addition to the enzyme and substrate, each reaction mixture was comprised of 0.5 mM NADH (Sigma-Aldrich), 10 mM α-ketoglutaric acid (Sigma-Aldrich), 1.4 μM bovine serum albumin (Sigma-Aldrich), and 12 μM glutamate dehydrogenase (Sigma-Aldrich). Vmax and the Michaelis-Menten constant were obtained by nonlinear regression analysis and applying a robust fit in GraphPad Prism (version 6) software (GraphPad Software, Inc., CA).
The substrate (allophanate) and product (N-carboxycarbamate) were also analyzed by liquid chromatography-mass spectrometry (LC-MS). Enzyme reaction mixtures were comprised of 53.5 mM potassium allophanate in 100 mM ammonium acetate buffer, pH 9.0. Reaction mixtures contained one of the following: no enzyme, 0.48 μM AtzF, 0.48 μM AtzF468, or 0.96 μM AtzF468. The reactions were carried out on ice. LC-MS analyses were carried out on an Agilent 6550 iFunnel quadrupole time of flight LC-MS system, with samples being introduced by direct injection. The mobile phase was comprised of 10 mM ammonium acetate, pH 9.0, and acetonitrile. Samples were analyzed in negative ion mode, and m/z values ranging from 50 to 150 were scanned.
The effect of pH on enzyme stability was determined by obtaining residual activities after heating AtzF, AtzF467, and AtzF H488A in either pH 7 or pH 9 buffer for 5 min in an Eppendorf Mastercycler EP apparatus (Eppendorf, Hamburg, Germany). The reaction components and method were the same as those used in the kinetic assays, and 2.2 pmol of each protein was used in 100 μl of reaction mixture.
Identification of cyanuric acid mineralization complex.
A culture of Pseudomonas sp. strain ADP was grown at 28°C with cyanuric acid as a sole nitrogen source. The composition of the medium used is as follows: 26.1 mM Na2HPO4·7H2O, 22 mM KH2PO4, 8.5 mM NaCl, 200 μM MgSO4, 2.9 mM sucrose, 3.4 mM trisodium citrate, 4.4 μM CaCl2, 10 mM cyanuric acid, 20 ml vitamin stock, and 1 ml of 1,000× stock solution of trace elements per liter of culture medium. The 1,000× trace elements solution comprised 34.8 μM ZnSO4·7H2O, 15.2 μM MnCl2·4H2O, 485 μM H3BO3, 84 μM CoCl2·2H2O, 5.9 μM CuCl2·2H2O, 12.4 μM Na2MoO4·2H2O, and 8.4 μM NiCl2·6H2O, and the pH of the medium was adjusted to 7.3 by addition of sodium hydroxide. The composition of the vitamin stock solution was 5 mg of thiamine HCl, 2 mg of biotin, 2 mg of folic acid, 10 mg of nicotinamide, and 10 mg of pyridoxine HCl per liter of deionized water, and 20 ml of vitamin stock solution was added per liter of culture medium. The culture medium was made sterile by filtering through 0.2-μm-pore-size filters (VacuCap 60; Pall Life Sciences, NY, USA) before the medium was inoculated with a single colony of Pseudomonas sp. strain ADP that had been grown on nutrient agar plates.
The culture was grown until it reached an optical density at 600 nm of 0.6 to 0.7, after which the cells were harvested by centrifugation at 8,000 × g for 15 min. Harvested cells were resuspended in 50 mM HEPES, 100 mM NaCl (pH 7.5) and lysed using BugBuster protein extraction reagent (Novagen, Darmstadt, Germany) per the manufacturer's instructions. After cell lysis, the soluble fraction was collected by spinning the lysate at 21,000 × g for 15 min. The cell extract was passed through 0.45-μm-pore-size syringe-driven filters (Millex HV, Cork, Ireland), before it was loaded onto a 130-ml size-exclusion column (Superdex 200; GE, Uppsala, Sweden). The activities of AtzD, AtzE, and AtzF in each faction were assessed using 68 μM cyanuric acid and biuret and 560 μM allophanate. The rates of background hydrolysis were subtracted in each case.
The fraction in which the catalytic activities of AtzD, AtzE, and AtzF were detected was resolved by SDS-PAGE (4 to 20% Tris-HEPES-SDS gels; Thermo Scientific, Rockford, IL, USA) (see Fig. S2 in the supplemental material). Certain zones corresponding to the expected locations of AtzD, AtzE, and AtzF were excised from the gel. In-gel tryptic digestion (with the inclusion of ProteaseMAX surfactant [Promega, Madison, WI, USA]) and tandem mass spectral analysis were performed as previously described using an Agilent Chip Cube system coupled to an Agilent XCD ion trap mass spectrometer (57). Mass spectra corresponding to common contaminants, such as the added trypsin and keratin, were identified before the remaining mass spectral data were used to search against a database containing all nonredundant UniProtKB/Swiss-Prot protein sequences from Pseudomonas and the Atz sequences using SpectrumMill software (version Rev A.03.03.084 SR4; Agilent) with its stringent default autovalidation settings.
