

Abstract
Methanogenic archaea play a key role in biogas-producing anaerobic digestion and yet remain poorly taxonomically characterized. This is in part due to the limitations of low-throughput Sanger sequencing of a single (16S rRNA) gene, which in the past may have undersampled methanogen diversity. In this study, archaeal communities from three sludge digesters in Hong Kong and one wastewater digester in China were examined using high-throughput pyrosequencing of the methyl coenzyme M reductase (mcrA) and 16S rRNA genes. Methanobacteriales, Methanomicrobiales, and Methanosarcinales were detected in each digester, indicating that both hydrogenotrophic and acetoclastic methanogenesis was occurring. Two sludge digesters had similar community structures, likely due to their similar design and feedstock. Taxonomic classification of the mcrA genes suggested that these digesters were dominated by acetoclastic methanogens, particularly Methanosarcinales, while the other digesters were dominated by hydrogenotrophic Methanomicrobiales. The proposed euryarchaeotal order Methanomassiliicoccales and the uncultured WSA2 group were detected with the 16S rRNA gene, and potential mcrA genes for these groups were identified. 16S rRNA gene sequencing also recovered several crenarchaeotal groups potentially involved in the initial anaerobic digestion processes. Overall, the two genes produced different taxonomic profiles for the digesters, while greater methanogen richness was detected using the mcrA gene, supporting the use of this functional gene as a complement to the 16S rRNA gene to better assess methanogen diversity. A significant positive correlation was detected between methane production and the abundance of mcrA transcripts in digesters treating sludge and wastewater samples, supporting the mcrA gene as a biomarker for methane yield.
INTRODUCTION
Methane from anaerobic digestion is an important source of renewable energy that can circumvent the problems of dwindling fossil fuels and of atmospheric carbon dioxide (CO2) emissions due to fossil fuel combustion (1). Anaerobic digestion is becoming a key part of municipal wastewater treatment, as it allows recovery of energy (biogas) from waste streams to offset on-site energy consumption. The anaerobic digestion of heterogeneous organic substrates for methane production is a complex process involving four major sequential phases: hydrolysis, fermentation, acetogenesis, and methanogenesis (2). Methanogens are strictly anaerobic archaea that produce methane from a limited number of substrates, including hydrogen (H2), acetate, and some C1 compounds (2). Phylogenetically, methanogens belong to the Euryarchaeota with six established (Methanobacteriales, Methanococcales, Methanomicrobiales, Methanocellales, Methanopyrales, and Methanosarcinales) and one proposed (Methanomassiliicoccales) order(s) (3) and at least 31 genera (4). As well as being major functional components of anaerobic digester communities (5, 6), methanogens are found in other anoxic environments such as peatlands (7), landfills (8), rice paddy fields (9), and ruminant gut (10), all of which emit methane to the atmosphere. The diversity and abundance of methanogens in anaerobic digesters are critical to operating efficiency, since methanogenesis is usually the rate-limiting step (2). Evidence from the better-characterized bacterial component of digester communities suggests that a high level of functional redundancy despite variable taxonomic composition may be an important feature of these communities (11, 12). However, many of the major methanogen groups in anaerobic digesters remain unknown or poorly understood (5), and variation between digesters has been little examined.
Methanogen phylogeny can be determined by sequencing the 16S rRNA gene using archaea-specific (13) or methanogen-specific (14) primers, and/or the α subunit of the methyl coenzyme M reductase (mcrA) gene (14, 15). mcrA encodes the enzyme catalyzing the terminal step in methanogenesis and is ubiquitous among known methanogens (15). Methanogen phylogenies determined using the mcrA and 16S rRNA genes are largely congruent (8, 15), and both genes have been used to elucidate the diversity and phylogeny of methanogens in anaerobic digesters (14–17). Methanogens in mixed communities have traditionally been investigated either by cultivation-dependent methods (18, 19) or Sanger sequencing of 16S rRNA or mcrA gene clone libraries (14, 20). Thus far, pyrosequencing of the mcrA gene has only seen limited application, e.g., to examine methanogens in river sediments (21) and an algal fed anaerobic digester (22), and this method has been underutilized relative to the potential taxonomic diversity. An improved taxonomic understanding of the methanogens involved will aid the optimization of anaerobic digesters to increase methane yield and other industrially important parameters.
In the present study, we report the taxonomic diversity of methanogens in four full-scale anaerobic digesters by analyzing the mcrA and archaeal 16S rRNA genes with 454 pyrosequencing. Of the four digesters, three treat fresh or saline municipal sludge, and one treats industrial organic wastewater. Digesters with different operating conditions and input streams were selected to examine methanogen composition across a broad range of digester types. In addition, the correlation between mcrA transcription and methane production is experimentally assessed to evaluate the applicability of mcrA expression as a biomarker for methanogenesis.
MATERIALS AND METHODS
Sample collection and DNA extraction.
Samples were collected during October and November 2011 from three full-scale municipal anaerobic digesters that treat sludge following secondary treatment, located at Sha Tin (ST), Shek Wu Hui (SWH), and Yuen Long (YL) in Hong Kong, and from one industrial anaerobic digester that treats organic wastewater from a beverage manufacturing company located in Guangzhou (GZ), China. Detailed descriptions of the four digesters, including operational parameters measured by the plant operators using standard methods (23), are provided in Table 1. Multiple sludge samples were collected from each digester while the system was in stable operation. Samples for molecular analysis were centrifuged at 6,200 × g for 10 min at 4°C and stored at −80°C until processing. Samples for cultivation experiments were kept at 35°C and used as inocula within 48 to 72 h. Genomic DNA (gDNA) was extracted as described previously (24). Briefly, two independently collected ∼250-mg sludge samples from each digester were pooled and DNA extracted with a PowerSoil DNA extraction kit (MoBio Laboratories, CA). The final DNA concentration was ∼100 ng/μl, and the A260/A280 ratio was ∼1.90.
TABLE 1.
Operating conditions and performance of the digesters analyzed in this study
| Parametera | Treatment plantb |
|||
|---|---|---|---|---|
| ST | SWH | YL | GZ | |
| Salinity (ppt) | 9.7 | 0 | 0 | 0 |
| Daily methane production (m3) | 9,663 | 2,476 | 455 | 211 |
| Daily vol processed (m3) | 1,910 | 453 | 163 | 700 |
| Operating temp (°C) | 32−37 | 34−37 | 33−35 | 27−28 |
| pHc | 6.6 ± 0.1 | 7.2 ± 0.1 | 6.5 | 7.2 ± 0.2 |
| Retention time (days) | 11 | 23 | 40 | 0.5 |
| Removal of total solids (%) | 27 | 47 | 62 | |
| Removal of volatile solids (%) | 24 | 11 | 12 | |
| Removal of chemical oxygen demand (%) | 87.6 | |||
ppt, parts per thousand. Daily methane production measurements were obtained at ambient temperature and pressure.
ST, SWH, and YL are located in Hong Kong and treat sludge from secondary treatment; GZ is in Guangzhou, China, and treats organic wastewater. The typical operating parameters are shown. Removal percentages are averages of 1 month of data before and after sampling as provided by the respective plants.
Ranges are given for 1 month of data. For YL, only one pH measurement was taken by plant operators during the month.
PCR amplification and 454 pyrosequencing.
gDNA was amplified with primers specific to the mcrA gene (8) and the archaeal 16S rRNA gene (25) according to the PCR protocols indicated in these references. To enable sample multiplexing during sequencing, barcodes were incorporated between the adapter and forward primer. Triplicate PCRs were performed for each sample, and the amplicons were pooled and purified. Equimolar concentrations were sequenced on a Roche 454 GS FLX Titanium platform (Roche, NJ) as described previously (24).