Protein structure accession number.
The structure identified in this study has been deposited in the Protein Data Bank under accession number 4CP8.
RESULTS AND DISCUSSION
Structure of amidase domain of AtzF.
The X-ray crystal structure for AtzF467 is dimeric (Fig. 3A), consistent with observations from previous studies (30). There are three independent dimers found in the asymmetric unit of the crystal: dimers A-B, C-D, and E-F. The A-B and C-D dimers are significantly more ordered, with average temperature factors (B-factors) of 30 to 33 Å2. The E-F dimer has a weaker density than the other dimers and has an average B-factor about double the average B-factors of the other dimers, 65 to 75 Å2.
FIG 3.

The AtzF structure and comparison to related structures. (A) A cartoon diagram of the dimer of the AtzF structure with the secondary structure colored by magenta for the beta sheet, cyan for alpha helices, and orange for loop structures. AtzF has two main domains: the catalytic domain and a second all-alpha-helical domain that forms the dimer interface. This has been highlighted by coloring these helices in green. (B) The AtzF structure superposed with structures with PDB accession numbers 4ISS and 4GYS. The structure with PDB accession number 4ISS is colored in cyan and has an extra domain which extends away from the rest of the molecule; AtzF is colored green, and the structure with PDB accession number 4GYS is colored magenta. The structures superpose well, despite limited sequence identity, with RMSD values of 1.3 to 1.6 Å. (C) Comparison of the AtzF dimer and the dimers with PDB accession numbers 4ISS and 4GYS. The figure shows how the dimers are similar and how the dimer with the extra domain (PDB accession number 4ISS) helps the dimer formation for this protein. (D) The catalytic site of these proteins. In two cases we see substrate mimetics bound into the catalytic site: in the case of AtzF we see clear density for malonate, whereas for the structure with PDB accession number 4ISS, there is tartrate.
The overall structure is similar to the structures of the K. lactis allophanate hydrolase (AHKl; PDB accession number 4ISS) and the N terminus of the G. bethesdensis allophanate hydrolase (AHGb; PDB accession number 4GYS) and to the structures of other Ser–cis-Ser–Lys hydrolases, such as the amidase subunit of the Staphylococcus aureus glutamine amidotransferase (PDB accession number 2DQN) (48) used for molecular replacement in this study. The structure with PDB accession number 2DQN has a root mean square deviation (RMSD) of 1.8 Å with 16 gaps, and its sequence has less than 30% identity to the AtzF sequence. The dimer interface buries approximately 1,150 Å2 of surface per monomer (Fig. 2). The dimer interface is essentially the same as that found in AHKl (PDB accession number 4ISS) and AHGb, except that the AtzF structure is missing the C-terminal domain, which forms a separate interface between the two monomers in AHKl (Fig. 3A).
Superposition of the AtzF and AHKl dimers (Fig. 3B and C) gives an RMSD of 1.6 Å for 851 residues (out of 895 and 1,226 residues for the AtzF and AHKl dimers, respectively) with 21 gaps (superpositioning was done with the secondary-structure matching (SSM) algorithm implemented in Coot software). A comparison with the structure of AHGb gives an RMSD of 1.3 Å for 857 residues (out of 895 and 921 residues for the AtzF and AHGb dimers, respectively) with 15 gaps (Fig. 3B and C). The two structures are very similar, with only a slight shift in helices 2 and 3 at the N terminus and a C-terminal extension of residues 444 to 462 in AHGb that is not seen in the AtzF structure due to the difference in the position of the truncations of each of the proteins. The only other region of significant deviation between the structures is in a mobile loop region between residues 255 and 268 of AtzF (residues 238 to 247 of AHGb). Notably, the structure used as the molecular replacement model, the structure with PDB accession number 2DQN, also possessed differences from AtzF similar to those shown by AHGb, in addition to an extended loop (residues 322 to 348 in the structure with PDB accession number 2DQN and residues 333 to 340 in AtzF). The active sites of AtzF, AHGb, and AHKl are also essentially identical to the positions of the amino acids essential for catalysis (Ser165, Ser189, and Lys91) and substrate binding (Tyr320 and Arg328), conserved between the three enzymes (Fig. 3D).
SAXS data (Fig. 4A to F) indicate that purified, full-length AtzF is a homotetramer in solution (Fig. 4A), rather than a hexamer, as size-exclusion chromatography (SEC) had previously suggested (31, 45). This is evident both from the fit of the data to a model of tetrameric AtzF and from the molecular weight of the complex calculated from the SAXS data (see Table S1 in the supplemental material). While there is a reasonable agreement overall between the tetrameric model of AtzF and the SAXS data, it can be seen that there is some divergence between the two at a low angle and around an oscillation in the data at about 0.1 Å−1. This is most likely explained by some conformational flexibility in the C-terminal domains of the complex, though modeling of this structural feature is beyond the scope of this study. The tetramer itself is a cylinder of sorts that has dimensions of about 180 by 115 by 60 Å. The length of the long axis of the tetramer (180 Å) is longer than might be expected for a tetramer with the molecular weight of AtzF, and this is possibly the cause of this inconsistency between SEC and the more accurate observations from SAXS.