Sequence analysis.
A total of 16,810 raw mcrA reads were generated, and were processed using the mothur pipeline (v1.32) with default parameters (26). Denoising was performed using the mothur command shhh.flows, an implementation of the PyroNoise algorithm (27) using the default parameters (maximum of 1,000 iterations; minimum change between iterations before stopping, 10−6; cutoff, 0.01; sigma, 0.06; flow order, TACG). Chimeras were identified with the mothur command chimera.uchime, a wrapper for the UCHIME package (28), with default parameters, and likely chimeric sequences were removed. Low-quality reads that likely resulted from pyrosequencing errors (>2-bp difference from primer sequence in primer region; >1-bp difference from barcode sequence in barcode region; >8-bp homopolymer runs; <300-bp length) were removed from further analysis. Barcode and primer sequences were removed. The remaining reads were compared to the NCBI nonredundant (nr) database using BLAST to ensure the top hit (sequence similarity) was a mcrA gene. After quality control, 16,634 reads were retained with 3,388 in the GZ digester sample, 4,947 in ST, 3,717 in SWH, and 4,582 in YL. These reads were then clustered into operational taxonomic units at 97% sequence similarity (OTUmcrA).
A custom database of mcrA sequences was constructed by downloading all mcrA sequences from the Functional Gene Repository v.7.3 (29). The sequences were checked against the NCBI nr database, and 939 sequences that attracted matches with taxonomic information from the domain to family levels were retained. The database was manually curated to ensure all major methanogen families were represented. The OTUmcrA were taxonomically classified against this database using the Wang algorithm (30) implemented in mothur.
In order to account for differences in sequencing depth between samples, the read sets were normalized by randomly selecting 3,388 sequences (the number in the smallest sample, GZ) from each sample. Weighted UniFrac distances were calculated in mothur, and Principal Coordinates Analysis (PCoA) was performed with the cmdscale function in the R package vegan (31, 32). A Venn diagram was constructed using the R package VennDiagram (33) to evaluate the number of OTUmcrA shared between the four digesters. To visualize the relationship between the most abundant OTUmcrA and the differences between each digester, Pearson correlation coefficient was calculated between the rarefied abundances for the 20 most abundant OTUmcrA (abundance averaged over the four digesters) and the PCoA axes using the add.spec.scores function from BiodiversityR (34). OTUmcrA richness (Chao1 estimator), evenness (Pielou index, J′), and diversity (Shannon-Weaver index, H′) were calculated in vegan. Rarefaction curves were generated using mothur. To investigate the detailed phylogenetic affiliations of the most abundant OTUmcrA, representative sequences for the 20 most abundant OTUmcrA (abundance averaged over the four digesters) and 28 additional sequences (selected representatives of each methanogen order and mcrA sequences from the NCBI nr database with high BLAST similarity to the abundant OTUmcrA) were aligned using MUSCLE (35), and a neighbor-joining (NJ) tree with Jukes-Cantor correction constructed in MEGA v.5.2.2 (36). The tree was rooted to accurately represent the evolutionary relationship between methanogen groups and allow comparison with 16S rRNA-based phylogeny. The Methanopyrus kandleri mcrA gene was selected as the outgroup sequence as it is deeply branching; no nonmethanogen sequence could be used since only methanogens carry the mcrA gene (8).
Quality checks and sequence clustering were performed as described above via the mothur pipeline for the archaeal 16S rRNA gene sequences to form OTU16S. OTU16S were taxonomically classified against the Greengenes database (37) using the Wang algorithm as described above. In total, 8,446 high-quality archaeal 16S rRNA sequences were used for downstream analyses, with 1,159 in the GZ digester sample, 2,849 in ST, 2,682 in SWH, and 1,756 in YL. The subsampling depth for normalization was set at 1,159 reads. Calculation of diversity indices and weighted UniFrac distances, PCoA ordination, regression of shared OTU onto PCoA axes, and Venn diagram construction were performed as described above.
mcrA transcription.
Sludge samples collected from the GZ and SWH digesters were incubated with food waste as described previously (24). Briefly, 50 ml of sludge was incubated with 5 g of volatile solids/liter of food waste as a heterogeneous substrate and incubated at 35°C. Each experiment was run in duplicate, and control experiments without substrate were also prepared. Methane concentration was measured by a gas chromatograph equipped with a flame ionization detector (GC-2010 Plus; Shimadzu, Japan) and methane production rate was calculated by the difference in methane yield between time points. After a linear increase in methane concentration commenced, 1 ml of culture was periodically collected from each replicate, pooled, and centrifuged at 13,800 × g for 6 min at 4°C, and the cell pellet was stored at −80°C until processing.
Total RNA was extracted from the frozen cell pellet using the RNeasy minikit (Qiagen, CA) and residual DNA was removed with the Qiagen RNase-free DNase set. Total RNA was quantified using a NanoDrop 2000 UV-Vis Spectrophotometer (NanoDrop Products, DE). A portion (2 μl) of total RNA was reverse transcribed to cDNA with random hexamers according to the manufacturer's protocol using the SuperScript III first-strand synthesis system (Invitrogen, CA). After reverse transcription (RT), mcrA transcript abundances were determined on a StepOnePlus quantitative PCR (qPCR) system (Applied Biosystems, CA) with 1× SYBR green PCR master mix (Applied Biosystems), 0.3 μM mcrA-F/mcrA-R primers (8), and 2 μl of cDNA template in a 25-μl reaction volume. Triplicate RT-qPCRs were performed for each sample along with a control reaction without reverse transcriptase.
Absolute quantification of mcrA transcripts was based on RNA standards transcribed in vitro using the MEGAscript T7 kit (Ambion, Austin, TX) according to the manufacturer's protocol with mcrA PCR product as the template. The synthesized RNA was treated with DNase to remove DNA contaminants and complete DNA removal was confirmed by triplicate RT-qPCRs without reverse transcriptase in which no fluorescence signal was detected after 40 cycles. Copy number was calculated from the size of the input PCR product and an average molecular mass of 340 Da per RNA nucleotide.
Accession number.
Sequences obtained in the present study have been deposited in the NCBI Sequence Read Archive (SRA) (BioProject accession number PRJNA245382).
RESULTS
Characteristics of the anaerobic digesters.
The four digesters examined in the present study are operated under different conditions and are geographically separated, with three (ST, SWH, and YL) located in Hong Kong and one (GZ) about 200 km to the north (details on digester operating conditions and performance provided in Table 1). Among the four digesters, only ST treats saline wastewater (9.7 ppt salinity). ST, SHW, and YL treat concentrated sludge following secondary treatment and operate at similar temperatures, but the processing capacity and volume of methane produced vary in the order ST > SHW > YL. The retention time of the three digesters also differs, with ST having the shortest (11 days) and YL the longest (40 days). In contrast to the other three digesters, GZ treats organic wastewater from industrial manufacturing and is small compared to other industrial wastewater systems (700 m3/day) with a short retention time (12 h). GZ's operating temperature is ∼5°C lower than the other digesters (Table 1).
Sequencing statistics and diversity estimates.
After the low-quality reads were filtered out, a total of 16,634 mcrA and 8,446 16S rRNA gene reads were retained for downstream analyses (Table 2). Rarefaction curves for both OTUmcrA and OTU16S did not plateau in any sample, indicating additional sampling effort would be required to completely assess community diversity (see Fig. S1 in the supplemental material), and neither gene had a clear advantage in capturing overall diversity, with greater diversity captured by the 16S rRNA gene in samples GZ and YL but with greater diversity captured by the mcrA gene in samples SWH and ST. This was supported by the Shannon-Weaver diversity index (H′, Table 2). The archaeal (16S rRNA gene) and methanogenic (mcrA) communities were highly uneven, with fewer than six OTUmcrA or five OTU16S from any sample comprising more than 100 sequences (see Fig. S2 in the supplemental material).