FIG 4.

Analysis of the multimerization of AtzF and Atzf467 by SAXS. (A) High-resolution model of an AtzF tetramer (green and blue) superimposed on a dummy atom model derived from the SAXS data (gray density). (B) High-resolution model of dimeric AtzF467 (green and blue) superimposed on a dummy atom model derived from the SAXS data (gray density). (C) Fit between the SAXS data and the AtzF tetramer model shown in panel A calculated using CRYSOL. (D) Fit between the measured AtzF467 protein and a model of tetrameric AtzF467 based on the AtzF tetramer model calculated using CRYSOL. (E) Fit between dimeric AtzF and the measured AtzF SAXS data calculated using CRYSOL. (F) Fit between the dimeric AtzF467 model shown in panel B and the measured AtzF467 SAXS data.
Previously, we had shown that removal of the C terminus of AtzF resulted in a 48-kDa protein (AtzF467) which was kinetically indistinguishable from AtzF (45). However, the removal of the C-terminal domain altered the quaternary structure of AtzF, as indicated by the reduction in apparent molecular mass of the enzyme during SEC from ∼360 kDa for the wild-type protein to ∼110 kDa (dimeric) for the truncated protein (45). SAXS of the amidase domain of AtzF (AtzF467) confirmed that the truncated variant is indeed a dimer in solution (Fig. 4B). These observations suggest that the C terminus of AtzF is required for homotetramer formation but not homodimer formation. In contrast to the data presented by Fan et al. for the AHKl protein (37), which they showed was a monomer in solution when the C-terminal domain was removed (or disrupted by mutations), we found that the truncated AtzF migrates as a dimer in solution, as shown by both SAXS and chromatographic (SEC) data. As the dimeric interfaces are similar between the two proteins, it is not clear why the truncated form of AHKl would behave differently from the truncated form of AtzF.
It is interesting that the kinetic parameters for AtzF characterized here are different from those reported by Shapir et al. in 2005 (35); the kcat and Km values for AtzF reported here are ∼4 to 13 s−1 and ∼120 to 300 μM, respectively (see Table S2 in the supplemental material), whereas they were 16 s−1 and 1,500 μM, respectively, in the previous study (35). While it is unclear why there is such a large difference in Km, it is noteworthy that the lowest substrate concentration tested in the study of Shapir et al. (34) was 200 μM (i.e., close to the Km determined in the work reported here). It is also of note that the purity of the allophanate used in the work by Shapir et al. (35) was not reported, and so the reported value of 150 μM for the Km reflects the highest possible Km (assuming 100% substrate purity).
There is no catalytic advantage conferred by the C terminus of AtzF in vitro.
The N-terminal domain of allophanate hydrolase deaminates allophanate to produce ammonia and N-carboxycarbamate (Fig. 1). The role of the smaller C-terminal domain is less clear, however. Fan et al. (37) suggested that the C-terminal domain of AHKl is catalytic, decarboxylating N-carboxycarbamate to form carbamate and carbon dioxide. On the basis of in silico substrate docking and mutagenesis studies, it was proposed that a histidine residue in the C-terminal domain (His492; Fig. 2) acts as a catalytic residue. If this is also true for AtzF, removal of the C-terminal domain or replacement of the catalytic histidine by alanine would be expected to reduce the rate of ammonia production by AtzF and the accumulation of the intermediate (N-carboxycarbamate) during the reaction.
We were unable to detect N-carboxycarbamate accumulation in reactions catalyzed by either the full-length AtzF or AtzF467 by LC-MS analysis (see Fig. S2 in the supplemental material), which may indicate that the C terminus of AtzF is not catalytic. However, N-carboxycarbamate is highly unstable in an aqueous environment, decaying by spontaneous decarboxylation to form carbamate and carbon dioxide. As the stability of carbamates is known to be temperature and pH dependent (58), the pH of the AtzF-catalyzed reactions was raised to 9 and the temperature was reduced to 4°C. Despite the conditions favoring the accumulation of N-carboxycarbamate, we were unable to detect this product in reactions catalyzed by either full-length AtzF or AtzF467. Although the absence of the accumulation of N-carboxycarbamate in reactions is not direct evidence for a lack of the hypothesized catalytic activity in the C terminus, it does suggest that such a function would have little physiological relevance.
To be sure that our inability to detect N-carboxycarbamate in the reactions was due to the absence of this reaction intermediate rather than a flaw in our detection method, we also assessed the rate of ammonia production by full-length AtzF or AtzF467 and by an AtzF variant in which the catalytic histidine residue had been replaced by an alanine residue. The enzymes were tested at a range of pHs and temperatures, to account for the difference in N-carboxycarbamate stability at different temperatures and pHs.