TABLE 2.
Read counts, OTU counts, and alpha diversity indices for mcrA and 16S genesa
| Treatment plant |
mcrA |
16S |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| No. of reads (no. rarefied) | No. of OTU (no. rarefied) | Chao1 | H′ | J′ | No. of reads (no. rarefied) | No. of OTU (no. rarefied) | Chao1 | H′ | J′ | |
| GZ | 3,388 (3,388) | 119 (119) | 371 | 1.2 | 0.36 | 1,159 (1,159) | 85 (88) | 194 | 2.0 | 0.59 |
| ST | 4,947 (3,388) | 214 (167) | 538 | 1.6 | 0.46 | 2,849 (1,159) | 119 (58) | 317 | 0.97 | 0.34 |
| SWH | 3,717 (3,388) | 221 (215) | 580 | 1.7 | 0.50 | 2,682 (1,159) | 105 (67) | 202 | 1.5 | 0.49 |
| YL | 4,582 (3,388) | 210 (180) | 592 | 1.2 | 0.38 | 1,756 (1,159) | 138 (109) | 272 | 2.1 | 0.63 |
Alpha diversity indices were calculated from rarefied read sets. The numbers of reads refer to reads that passed quality checks.
Community composition of methanogens and archaea.
Methanogens (OTUmcrA) from the order Methanomicrobiales were dominant in the ST (63% of sequences) and GZ (79%) digesters, while Methanosarcinales dominated SWH (43%) and YL (52%) (Fig. 1). Each of the two Methanobacteriales families Methanobacteriaceae and Methanothermaceae was also detected, with the former comprising 40% of mcrA sequences in YL, 17% in SWH, 2.2% in GZ, and 1.5% in ST, and the latter found only in ST (0.040%). The majority of Methanomicrobiales OTUmcrA were unclassified, although the families Methanomicrobiaceae (0.22 to 1.0%), Methanoregulaceae (0.020 to 2.7%), and Methanospirillaceae (GZ 0.12%, ST 16%, SWH 10%, YL 0.48%) were identified in all four digesters and Methanocorpusculaceae in ST (0.061%). The failure to classify most Methanomicrobiales to the family level may be due to the paucity of available mcrA sequences for candidate or recently described Methanomicrobiales genera (e.g., Methanolinea and Methanoregula) (38). Of the order Methanosarcinales, the family Methanosaetaceae was most abundant (GZ 8.3%, ST 1.0%, SWH 42%, YL 52%), though Methanosarcinaceae were also detected (0.27 to 2.3%). Finally, a small proportion of OTUmcrA from family Methanopyraceae (order Methanopyrales) were detected in GZ (5.1%), SWH (0.027%), and YL (0.48%) (although the dominant Methanopyrales OTUmcrA may be better classified as a member of the Methanomassiliicoccales; see Discussion). No OTUmcrA were classified as members of the methanogenic orders Methanococcales or Methanocellales, despite there being representative mcrA sequences for these orders in the reference database.
FIG 1.

Taxonomic composition of methanogen (mcrA) and archaea (16S) communities. Selected nonmethanogen, low-abundance, and unclassified taxa (including OTU assigned by the Wang algorithm to poorly defined taxa) have been aggregated for clarity. Note that a large proportion of Methanopyrales OTUmcrA may be better classified as Methanomassiliicoccales, and a large proportion of unclassified OTUmcrA may represent the WSA2 group (see Discussion).
Overall, the OTUmcrA profile of each digester could be characterized as either Methanomicrobiales dominated (GZ and ST) or Methanosaetaceae and Methanobacteriaceae dominated (SWH and YL), with each also containing a minority population of the nondominant order(s) as well as Methanopyrales and unclassified OTUmcrA. However, these taxonomic similarities may break down at the OTU level (see “Comparison of digesters,” below).
In the archaeal community, OTU16S affiliated with both the Crenarchaeota and Euryarchaeota were detected in addition to some OTU16S unclassifiable past the domain level (0.21 to 0.74%) (Fig. 1). A large proportion of OTU16S (GZ 5.2%, ST 34%, SWH 38%, YL 7.9%) were assigned to the uncultured group WSA2, classified in the Greengenes v.13_5 taxonomy as a family of the order Methanobacteriales. WSA2 (sometimes named “ArcI” or “Arc I” after reference 39) has been previously detected at high abundance in anaerobic digesters and found to form a class-level monophyletic lineage within the Euryarchaeota distinct from the Methanobacteriales (16, 39), a phylogeny supported by a meta-analysis of anaerobic digester 16S rRNA gene sequences (5). Accordingly, we treat the WSA2 OTU16S here as a class of the Euryarchaeota separate from the Methanobacteriales. Excluding WSA2 thus, very few Methanobacteriales OTU16S were detected (GZ 2.2%, ST 0.35%, SWH 0.48%, YL 1.2%), with all from the family Methanobacteriaceae except for 0.057% of YL sequences assigned to candidate division MSBL1 (40).
Other methanogenic OTU16S were of the order Methanomicrobiales (GZ 44%, ST 1.0%, SWH 1.2%, YL 13%), dominated in GZ by the Methanospirillaceae (39%) and YL by the Methanoregulaceae (11%); and the Methanosarcinales (GZ 4.9%, ST 1.9%, SWH 6.9%, YL 60%), dominated by the Methanosaetaceae (GZ 4.7%, ST 1.5%, SWH 6.8%, YL 50%). In ST only, a small population (0.035%) of Methanococcales was found, all of the family Methanococcaceae. No Methanocellales or Methanopyrales OTU16S were detected, although there were representative sequences for both in the reference database.
Nonmethanogenic Euryarchaeota were also identified in all digesters, including OTU of the classes Halobacteria (0.070 to 0.86%), Thermoplasmata (0.06 to 0.49%), and miscellaneous unclassified sequences and candidate divisions (1.7 to 7.0%). Most of the remaining OTU16S were classified to the Crenarchaeota, divided between the families Nitrososphaeraceae (0.21 to 11%), Cenarchaeaceae (GZ 0.26%, ST 59%, SWH 1.5%, YL 0.63%), class Thermoprotei (GZ 3.5%, ST 0.21%, SWH 35%, YL 2.8%), the uncultured Miscellaneous Crenarchaeotal Group (MCG) (GZ 22%, ST 0.35%, SWH 0.67%, YL 1.1%), and the Marine Benthic Group B (0 to 0.086%) with the remainder unclassified Thaumarchaeota (0.075 to 0.11%) (in the Greengenes v.13_5 taxonomy, the Thaumarchaeota are classified under the phylum Crenarchaeota).
Phylogeny of abundant methanogens.
To investigate the detailed phylogenetic affiliation of the most abundant OTUmcrA, the 20 most abundant OTUmcrA (averaged across the four digesters) were aligned with 28 reference sequences and a NJ tree constructed (Fig. 2). Three OTUmcrA clustered with reference Methanobacteriales sequences, six with Methanomicrobiales, four with Methanosarcinales, one with Methanococcales, and one with representatives of the proposed order Methanomassiliicoccales (3). The remaining five OTUmcrA were placed in two clusters branching deeply within the Methanomicrobia. These OTUmcrA included two highly abundant sequences from ST (OTU006) and SWH (OTU007) and, while clustering closely with a number of unclassified mcrA sequences from previous studies, were not affiliated with any cultured isolates.
FIG 2.

Neighbor-joining tree of 20 most abundant OTUmcrA, with selected reference sequences. Relative abundances (%) for each digester are given to the right of each OTUmcrA. Bootstrap values >50% (500 repetitions) are shown on nodes. The scale bar indicates sequence dissimilarity between nodes. For each OTUmcrA, the Wang algorithm's taxonomic assignment (class or finer) is indicated in parentheses.
Consistent with the taxonomic classifications assigned by mothur, the most abundant OTUmcrA in GZ (OTU001) and ST (OTU004) were placed in the Methanomicrobiales cluster, while those in SWH (OTU005) and YL (OTU003) were associated with the Methanosarcinales. Only two of the most abundant OTUmcrA, OTU005 (Methanosarcinales) and OTU002 (Methanobacteriales) were present in all four digesters, reflecting the overall low level of community overlap (see Fig. S3 in the supplemental material).
Comparison of digesters.
The digesters' methanogen and archaeal communities were compared by PCoA ordination of the weighted UniFrac distances between samples. For both communities, the SWH and YL digesters were closely clustered and separated from the other two digesters (Fig. 3). Correlations between dominant OTU and the PCoA axes showed that the majority of dominant OTU were strongly associated with either a single digester or SWH+YL. Venn diagrams for both methanogenic and archaeal communities showed little overlap between the digesters, with 93% of OTUmcrA and 90% of OTU16S found only in a single digester (see Fig. S3 in the supplemental material).
FIG 3.
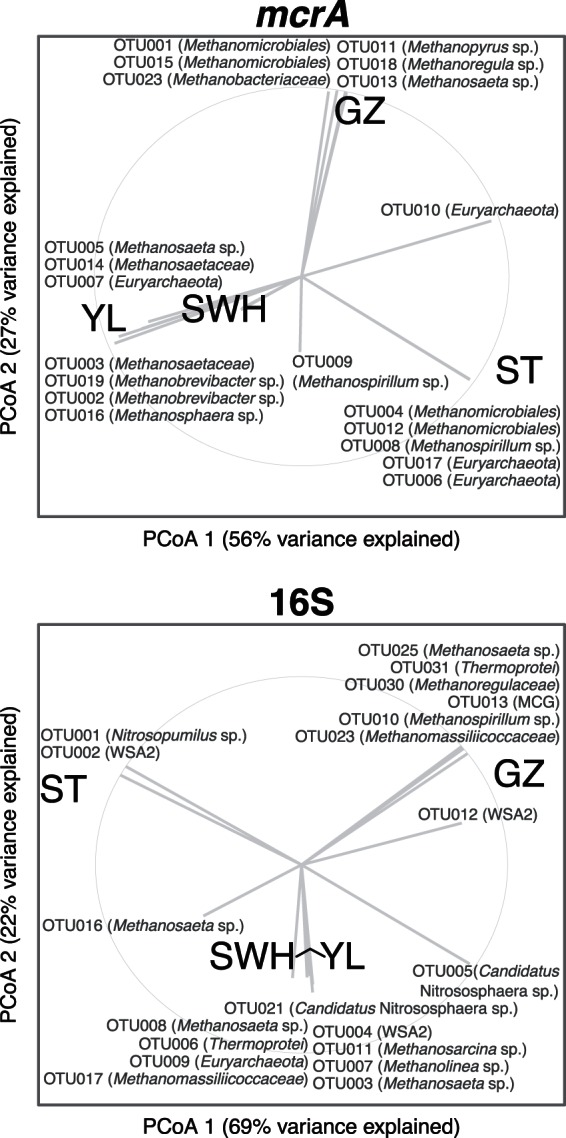
Principle coordinates analyses (PCoA) of methanogen (mcrA) and archaea (16S) communities. Vectors indicate correlation (ρ) between the abundances of the 20 most abundant OTU and the PCoA axes and should not be interpreted as identifying OTU that explain the variance between digesters. The light gray circle represents ρ = 1.
Correlation between abundance of mcrA transcripts and methane production.
The mcrA gene is a good candidate for a biomarker of methanogenesis rates in anaerobic digesters. Inocula from the GZ and SWH digesters were incubated on a food waste substrate to investigate the relationship between the methane production rate and transcription of the mcrA gene. Significant linear correlations (R2 > 0.87) were found between the methane production rates and abundance of mcrA transcripts for both digester inocula (Fig. 4). A negligible amount of methane was produced from the control samples without substrate.
FIG 4.
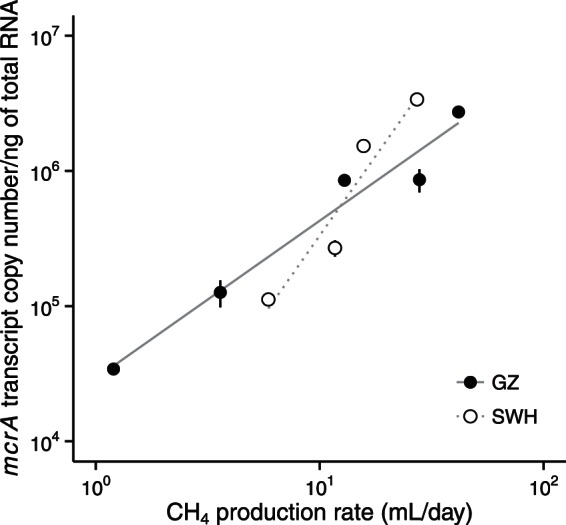
Relationship between mcrA transcription and the rate of methane (CH4) production in anaerobic cultures inoculated with material from the GZ and SHX digesters. Each point represents one time point during the incubation. Error bars represent standard deviation for triplicate RT-qPCRs (vertical axis) and the 95% confidence intervals for CH4 production (horizontal axis, smaller than the point in most cases).
DISCUSSION
Methanogenesis is a major function of anaerobic digesters in wastewater treatment plants. However, the taxonomic identity of the methanogen groups involved and their community composition under different operating conditions are still not well understood (5, 6). Although the 16S rRNA gene is a common target for community analysis, the mcrA gene has been used for the taxonomic classification of methanogens either independently (41, 42) or in concert with the 16S rRNA gene (14, 20). In the present study, pyrosequencing of both genes was applied to investigate the community composition of methanogens from four different anaerobic digesters.
Previous investigations mainly relied on Sanger sequencing of 16S rRNA gene clone libraries (16, 20, 41, 42), which can underestimate richness and diversity due to lack of sequencing depth. The development of high-throughput pyrosequencing has expanded our view of the diversity, abundance and structure of microbial communities in many environments (43). In the present study, 694 OTUmcrA (from 16,634 reads) and 391 OTU16S (8,446 reads) were recovered at the 97% sequence similarity level (Table 2), and rarefaction suggests richness was not sampled to exhaustion (see Fig. S1 in the supplemental material). A meta-analysis of archaeal 16S rRNA gene sequences from anaerobic digesters found that ∼90% of 97% similar OTU were identified with <3,000 sequence reads and estimated through rarefaction a total richness of 327 OTU across all digesters studied (5). Although the greater sampling depth enabled by high-throughput sequencing may explain the higher observed richness here, it does not account for the higher estimated total richness. The higher OTU16S richness in the present study may be due to the use of the hypervariable V1 region as sequencing target, resulting in a finer phylogenetic resolution compared to more conserved targets. However, OTUmcrA richness was also high relative to comparable studies using the 97% sequence similarity threshold. For example, a study of 118 mcrA sequences from an anaerobic batch reactor identified 21 OTUmcrA at an estimated 90% coverage of total richness (20), whereas a study of 123 mcrA clones from an anaerobic biogas reactor identified 28 OTUmcrA at 89% estimated coverage (42). It is therefore possible that the digesters selected for the present study are more diverse than others previously reported on. Alternatively, previous clone library-based studies may have underestimated OTU richness due to low evenness combined with undersampling (44). In this investigation, Pielou's evenness J′ was generally low (Table 2), an observation consistent with the rank-abundance curves (see Fig. S2 in the supplemental material), which suggests the digesters are dominated by a small number of abundant OTU. The majority of both OTUmcrA and OTU16S were represented by a single sequence (singleton OTU). An important caveat is that PCR and sequencing error can inflate richness estimates by generating spurious OTU. Appropriate denoising and quality control steps were applied (see Materials and Methods), and the OTU identity threshold used in the present study (97%) should be sufficient to compensate for typical 454 sequencing error (45). When OTU containing only one read across all samples (global singletons) are excluded, a conservative filter given that only one sample was taken from each digester, 201 OTUmcrA remain, still exceeding previous reports, although only 114 OTU16S are retained. Despite this and the quality control steps taken, it is still possible that spurious OTU contributed in part to the richness observed in this study.