For reactions at 28°C, the full-length AtzF was indistinguishable from the two variants at pH values of 8 and below. However, at pH values above 8, the full-length enzyme had kcat values greater than those for either AtzF467 or AtzF H488A. The difference for AtzF H488A was slight, but the reduction in activity for AtzF467 was considerable (Fig. 5A). For reactions at 4°C, the kcat value of full-length AtzF was indistinguishable from those of the two variants at pH values of 7.5 and below; however, at pH values above 7.5, the full-length enzymes had kcat values greater than those for AtzF467, albeit the effect on the catalytic rate was less extensive than that at 28°C. At 4°C, there was no difference in the kcat values for AtzF and the AtzF H488A variant (Fig. 5B).
FIG 5.

Activity of AtzF (and fragments) at different pHs and temperatures. kcat values at different pH levels obtained at 28°C (A) and 4°C (B) for AtzF, AtzF467, and AtzF H488A were compared. All the data points plotted had standard errors of less than 5.3%, and standard errors are presented in Table S2 in the supplemental material.
If the differences in apparent kcat values were solely a result of a reduction in catalytic efficiency, it would be expected that the AtzF H488A and AtzF467 variants would be indistinguishable from each other, which they are not. Moreover, the effect on the catalytic rate in the variants is reduced at low temperatures, where the stabilization of N-carboxycarbamate should make the effects more pronounced and not less so. Indeed, the kcat values of AtzF H488A were essentially identical to those of the wild-type AtzF protein at 4°C.
As the pH-dependent differences between full-length AtzF and its variants were more pronounced at room temperature than at 4°C, it is possible that they are a result of differences in the thermal stability of the three enzymes. The residual activities of AtzF, AtzF467, and AtzF H488A were therefore tested after incubation for 5 min at temperatures between 30°C and 70°C and were assayed at pH 7, where no difference in the apparent kcat values for the three enzymes had been observed, and pH 9, where differences in apparent kcat values for the three enzymes had been observed (Fig. 6A and B).
FIG 6.

Effect of pH on residual activity of AtzF and its variants. Differences in the residual activity of AtzF, AtzF467, and AtzF H488A at pH 7 (A) and pH 9 (B) were compared. Enzymes were incubated in pH 7 or pH 9 buffers for 5 min at temperatures ranging from 30 to 70°C before testing for residual activity.
At pH 7, there was no significant difference in the residual activities recovered for AtzF, AtzF467, and AtzF H488A, but at pH 9, the apparent melting temperature (Tm) for AtzF467 was reduced from ∼44°C to ∼41°C, and although the apparent Tm for AtzF H488A was ∼45°C, it was lower than that for AtzF by nearly 1.5°C. It is therefore plausible that at least a component of the pH-dependent difference in apparent kcat values was a result of a pH-dependent reduction in stability for AtzF when the C terminus was removed or H488 was mutated to an alanine.
Regardless of the rate differences observed at pHs higher than the physiological pH (i.e., 6.5 to 7.5), we were unable to observe a difference in the rate of ammonia release between the wild-type and variant AtzF enzymes tested in vitro under physiological pH conditions.
AtzF appears to form a large complex with AtzD and AtzE.
Allophanate is unstable and has been reported to decompose rapidly to form urea and carbon dioxide under physiological conditions (Fig. 1) (30). For both the ureolytic pathway and triazine degradation pathway, the spontaneous decomposition of allophanate is undesirable, causing an ATP-consuming futile cycle for the ureolytic enzymes and forming a dead-end product in triazine hydrolysis that results in the metabolic loss of two of the three nitrogens of the heterocycle. It is likely that the formation of a urea carboxylase/allophanate hydrolase complex in the ureolytic system protects allophanate from decarboxylation by channeling the substrate from one active site to another. It is reasonable to assume that a similar relationship may exist between AtzF and the biuret hydrolase, AtzE, which prevents the production of urea. As AtzF is a relatively inefficient enzyme, with kcat and Km values of 8 s−1 and 120 μM, respectively (see Table S2 in the supplemental material), it is likely that the nitrogen from the degradation of the triazine ring is lost to the competing abiotic decarboxylation rather than channeled through allophanate hydrolase in the absence of the formation of a substrate-channeling complex.
SEC was used to fractionate a Pseudomonas sp. strain ADP cell extract obtained from a culture that had been induced for AtzD, AtzE, and AtzF production by incubation in a medium containing cyanuric acid as the sole carbon source. AtzD, AtzE, and AtzF activities were found to coelute in a fraction with an estimated molecular mass of ∼660 kDa. The activities of these three enzymes were not found in other fractions. This observation is consistent with a complex that consists of four AtzD monomers, four AtzF monomers, and two AtzE monomers, albeit deriving the stoichiometry of the three enzymes from SEC alone may be misleading.
The SEC fraction with AtzD, AtzE, and AtzF activities was resolved by SDS-PAGE and with gel sections taken from where the three proteins should migrate (on the basis of molecular mass; see Fig. S3 in the supplemental material). LC-MS of the tryptic digestion products from these sections showed that peptides that were consistent with a tryptic fragmentation pattern of AtzD, AtzE, and AtzF were present. The sequence coverage for AtzD, AtzE, and AtzF was 21% by 9 unique peptides, 25% by 9 unique peptides, and 3% by 2 unique peptides, respectively. Unsurprisingly, other proteins identified in the samples were abundant cellular components of Pseudomonas, such as translation elongation factors (GI 169761912, GI 169761911, and GI 169760553) and heat shock proteins (GI 169760669). When considered along with the enzyme activity data from SEC, these data support the presence of a large protein complex consisting of AtzD, AtzE, and AtzF.
Production of ammonia was observed upon addition of cyanuric acid to this fraction, with 3 mol of ammonia being produced for every mole of cyanuric acid added, suggesting that the complex mineralized cyanuric acid. Interestingly, the rate of ammonia production was much greater upon addition of cyanuric acid than it was when biuret (the AtzE substrate) was added (Table 3), which may imply that access to the AtzE active site from solvent may be restricted and passage of substrate from the AtzD active site to the AtzE active site may occur via substrate tunneling or channeling. If the AtzD product (1-carboxybiuret; Fig. 1) is protected from extended contact with the solvent, it is possible that AtzE is a 1-carboxybiuret hydrolase, rather than a biuret hydrolase. Unfortunately, despite considerable effort, we were unable to produce soluble AtzE in a quantity sufficient to reconstruct the complex in vitro for further study and so were unable to probe this hypothesis further.
TABLE 3.
Rates of ammonia release by the SEC fraction containing AtzD, AtzE, and AtzF activitiesa
| Substrate | Substrate/ammonia ratio | Rate of ammonia release (nM/s) |
|---|---|---|
| Cyanuric acid | 3.2 | 45.4 |
| Biuret | ND | 1 |
| Allophanate | 1.1 | 180 |
Cyanuric acid, biuret, and allophanate were added as substrates. The SEC fraction was estimated to contain proteins with a molecular mass of ∼660 kDa. ND, the stoichiometry of substrate to ammonia was not determined for biuret, because of the low rate of ammonia production.
Conclusion.
AtzF is a component of a large (∼0.7-MDa) cyanuric acid-mineralizing complex, comprised of AtzD, AtzE, and AtzF. We postulate that this complex protects the unstable intermediate allophanate from spontaneous decarboxylation under physiological conditions by substrate channeling and/or by precluding water from reaching the reactive centers, strategies used in other enzymes that catalyze water-sensitive reactions, such as carbamoyl phosphate synthase and the GatCAB glutamine amidotransferase (48). Interestingly, ureolytic allophanate hydrolases also form complexes with urea carboxylase. This suggests either that allophanate hydrolases have evolved to form multiprotein complexes more than once or that they have switched partners over the course of their evolution.
The biochemical data that we have presented (accumulation of N-carboxycarbamate and the rate of ammonia production by AtzF and its variants) fail to support a physiologically relevant catalytic function for the C terminus of AtzF. It is possible that the proposed N-carboxycarbamate decarboxylase activity is relevant under conditions that stabilize N-carboxycarbamate, such as a water-limited environment. These conditions could be present in the cyanuric acid-mineralizing complex, albeit further work will be required to elucidate structure-function relationships within the complex.
Supplementary Material
ACKNOWLEDGMENTS
We thank the beamline scientists of the Australian Synchrotron and the CSIRO Collaborative Crystallisation Centre, Melbourne, Australia. We also thank Chris Blake (Research School of Chemistry, Australian National University) for his help with the NMR studies and Carol Hartley and Robyn Russell (CSIRO) for helpful discussions.
We do not have a conflict of interest to declare.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02783-14.