This study also provides an opportunity to compare the 16S rRNA and mcrA genes as markers for characterizing methanogen assemblages. Previous studies have found greater diversity using mcrA primers than archaeon-specific 16S rRNA gene primers at 97% sequence similarity (20) or by restriction fragment length polymorphism fingerprint (46), but similar diversity when sequence similarity thresholds are calibrated to taxonomic rank (14). In the present study, rarefaction did not suggest a systematic difference in the total richness revealed by the two genes at the 97% level, with more OTU16S identified in samples GZ and YL but more OTUmcrA in samples SWH and ST. This pattern was reflected by the Shannon-Weaver diversity index (Table 2), although the Chao1 index was consistently higher for OTUmcrA, likely due to the higher sequencing depth. It is difficult to meaningfully compare OTU richness between the two genes since even at the same sequence similarity threshold they likely represent different levels of taxonomic resolution (14) and since the OTU16S include nonmethanogens. There is no widely used mcrA similarity threshold for delineating methanogen species, although studies generally agree that interspecies mcrA gene similarity is much lower than 16S rRNA gene similarity (14, 47).
The two gene targets also gave strikingly different pictures of the digesters' taxonomic compositions (Fig. 1). The same methanogen families were identified with both genes, except for the Methanopyraceae which was identified only by mcrA despite several representative sequences in the 16S rRNA gene reference database, the WSA2 group for which no mcrA sequence is available, and some minor (<3 sequences) families. However, there were large differences between the genes in the relative abundances of methanogen groups. Methanogens of the order Methanobacteriales, while abundant in the SWH and YL OTUmcrA, were only a small fraction of the OTU16S. Similarly, while the mcrA-based abundances suggested GZ and ST methanogens are dominated by Methanomicrobiales and SWH and YL by Methanosarcinales, in ST and SWH the 16S rRNA gene abundances suggested only small populations of these orders with the difference made up by WSA2 OTU. A known problem with some mcrA-targeting primers is that they also amplify the paralogous mrtA gene (encoding methyl coenzyme M reductase II) found in members of the Methanobacteriales and Methanococcales, increasing the observed proportions of these groups (8, 15). However, the observed differences are not explained by a systematic overrepresentation of the Methanobacteriales. Some mcrA primer sets are also known to exclude certain methanogen groups, with, e.g., the mcrA3 set unable to detect members of the genus Methanosaetaceae (20, 46), although the primer set used in the present study does not appear to exclude any major methanogen groups (8). Differences in 16S rRNA (48) and mcrA (15) copy numbers between organisms may also have biased the taxonomic profiles. Overall, our results provide further support for the conclusion that a combination of the two genes is valuable in assessing the full spectrum of methanogen diversity (20).
While the digesters harbored distinct methanogen and archaeal communities, the SWH and YL digesters were most similar in overall taxonomic composition (Fig. 1 and 3). This reflects the similarity between the SWH and YL treatment plants, which treat secondary sludge from municipal sewage (unlike GZ) at low salinity (unlike ST), although at different scales (Table 1). SWH and YL shared more OTU from both communities than all other digester combinations except the GZ+YL methanogens (see Fig. S3 in the supplemental material). Overall, however, there was little overlap between the communities; among the most abundant OTU, the majority were strongly associated with either a single digester or SWH+YL (Fig. 3). This lack of overlap is probably not solely attributable to undersampling or low evenness, as even among the most abundant OTU in each community the overlap was low. It may reflect small differences in operating conditions and feedstock composition between the digesters. As a metagenomic study of the ST and SWH digesters found that they did not significantly differ in genomically encoded metabolic functions (49), it may also be due to stochastic occupation by closely related and functionally interchangeable taxa of the limited set of biochemical niches within the anaerobic digestion process. A high level of functional redundancy is commonly reported in anaerobic digester communities (12).
The NJ tree of abundant OTUmcrA sequences produced class-level clades generally congruent with the accepted phylogeny of the Methanomicrobia (Fig. 2) and supported the Wang algorithm's taxonomic classifications with two exceptions. The first exception was OTU016, which clustered with the Methanococcales despite being classified to genus Methanosphaera by the Wang algorithm. Given the lack of closely related reference sequences and the deep branching of this OTU relative to the Methanococcales and the very low proportion of 16S rRNA gene sequences classified to the Methanococcales, this may be a misplacement of a Methanobacteriales sequence. The second exception was OTU011, most abundant in GZ (5.1%) but also present in SWH and YL (<0.5%), which had been classified to the genus Methanopyrus but did not have high sequence similarity with the Methanopyrus kandleri sequence subsequently used to root the tree. OTU011 was instead affiliated with the mcrA genes of “Candidatus Methanomethylophilus alvus” and “Candidatus Methanogranum caenicola,” members of the proposed methanogen order Methanomassiliicoccales (3, 50, 51), as well as an unclassified reference sequence. Nine OTU16S were classified by the Wang algorithm to the family Methanomassiliicoccaceae, with the highest collective abundance in GZ (GZ 4.9%, ST 0.18%, SWH 2.3%, and YL 1.0%), supporting the presence of Methanomassiliicoccaceae in the digesters and at highest abundance in GZ. “Ca. Methanomethylophilus alvus” is a putative obligate hydrogen-dependent methylotrophic methanogen (52) with the genomic potential to utilize a wide range of methylated compounds, a metabolic ability which may be common to the proposed order (3). Similarly, “Ca. Methanogranum caenicola” was isolated from anaerobic packed-bed reactor sludge and determined in culture to produce methane through hydrogen-dependent reduction of methanol (50). Notably, OTU011 was only identified at significant abundance in GZ (5.1%), which treats organic wastewater from industrial manufacturing, as opposed to the other three digesters (<0.5%) which treat concentrated sludge following secondary treatment of sewage. The high abundance of OTU011 in GZ may therefore reflect the presence of methanol or other methylated compounds in this industrial waste stream. Since OTU011 comprised almost all of the mcrA sequences assigned by the Wang algorithm to the Methanopyrales (the exception being a single YL sequence), reassigning this OTU to the Methanomassiliicoccaceae is consistent with the lack of Methanopyrales OTU16S and suggests the Methanopyrales are either present at extremely low abundance in, or absent from, the digesters.