REFERENCES
- 1.Tomlin C. (ed). 2006. The pesticide manual: a world compendium, 14th ed. BCPC, Alton, United Kingdom. [Google Scholar]
- 2.Thurman EM, Meyer MT. 1996. Herbicide metabolites in surface water and groundwater: introduction and overview, p 1–15. In Meyer MT, Thurman EM (ed), Herbicide metabolites in surface water and groundwater, vol 630 American Chemical Society, Washington, DC. [Google Scholar]
- 3.Gavrilescu M. 2005. Fate of pesticides in the environment and its bioremediation. Eng Life Sci 5:497–526. doi: 10.1002/elsc.200520098. [DOI] [Google Scholar]
- 4.Hayes TB, Khoury V, Narayan A, Nazir M, Park A, Brown T, Adame L, Chan E, Buchholz D, Stueve T, Gallipeau S. 2010. Atrazine induces complete feminization and chemical castration in male African clawed frogs (Xenopus laevis). Proc Natl Acad Sci U S A 107:4612–4617. doi: 10.1073/pnas.0909519107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hayes T, Haston K, Tsui M, Hoang A, Haeffele C, Vonk A. 2003. Atrazine-induced hermaphroditism at 0.1 ppb in American leopard frogs (Rana pipiens): laboratory and field evidence. Environ Health Perspect 111:568–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hayes TB, Collins A, Lee M, Mendoza M, Noriega N, Stuart AA, Vonk A. 2002. Hermaphroditic, demasculinized frogs after exposure to the herbicide atrazine at low ecologically relevant doses. Proc Natl Acad Sci U S A 99:5476–5480. doi: 10.1073/pnas.082121499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Udikovic-Kolic N, Scott C, Martin-Laurent F. 2012. Evolution of atrazine-degrading capabilities in the environment. Appl Microbiol Biotechnol 96:1175–1189. doi: 10.1007/s00253-012-4495-0. [DOI] [PubMed] [Google Scholar]
- 8.Russell RJ, Scott C, Jackson CJ, Pandey R, Pandey G, Taylor MC, Coppin CW, Liu JW, Oakeshott JG. 2011. The evolution of new enzyme function: lessons from xenobiotic metabolizing bacteria versus insecticide-resistant insects. Evol Appl 4:225–248. doi: 10.1111/j.1752-4571.2010.00175.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shapir N, Mongodin EF, Sadowsky MJ, Daugherty SC, Nelson KE, Wackett LP. 2007. Evolution of catabolic pathways: genomic insights into microbial s-triazine metabolism. J Bacteriol 189:674–682. doi: 10.1128/JB.01257-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wackett LP. 2004. Evolution of enzymes for the metabolism of new chemical inputs into the environment. J Biol Chem 279:41259–41262. doi: 10.1074/jbc.R400014200. [DOI] [PubMed] [Google Scholar]
- 11.Noor S, Changey F, Oakeshott JG, Scott C, Martin-Laurent F. 2014. Ongoing functional evolution of the bacterial atrazine chlorohydrolase AtzA. Biodegradation 25:21–30. doi: 10.1007/s10532-013-9637-2. [DOI] [PubMed] [Google Scholar]
- 12.Noor S, Taylor MC, Russell RJ, Jermiin LS, Jackson CJ, Oakeshott JG, Scott C. 2012. Intramolecular epistasis and the evolution of a new enzymatic function. PLoS One 7:e39822. doi: 10.1371/journal.pone.0039822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raillard S, Krebber A, Chen YC, Ness JE, Bermudez E, Trinidad R, Fullem R, Davis C, Welch M, Seffernick J, Wackett LP, Stemmer WPC, Minshull J. 2001. Novel enzyme activities and functional plasticity revealed by recombining highly homologous enzymes. Chem Biol 8:891–898. doi: 10.1016/S1074-5521(01)00061-8. [DOI] [PubMed] [Google Scholar]
- 14.Seffernick JL, de Souza ML, Sadowsky MJ, Wackett LP. 2001. Melamine deaminase and atrazine chlorohydrolase: 98 percent identical but functionally different. J Bacteriol 183:2405–2410. doi: 10.1128/JB.183.8.2405-2410.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peat TS, Balotra S, Wilding M, French NG, Briggs LJ, Panjikar S, Cowieson N, Newman J, Scott C. 2013. Cyanuric acid hydrolase: evolutionary innovation by structural concatenation. Mol Microbiol 88:1149–1163. doi: 10.1111/mmi.12249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lima D, Viana P, Andre S, Chelinho S, Costa C, Ribeiro R, Sousa JP, Fialho AM, Viegas CA. 2009. Evaluating a bioremediation tool for atrazine contaminated soils in open soil microcosms: the effectiveness of bioaugmentation and biostimulation approaches. Chemosphere 74:187–192. doi: 10.1016/j.chemosphere.2008.09.083. [DOI] [PubMed] [Google Scholar]
- 17.Scott C, Lewis SE, Milla R, Taylor MC, Rodgers AJW, Dumsday G, Brodie JE, Oakeshott JG, Russell RJ. 2010. A free-enzyme catalyst for the bioremediation of environmental atrazine contamination. J Environ Manage 91:2075–2078. doi: 10.1016/j.jenvman.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 18.Scott C, Pandey G, Hartley CJ, Jackson CJ, Cheesman MJ, Taylor MC, Pandey R, Khurana JL, Teese M, Coppin CW, Weir KM, Jain RK, Lal R, Russell RJ, Oakeshott JG. 2008. The enzymatic basis for pesticide bioremediation. Indian J Microbiol 48:65–79. doi: 10.1007/s12088-008-0007-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Topp E, Martin-Laurent F, Hartmann A, Soulas G. 2004. Bioremediation of atrazine-contaminated soil, p 141–154. In Gan JJ, Zhu PC, Aust SD, Lemley AT (ed), Pesticide decontamination and detoxification, vol 863 American Chemical Society, Washington, DC. [Google Scholar]
- 20.de Souza ML, Wackett LP, Boundymills KL, Mandelbaum RT, Sadowsky MJ. 1995. Cloning, characterization and expression of a gene region from Pseudomonas sp. strain ADP involved in the dechlorination of atrazine. Appl Environ Microbiol 61:3373–3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Souza ML, Sadowsky MJ, Wackett LP. 1996. Atrazine chlorohydrolase from Pseudomonas sp. strain ADP: gene sequence, enzyme purification, and protein characterization. J Bacteriol 178:4894–4900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boundy-Mills KL, deSouza ML, Mandelbaum RT, Wackett LP, Sadowsky MJ. 1997. The atzB gene of Pseudomonas sp. strain ADP encodes the second enzyme of a novel atrazine degradation pathway. Appl Environ Microbiol 63:916–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seffernick JL, Aleem A, Osborne JP, Johnson G, Sadowsky MJ, Wackett LP. 2007. Hydroxyatrazine N-ethylaminohydrolase (AtzB): an amidohydrolase superfamily enzyme catalyzing deamination and dechlorination. J Bacteriol 189:6989–6997. doi: 10.1128/JB.00630-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sadowsky MJ, Tong ZK, de Souza M, Wackett LP. 1998. AtzC is a new member of the amidohydrolase protein superfamily and is homologous to other atrazine-metabolizing enzymes. J Bacteriol 180:152–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shapir N, Osborne JP, Johnson G, Sadowsky MJ, Wackett LP. 2002. Purification, substrate range, and metal center of AtzC: the N-isopropylammelide aminohydrolase involved in bacterial atrazine metabolism. J Bacteriol 184:5376–5384. doi: 10.1128/JB.184.19.5376-5384.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fruchey I, Shapir N, Sadowsky MJ, Wackett LP. 2003. On the origins of cyanuric acid hydrolase: purification, substrates, and prevalence of AtzD from Pseudomonas sp. strain ADP. Appl Environ Microbiol 69:3653–3657. doi: 10.1128/AEM.69.6.3653-3657.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seffernick JL, Erickson JS, Cameron SM, Cho S, Dodge AG, Richman JE, Sadowsky MJ, Wackett LP. 2012. Defining sequence space and reaction products within the cyanuric acid hydrolase (AtzD)/barbiturase protein family. J Bacteriol 194:4579–4588. doi: 10.1128/JB.00791-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cameron SM, Durchschein K, Richman JE, Sadowsky MJ, Wackett LP. 2011. New family of biuret hydrolases involved in s-triazine ring metabolism. ACS Catal 1:1075–1082. doi: 10.1021/cs200295n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shapir N, Cheng G, Sadowsky MJ, Wackett LP. 2006. Purification and characterization of TrzF: biuret hydrolysis by allophanate hydrolase supports growth. Appl Environ Microbiol 72:2491–2495. doi: 10.1128/AEM.72.4.2491-2495.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheng G, Shapir N, Sadowsky MJ, Wackett LP. 2005. Allophanate hydrolase, not urease, functions in bacterial cyanuric acid metabolism. Appl Environ Microbiol 71:4437–4445. doi: 10.1128/AEM.71.8.4437-4445.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shapir N, Sadowsky MJ, Wackett LP. 2005. Purification and characterization of allophanate hydrolase (AtzF) from Pseudomonas sp. strain ADP. J Bacteriol 187:3731–3738. doi: 10.1128/JB.187.11.3731-3738.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jackson CJ, Coppin CW, Carr PD, Aleksandrov A, Wilding M, Sugrue E, Ubels J, Paks M, Newman J, Peat TS, Russell RJ, Field M, Weik M, Oakeshott JG, Scott C. 2014. 300-fold increase in production of the Zn2+-dependent dechlorinase TrzN in soluble form via apoenzyme stabilization. Appl Environ Microbiol 80:4003–4011. doi: 10.1128/AEM.00916-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seffernick JL, Reynolds E, Fedorov AA, Fedorov E, Almo SC, Sadowsky MJ, Wackett LP. 2010. X-ray structure and mutational analysis of the atrazine chlorohydrolase TrzN. J Biol Chem 285:30606–30614. doi: 10.1074/jbc.M110.138677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shapir N, Pedersen C, Gil O, Strong L, Seffernick J, Sadowsky MJ, Wackett LP. 2006. TrzN from Arthrobacter aurescens TC1 is a zinc amidohydrolase. J Bacteriol 188:5859–5864. doi: 10.1128/JB.00517-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shapir N, Rosendahl C, Johnson G, Andreina M, Sadowsky MJ, Wackett LP. 2005. Substrate specificity and colorimetric assay for recombinant TrzN derived from Arthrobacter aurescens TC1. Appl Environ Microbiol 71:2214–2220. doi: 10.1128/AEM.71.5.2214-2220.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mulbry WW, Zhu H, Nour SM, Topp E. 2002. The triazine hydrolase gene trzN from Nocardioides sp. strain C190: cloning and construction of gene-specific primers. FEMS Microbiol Lett 206:75–79. doi: 10.1111/j.1574-6968.2002.tb10989.x. [DOI] [PubMed] [Google Scholar]