The WSA2 group was a major component of the OTU16S assemblages, particularly in ST and SWH (Fig. 1). Although classified in Greengenes as a member of the Methanobacteriales, it has been identified at high abundance in other anaerobic digesters and consistently found to form a class-level monophyletic lineage within the Euryarchaeota distinct from the Methanobacteria (5, 16, 39). Based on this phylogenetic placement, growth in culture on formate and H2/CO2 (39), and possible competition with Methanosarcinales for acetate (16), WSA2 group organisms are very likely methanogens. The mcrA tree constructed here contained two groups of deeply branching, abundant unclassified OTUmcrA designated unclassified group (UG) I and UGII (Fig. 2). Two UGI OTUmcrA were highly abundant in SWH (OTU007, 23%) and ST (OTU006, 26%), in similar proportions to those of WSA2 OTU16S in those digesters (Fig. 1) and accounting for the majority (77%) of the unclassified mcrA sequences observed here. Although systematic biases due to PCR bias and copy number variation very likely influence the observed relative abundances, the similarity in proportions across the four digesters is consistent with the hypothesis that the genes originate from the same organism(s). It is thus plausible the UGI and possibly UGII mcrA sequences originate from the WSA2 group, for which no representative mcrA sequences yet exist. Future experiments, e.g., dual-probe fluorescence in situ hybridization or construction of a draft WSA2 genome from metagenomic sequences, will be useful to confirm this hypothesis.
Both Crenarchaeota and Euryarchaeota OTU16S were detected in all digesters. ST Crenarchaeota consisted almost exclusively of a single OTU16S classified to the genus Nitrosopumilus, the only species of which is the abundant marine archaeon N. maritimus, likely reflecting the widespread use of seawater for toilet flushing in the region served by the ST wastewater treatment plant. Crenarchaeota in the other three digesters were predominantly from the class Thermoprotei, common in anaerobic digesters (5), the ammonia-oxidizing family Nitrososphaeraceae, and the poorly characterized Miscellaneous Crenarchaeotal Group (MCG). Although Crenarchaeota are frequent components of anaerobic digester communities (5), their role in the digestion process is largely unknown, although they have been found to collocate with active Methanosaeta cells in a granular digester biofilm (53), suggesting some metabolic interaction and potentially syntrophy with methanogens.
Methanogens of the orders Methanobacteriales, Methanomicrobiales, and Methanosarcinales have previously been reported as abundant in anaerobic digesters treating cow manure (20, 41), wastewater algae (22), and municipal solid waste (54). Methanobacteriales and Methanomicrobiales produce methane by reduction of CO2 with H2 as an electron donor (hydrogenotrophic methanogenesis), while Methanosarcinales directly cleave acetate to methane and CO2 (acetoclastic methanogenesis) (55). Stoichiometric modeling (56, 57) and measurements of natural methanogenic systems (58) have been used to predict acetate accounts for ∼70% of methane production, and a meta-analysis of anaerobic digester studies found that the acetoclastic genus Methanosaeta accounted for 55% of archaea (although a large proportion of sequences were unclassifiable to genus) (5). However, several studies of biogas reactors have identified a dominance of hydrogenotrophic methanogens, though the relative proportions of Methanobacteriales and Methanomicrobiales vary (41, 42, 46). In the present study, mcrA-based taxonomic classification suggested the SWH and YL digesters had large or dominant populations of acetoclastic Methanosarcinales, while GZ and ST were dominated by hydrogenotrophic Methanomicrobiales. Within the Methanosarcinales, Methanosaetaceae far outnumbered Methanosarcinaceae in all digesters and with both genes (Fig. 1). Methanosaetaceae dominance is characteristic of digesters with low acetate concentrations and moderate retention times (20, 59, 60). Acetate was not detected in any of the sampled digesters (data not shown), consistent with the dominance of Methanosaetaceae and indicating that syntrophic acetoclastic methanogenesis in these reactors keeps pace with acetogenesis. A previous metagenomic study of the ST and SWH digesters similarly found the Methanosaeta to be dominant in all but one sample (49), suggesting this pattern is stable over time.
In addition to its use in determining methanogenic diversity, the mcrA gene is a potential biomarker of methane yield from methanogenesis. Using qPCR with broad-specificity mcrA primers, the mcrA gene copy number has been found to correlate with methanogen abundance in sludge from anaerobic digesters and in methane seep sediments (61) and with methane production rates in a biogas reactor (17). However, gene copy number is not a reliable predictor of metabolic activity and is unable to capture the real-time responses of methanogens to changing operating conditions. In the present study, we used RT-qPCR to measure mcrA transcription and demonstrated a significant linear correlation between mcrA transcript quantity and methane production rate in two digester microbial communities (Fig. 4). This result confirms that the physiological activity of methanogens in a complex system can be gauged by analyzing the expression of the mcrA gene, and the expression level can also be used to predict methane yield.
Anaerobic digestion is widely used in the treatment of wastewater sludge, and yet the microbial consortia performing this process are poorly understood. A thorough understanding of the methanogens and other archaea in biogas-producing reactors is essential to improving methane yield and other industrially important parameters. In the present study, pyrosequencing of the mcrA gene detected both hydrogenotrophic and acetoclastic methanogens in each digester and found an overall taxonomic composition quite different to that found with the 16S rRNA gene. The identification of the proposed order Methanomassiliicoccales and the uncultured but ubiquitous WSA2 lineage suggest these groups may play roles in the anaerobic digestion process and are important targets for future investigation. Finally, a significant positive correlation was identified between methane production rates and the abundance of mcrA transcripts in digesters despite very different methanogen compositions. Overall, our study confirmed the efficacy of the mcrA gene as a marker for both methanogen taxonomy and metabolic activity and also reinforced the value of using multiple genes to assess microbial diversity.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by a grant from the Research Grants Council of Hong Kong through project 116111 and grant 7008131 of the City University of Hong Kong.
We thank the Hong Kong Drainage Services Department and the operators at the GZ plant for sampling assistance. We also express our gratitude to Hongmei Jing for her assistance in this study.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02566-14.