- 37.Fan C, Li Z, Yin HY, Xiang S. 2013. Structure and function of allophanate hydrolase. J Biol Chem 288:21422–21432. doi: 10.1074/jbc.M113.453837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin Y, St Maurice M. 2013. The structure of allophanate hydrolase from Granulibacter bethesdensis provides insights into substrate specificity in the amidase signature family. Biochemistry 52:690–700. doi: 10.1021/bi301242m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Whitney PA, Cooper TG, Magasani B. 1973. Induction of urea carboxylase and allophanate hydrolase in Saccharomyces cerevisiae. J Biol Chem 248:6203–6209. [PubMed] [Google Scholar]
- 40.Whitney PA, Cooper TG. 1972. Urea carboxylase and allophanate hydrolase—2 components of a multienzyme complex in Saccharomyces cerevisiae. Biochem Biophys Res Commun 49:45–51. doi: 10.1016/0006-291X(72)90007-1. [DOI] [PubMed] [Google Scholar]
- 41.Whitney PA, Cooper TF. 1972. Urea carboxylase and allophanate hydrolase—2 components of adenosine triphosphate-urea amidohydrolase in Saccharomyces cerevisiae. J Biol Chem 247:1349–1353. [PubMed] [Google Scholar]
- 42.Kanamori T, Kanou N, Kusakabe S, Atomi H, Imanaka T. 2005. Allophanate hydrolase of Oleomonas sagaranensis involved in an ATP-dependent degradation pathway specific to urea. FEMS Microbiol Lett 245:61–65. doi: 10.1016/j.femsle.2005.02.023. [DOI] [PubMed] [Google Scholar]
- 43.Shin S, Yun YS, Koo HM, Kim YS, Choi KY, Oh BH. 2003. Characterization of a novel Ser-cisSer-Lys catalytic triad in comparison with the classical Ser-His-Asp triad. J Biol Chem 278:24937–24943. doi: 10.1074/jbc.M302156200. [DOI] [PubMed] [Google Scholar]
- 44.Shin S, Lee TH, Ha NC, Koo HM, Kim SY, Lee HS, Kim YS, Oh BH. 2002. Structure of malonamidase E2 reveals a novel Ser-cisSer-Lys catalytic triad in a new serine hydrolase fold that is prevalent in nature. EMBO J 21:2509–2516. doi: 10.1093/emboj/21.11.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Balotra S, Newman J, French NG, Briggs LJ, Peat TS, Scott C. 2014. Crystallization and preliminary X-ray diffraction analysis of the amidase domain of allophanate hydrolase from Pseudomonas sp. strain ADP. Acta Crystallogr F Struct Biol Cryst Commun 70:310–315. doi: 10.1107/S2053230X13034705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain-reaction. Gene 77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 47.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J Appl Crystallogr 40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nakamura A, Yao M, Chimnaronk S, Sakai N, Tanaka I. 2006. Ammonia channel couples glutaminase with transamidase reactions in GatCAB. Science 312:1954–1958. doi: 10.1126/science.1127156. [DOI] [PubMed] [Google Scholar]
- 49.Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66(Pt 4):486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. 2011. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr 67(Pt 4):355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI. 2003. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J Appl Crystallogr 36:1277–1282. doi: 10.1107/S0021889803012779. [DOI] [Google Scholar]
- 52.Svergun D, Barberato C, Koch MHJ. 1995. CRYSOL—a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J Appl Crystallogr 28:768–773. doi: 10.1107/S0021889895007047. [DOI] [Google Scholar]
- 53.Franke D, Svergun DI. 2009. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J Appl Crystallogr 42:342–346. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kozin MB, Svergun DI. 2001. Automated matching of high- and low-resolution structural models. J Appl Crystallogr 34:33–41. doi: 10.1107/S0021889800014126. [DOI] [Google Scholar]
- 55.Mylonas E, Svergun DI. 2007. Accuracy of molecular mass determination of proteins in solution by small-angle X-ray scattering. J Appl Crystallogr 40:S245–S249. doi: 10.1107/S002188980700252X. [DOI] [Google Scholar]
- 56.Whitten AE, Cai SZ, Trewhella J. 2008. MULCh: modules for the analysis of small-angle neutron contrast variation data from biomolecular assemblies. J Appl Crystallogr 41:222–226. doi: 10.1107/S0021889807055136. [DOI] [Google Scholar]
- 57.Campbell PM, Trueman HE, Zhang Q, Kojima K, Kameda T, Sutherland TD. 2014. Cross-linking in the silks of bees, ants and hornets. Insect Biochem Mol Biol 48:40–50. doi: 10.1016/j.ibmb.2014.02.009. [DOI] [PubMed] [Google Scholar]
- 58.Frahn JL, Mills JA. 1964. Paper ionophoresis of amino compounds: formation of carbabmates and related reactions. Aust J Chem 17:256–260. doi: 10.1071/CH9640256. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.