REFERENCES
- 1.Chynoweth DP, Owens JM, Legrand R. 2001. Renewable methane from anaerobic digestion of biomass. Renew Energ 22:1–8. doi: 10.1016/S0960-1481(00)00019-7. [DOI] [Google Scholar]
- 2.Zinder SH. 1993. Physiological ecology of methanogens, p 128–206. In Ferry JG. (ed), Methanogenesis: ecology, physiology, biochemistry, and genetics. Chapman and Hall, New York, NY. [Google Scholar]
- 3.Borrel G, O'Toole PW, Harris HMB, Peyret P, Brugère JF, Gribaldo S. 2013. Phylogenomic data support a seventh order of methylotrophic methanogens and provide insights into the evolution of methanogenesis. Genome Biol Evol 5:1769–1780. doi: 10.1093/gbe/evt128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu Y, Whitman WB. 2008. Metabolic, phylogenetic, and ecological diversity of the methanogenic archaea. Ann N Y Acad Sci 1125:171–189. doi: 10.1196/annals.1419.019. [DOI] [PubMed] [Google Scholar]
- 5.Nelson MC, Morrison M, Yu Z. 2011. A meta-analysis of the microbial diversity observed in anaerobic digesters. Bioresour Technol 102:3730–3739. doi: 10.1016/j.biortech.2010.11.119. [DOI] [PubMed] [Google Scholar]
- 6.Narihiro T, Sekiguchi Y. 2007. Microbial communities in anaerobic digestion processes for waste and wastewater treatment: a microbiological update. Curr Opin Biotechnol 18:273–278. doi: 10.1016/j.copbio.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 7.Galand PE, Fritze H, Conrad R, Yrjälä K. 2005. Pathways for methanogenesis and diversity of methanogenic archaea in three boreal peatland ecosystems. Appl Environ Microbiol 71:2195–2198. doi: 10.1128/AEM.71.4.2195-2198.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Luton PE, Wayne JM, Sharp RJ, Riley PW. 2002. The mcrA gene as an alternative to 16S rRNA in the phylogenetic analysis of methanogen populations in landfill. Microbiology 148:3521–3530. [DOI] [PubMed] [Google Scholar]
- 9.Iino T, Mori K, Suzuki K. 2010. Methanospirillum lacunae sp. nov., a methane-producing archaeon isolated from a puddly soil, and emended descriptions of the genus Methanospirillum and Methanospirillum hungatei. Int J Syst Evol Microbiol 60:2563–2566. doi: 10.1099/ijs.0.020131-0. [DOI] [PubMed] [Google Scholar]
- 10.Frey JC, Pell AN, Berthiaume R, Lapierre H, Lee S, Ha JK, Mendell JE, Angert ER. 2010. Comparative studies of microbial populations in the rumen, duodenum, ileum and faeces of lactating dairy cows. J Appl Microbiol 108:1982–1993. doi: 10.1111/j.1365-2672.2009.04602.x. [DOI] [PubMed] [Google Scholar]
- 11.Werner JJ, Knights D, Garcia ML, Scalfone NB, Smith S, Yarasheski K, Cummings TA, Beers AR, Knight R, Angenent LT. 2011. Bacterial community structures are unique and resilient in full-scale bioenergy systems. Proc Natl Acad Sci U S A 108:4158–4163. doi: 10.1073/pnas.1015676108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vanwonterghem I, Jensen PD, Ho DP, Batstone DJ, Tyson GW. 2014. Linking microbial community structure, interactions and function in anaerobic digesters using new molecular techniques. Curr Opin Biotechnol 27:55–64. doi: 10.1016/j.copbio.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 13.Kröber M, Bekel T, Diaz NN, Goesmann A, Jaenicke S, Krause L, Miller D, Runte KJ, Viehöver P, Pühler A, Schlüter A. 2009. Phylogenetic characterization of a biogas plant microbial community integrating clone library 16S-rDNA sequences and metagenome sequence data obtained by 454 pyrosequencing. J Biotechnol 142:38–49. doi: 10.1016/j.jbiotec.2009.02.010. [DOI] [PubMed] [Google Scholar]
- 14.Steinberg LM, Regan JM. 2008. Phylogenetic comparison of the methanogenic communities from an acidic, oligotrophic fen and an anaerobic digester treating municipal wastewater sludge. Appl Environ Microbiol 74:6663–6671. doi: 10.1128/AEM.00553-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Friedrich MW. 2005. Methyl-coenzyme M reductase genes: unique functional markers for methanogenic and anaerobic methane-oxidizing Archaea. Methods Enzymol 397:428–442. doi: 10.1016/S0076-6879(05)97026-2. [DOI] [PubMed] [Google Scholar]
- 16.Rivière D, Desvignes V, Pelletier E, Chaussonnerie S, Guermazi S, Weissenbach J, Li T, Camacho P, Sghir A. 2009. Towards the definition of a core of microorganisms involved in anaerobic digestion of sludge. ISME J 3:700–714. doi: 10.1038/ismej.2009.2. [DOI] [PubMed] [Google Scholar]
- 17.Traversi D, Villa S, Lorenzi E, Degan R, Gilli G. 2012. Application of a real-time qPCR method to measure the methanogen concentration during anaerobic digestion as an indicator of biogas production capacity. J Environ Manage 111:173–177. doi: 10.1016/j.jenvman.2012.07.021. [DOI] [PubMed] [Google Scholar]
- 18.Leigh JA. 2011. Growth of methanogens under defined hydrogen conditions. Methods Enzymol 494:111–118. doi: 10.1016/B978-0-12-385112-3.00006-8. [DOI] [PubMed] [Google Scholar]
- 19.Ver Eecke HC, Butterfield DA, Huber JA, Lilley MD, Olson EJ, Roe KK, Evans LJ, Merkel AY, Cantin HV, Holden JF. 2012. Hydrogen-limited growth of hyperthermophilic methanogens at deep-sea hydrothermal vents. Proc Natl Acad Sci U S A 109:13674–13679. doi: 10.1073/pnas.1206632109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma J, Zhao B, Frear C, Zhao Q, Yu L, Li X, Chen S. 2013. Methanosarcina domination in anaerobic sequencing batch reactor at short hydraulic retention time. Bioresour Technol 137:41–50. doi: 10.1016/j.biortech.2013.03.101. [DOI] [PubMed] [Google Scholar]
- 21.Zeleke J, Lu S-L, Wang J-G, Huang J-X, Li B, Ogram AV, Quan Z-X. 2013. Methyl coenzyme M reductase A (mcrA) gene-based investigation of methanogens in the mudflat sediments of Yangtze River estuary, China. Microb Ecol 66:257–267. doi: 10.1007/s00248-012-0155-2. [DOI] [PubMed] [Google Scholar]
- 22.Ellis JT, Tramp C, Sims RC, Miller CD. 2012. Characterization of a methanogenic community within an algal fed anaerobic digester. ISRN Microbiol 2012:753892. doi: 10.5402/2012/753892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clesceri LS, Greenberg AE, Trussell RR, APHA, AWWA, WEF . 1989. Standard methods for the examination of water and wastewater, 17th ed American Public Health Association, Washington, DC. [Google Scholar]
- 24.Lu X, Rao S, Shen Z, Lee PKH. 2013. Substrate induced emergence of different active bacterial and archaeal assemblages during biomethane production. Bioresour Technol 148:517–524. doi: 10.1016/j.biortech.2013.09.017. [DOI] [PubMed] [Google Scholar]
- 25.Kan J, Clingenpeel S, Macur RE, Inskeep WP, Lovalvo D, Varley J, Gorby Y, McDermott TR, Nealson K. 2011. Archaea in Yellowstone Lake. ISME J 5:1784–1795. doi: 10.1038/ismej.2011.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Quince C, Lanzén A, Curtis TP, Davenport RJ, Hall N, Head IM, Read LF, Sloan WT. 2009. Accurate determination of microbial diversity from 454 pyrosequencing data. Nat Methods 6:639. doi: 10.1038/nmeth.1361. [DOI] [PubMed] [Google Scholar]
- 28.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. 2011. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fish JA, Chai B, Wang Q, Sun Y, Brown CT, Tiedje JM, Cole JR. 2013. FunGene: the functional gene pipeline and repository. Front Microbiol 4:291. doi: 10.3389/fmicb.2013.00291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Q, Garrity GM, Tiedje JM, Cole JR. 2007. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.R Core Team. 2013. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 32.Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O'Hara RB, Simpson GL, Solymos P, Stevens MHH, Wagner H. 2013. vegan: community ecology package. R package version 2.0-10. http://CRAN.R-project.org/package=vegan. [Google Scholar]
- 33.Chen H. 2014. VennDiagram: generate high-resolution Venn and Euler plots. R package version 1.6.7. http://CRAN.R-project.org/package=VennDiagram. [Google Scholar]
- 34.Kindt R, Coe R. 2005. Tree diversity analysis: a manual and software for common statistical methods for ecological and biodiversity studies. World Agroforestry Centre (ICRAF), Nairobi, Kenya. [Google Scholar]
- 35.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum-parsimony methods. Mol Biol Evol 28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. 2006. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72:5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Imachi H, Sakai S, Sekiguchi Y, Hanada S, Kamagata Y, Ohashi A, Harada H. 2008. Methanolinea tarda gen. nov., sp. nov., a methane-producing archaeon isolated from a methanogenic digester sludge. Int J Syst Evol Microbiol 58:294–301. doi: 10.1099/ijs.0.65394-0. [DOI] [PubMed] [Google Scholar]
- 39.Chouari R, Le Paslier D, Daegelen P, Ginestet P, Weissenbach J, Sghir A. 2005. Novel predominant archaeal and bacterial groups revealed by molecular analysis of an anaerobic sludge digester. Environ Microbiol 7:1104–1115. doi: 10.1111/j.1462-2920.2005.00795.x. [DOI] [PubMed] [Google Scholar]
- 40.van der Wielen PWJJ, Bolhuis H, Borin S, Daffonchio D, Corselli C, Giuliano L, D'Auria G, de Lange GJ, Huebner A, Varnavas SP, Thomson J, Tamburini C, Marty D, McGenity TJ, Timmis KN, BioDeep Scientific Party . 2005. The enigma of prokaryotic life in deep hypersaline anoxic basins. Science 307:121–123. doi: 10.1126/science.1103569. [DOI] [PubMed] [Google Scholar]
- 41.Rastogi G, Ranade DR, Yeole TY, Patole MS, Shouche YS. 2008. Investigation of methanogen population structure in biogas reactor by molecular characterization of methyl-coenzyme M reductase A (mcrA) genes. Bioresour Technol 99:5317–5326. doi: 10.1016/j.biortech.2007.11.024. [DOI] [PubMed] [Google Scholar]
- 42.Zhu C, Zhang J, Tang Y, Zhengkai X, Song R. 2011. Diversity of methanogenic archaea in a biogas reactor fed with swine feces as the mono-substrate by mcrA analysis. Microbiol Res 166:27–35. doi: 10.1016/j.micres.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 43.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, Knight R. 2011. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A 108:4516–4522. doi: 10.1073/pnas.1000080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hughes JB, Hellmann JJ, Ricketts TH, Bohannan BJM. 2001. Counting the uncountable: statistical approaches to estimating microbial diversity. Appl Environ Microbiol 67:4399–4406. doi: 10.1128/AEM.67.10.4399-4406.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Glenn TC. 2011. Field guide to next-generation DNA sequencers. Mol Ecol Resour 11:759–769. doi: 10.1111/j.1755-0998.2011.03024.x. [DOI] [PubMed] [Google Scholar]
- 46.Nettmann E, Bergmann I, Mundt K, Linke B, Klocke M. 2008. Archaea diversity within a commercial biogas plant utilizing herbal biomass determined by 16S rDNA and mcrA analysis. J Appl Microbiol 105:1835–1850. doi: 10.1111/j.1365-2672.2008.03949.x. [DOI] [PubMed] [Google Scholar]
- 47.Springer E, Sachs MS, Woese CR, Boone DR. 1995. Partial gene sequences for the A subunit of methyl-coenzyme M reductase (mcrI) as a phylogenetic tool for the family Methanosarcinaceae. Int J Syst Evol Microbiol 45:554–559. [DOI] [PubMed] [Google Scholar]
- 48.Kembel SW, Wu M, Eisen JA, Green JL. 2012. Incorporating 16S gene copy number information improves estimates of microbial diversity and abundance. PLoS Comput Biol 8:e1002743. doi: 10.1371/journal.pcbi.1002743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang Y, Yu K, Xia Y, Lau FT, Tang DT, Fung WC, Fang HH, Zhang T. 2014. Metagenomic analysis of sludge from full-scale anaerobic digesters operated in municipal wastewater treatment plants. Appl Microbiol Biotechnol 98:5709–5718. doi: 10.1007/s00253-014-5648-0. [DOI] [PubMed] [Google Scholar]
- 50.Iino T, Tamaki H, Tamazawa S, Ueno Y, Ohkuma M, Suzuki K-I, Igarashi Y, Haruta S. 2013. Candidatus Methanogranum caenicola: a novel methanogen from the anaerobic digested sludge, and proposal of Methanomassiliicoccaceae fam. nov. and Methanomassiliicoccales ord. nov., for a methanogenic lineage of the class Thermoplasmata. Microb Environ 28:244–250. doi: 10.1264/jsme2.ME12189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Paul K, Nonoh JO, Mikulski L, Brune A. 2012. “Methanoplasmatales,” Thermoplasmatales-related archaea in termite guts and other environments, are the seventh order of methanogens. Appl Environ Microbiol 78:8245–8253. doi: 10.1128/AEM.02193-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Borrel G, Harris HMB, Tottey W, Mihajlovski A, Parisot N, Peyretaillade E, Peyret P, Gribaldo S, O'Toole PW, Brugère JF. 2012. Genome sequence of “Candidatus Methanomethylophilus alvus” Mx1201, a methanogenic archaeon from the human gut belonging to a seventh order of methanogens. J Bacteriol 194:6944–6945. doi: 10.1128/JB.01867-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Collins G, O'Connor L, Mahony T, Gieseke A, de Beer D, O'Flaherty V. 2005. Distribution, localization, and phylogeny of abundant populations of Crenarchaeota in anaerobic granular sludge. Appl Environ Microbiol 71:7523–7527. doi: 10.1128/AEM.71.11.7523-7527.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bareither CA, Wolfe GL, McMahon KD, Benson CH. 2013. Microbial diversity and dynamics during methane production from municipal solid waste. Waste Manage 33:1982–1992. doi: 10.1016/j.wasman.2012.12.013. [DOI] [PubMed] [Google Scholar]
- 55.Garcia J-L, Patel BKC, Ollivier B. 2000. Taxonomic, phylogenetic, and ecological diversity of methanogenic Archaea. Anaerobe 6:205–226. doi: 10.1006/anae.2000.0345. [DOI] [PubMed] [Google Scholar]
- 56.Conrad R. 1999. Contribution of hydrogen to methane production and control of hydrogen concentrations in methanogenic soils and sediments. FEMS Microbiol Ecol 28:193–202. doi: 10.1111/j.1574-6941.1999.tb00575.x. [DOI] [Google Scholar]
- 57.Siegrist H, Vogt D, Garcia-Heras JL, Gujer W. 2002. Mathematical model for meso- and thermophilic anaerobic sewage sludge digestion. Environ Sci Technol 36:1113–1123. doi: 10.1021/es010139p. [DOI] [PubMed] [Google Scholar]
- 58.Kotsyurbenko OR, Chin K-J, Glagolev MV, Stubner S, Simankova MV, Nozhevnikova AN, Conrad R. 2004. Acetoclastic and hydrogenotrophic methane production and methanogenic populations in an acidic West-Siberian peat bog. Environ Microbiol 6:1159–1173. doi: 10.1111/j.1462-2920.2004.00634.x. [DOI] [PubMed] [Google Scholar]
- 59.McHugh S, Carton M, Mahony T, O'Flaherty V. 2003. Methanogenic population structure in a variety of anaerobic bioreactors. FEMS Microbiol Lett 219:297–304. doi: 10.1016/S0378-1097(03)00055-7. [DOI] [PubMed] [Google Scholar]
- 60.Conklin A, Stensel HD, Ferguson J. 2006. Growth kinetics and competition between Methanosarcina and Methanosaeta in mesophilic anaerobic digestion. Water Environ Res 78:486–496. doi: 10.2175/106143006X95393. [DOI] [PubMed] [Google Scholar]
- 61.Nunoura T, Oida H, Miyazaki J, Miyashita A, Imachi H, Takai K. 2008. Quantification of mcrA by fluorescent PCR in methanogenic and methanotrophic microbial communities. FEMS Microbiol Ecol 64:240–247. doi: 10.1111/j.1574-6941.2008.00451.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.