

Abstract
The utilization of silicon by diatoms has both global and small-scale implications, from oceanic primary productivity to nanotechnological applications of their silica cell walls. The sensing and transport of silicic acid are key aspects of understanding diatom silicon utilization. At low silicic acid concentrations (<30 μM), transport mainly occurs through silicic acid transport proteins (SITs), and at higher concentrations it occurs through diffusion. Previous analyses of the SITs were done either in heterologous systems or without a distinction between individual SITs. In the present study, we examined individual SITs in Thalassiosira pseudonana in terms of transcript and protein abundance in response to different silicic acid regimes and examined knockdown lines to evaluate the role of the SITs in transport, silica incorporation, and lipid accumulation resulting from silicon starvation. SIT1 and SIT2 were localized in the plasma membrane, and protein levels were generally inversely correlated with cellular silicon needs, with a distinct response being found when the two SITs were compared. We developed highly effective approaches for RNA interference and antisense knockdowns, the first such approaches developed for a centric diatom. SIT knockdown differentially affected the uptake of silicon and the incorporation of silicic acid and resulted in the induction of lipid accumulation under silicon starvation conditions far earlier than in the wild-type cells, suggesting that the cells were artificially sensing silicon limitation. The data suggest that the transport role of the SITs is relatively minor under conditions with sufficient silicic acid. Their primary role is to sense silicic acid levels to evaluate whether the cell can proceed with its cell wall formation and division processes.
INTRODUCTION
Silicon, a member of group 14 of the periodic table containing carbon, germanium, and lead, is one of the most abundant elements (27.7%) of the Earth's crust second only to oxygen (46.6%) (1). Dissolved silica in the form of orthosilicic acid, Si(OH)4, derived from the weathering of the Earth's crust (2), is an essential nutrient for a few groups of organisms of marine and freshwater environments, such as diatoms, radiolaria, silicoflagellates, and sponges (3). Diatoms require a large quantity of silicon, which is biomineralized to form their unique siliceous cell wall (frustule) in various shapes and morphologies with highly intricate and ornate architectures. Diatoms contribute about 40% of the total ocean organic carbon production, and they dominate ocean Si production (4, 5). Though silicon does not appear to be directly involved in cellular metabolic processes, diatoms require silicon for progression of cell cycle events, such as cell division and DNA replication (5–8). Silicon requirements are tightly coupled to cell cycle progression, a unique feature of diatoms (7). Deprivation of Si arrests the cell cycle at G1, G2, or M phase, depending upon the diatom species. In Thalassiosira pseudonana, upon Si depletion the cell cycle arrests predominantly at the G1 phase. Cell cycle progression starts with subsequent silicon replenishment. Girdle band synthesis in G1 occurs at between 0 and 3 h, DNA replication and cytokinesis in S phase occur at between 3 and 4 h, new valve formation inside the silicon deposition vesicle and exocytosis in G2/M phases occur at between 5 and 8 h, and ultimately, cells separate into two daughter cells and begin a new G1 phase (8). Thus, Si incorporation occurs in two phases: initially into girdle bands that facilitate cell expansion and later into valves, which enable cell division.
The average concentration of dissolved silicic acid [Si(OH)4, referred to here as Si] in the world's oceans is about 70 μM, but the Si concentration of surface water, which is the diatom habitat, is generally less than 10 μM and sometimes less than 1 μM (5, 9). An exception is the Southern Ocean, which has a silicic acid concentration of 65 μM, a concentration at which the uncharged Si(OH)4 molecule passively diffuses through diatom cell membranes (10, 11). The average low Si content of diatom habitats makes an active silicic acid transport mechanism via transporters a necessity for the growth of diatoms in most areas. Specific silicon transporter proteins (SITs) were identified in a large number of evolutionarily diverse diatoms with a high degree of conservation (12–17), showing an absolute necessity of Si transporters for diatom survival. Even the genome of Phaeodactylum tricornutum, a diatom without an obligate need for silicon, encodes functional SITs (18, 19). Three SIT genes were identified in the T. pseudonana genome on the basis of sequence similarity (12, 15, 20, 21). Under conditions with low Si concentrations (≤30 μM silicic acid), SITs are involved in the uptake of Si(OH)4, as shown by saturable kinetics and competitive inhibition by germanium (10). An alternative model of silicon uptake during surge uptake to meet the requirement for a large amount of silicon for valve synthesis was based on pinocytosis, which is the nonspecific engulfment of extracellular fluid and dissolved solute particles (22). However, classical pinocytosis is not cost-effective in terms of bioenergetics, membrane recycling, and kinetics, and thus, it cannot be the primary mechanism of silicon uptake in diatoms (23). Lately, a mathematical model based on compartmental analysis supporting the macropinocytosis-mediated silicon uptake hypothesis during initial and early valve formation when a high quantity of silicon is required, albeit without morphological and molecular evidence, was presented (24).
So far, the function of individual SITs in the transport of Si has been analyzed heterologously (15, 25), or silicic acid uptake in the native organism has been monitored without distinguishing between particular SITs (10). The development of transgenic tools enables an investigation into the roles that individual SITs play in vivo. In the study described here, we examined both transcript- and protein-level changes in the SITs in T. pseudonana in response to different silicic acid concentrations. We also determined their spatial localization. In addition, two of the SITs were downregulated using antisense RNA and RNA interference (RNAi), which generated phenotypic changes that suggest a major regulatory or signaling role for the SITs.
MATERIALS AND METHODS
Culture conditions.
Thalassiosira pseudonana (CCMP1335) stock cultures were grown in NEPC medium (http://www3.botany.ubc.ca/cccm/NEPCC/esaw.html) containing 100 μM Si(OH)4. Cultures were grown on an orbital shaker under continuous illumination of cool-white fluorescent lamps at 100 μE m−2 s−1 and 18°C.
Vector construction. (i) Gateway destination vectors.
A T. pseudonana Gateway destination vector, pMHL_78, for antisense RNA and RNAi was constructed by cloning a Gateway frame B cassette (Life Technologies) between the fcp promoter and fcp terminator after removing enhanced green fluorescent protein (eGFP) in pTpFcpGFP (26). For expression under the control of native promoters, the Gateway frame B cassette was cloned into the EcoRV site of pBluescript, generating the destination vector pMHL_71. Genes of interest were PCR amplified using primers containing the corresponding Gateway att sequences (see Table S1 in the supplemental material) cloned into the destination vectors using a MultiSite Gateway Pro kit (Life Technologies).
(ii) Expression vector.
For expression under the control of native promoters, DNA fragments, including promoter and full-length coding sequences devoid of stop codons, were amplified by PCR from T. pseudonana genomic DNA and cloned into a Gateway donor vector, pDONR221 attP1-attP4. The PCR fragment of eGFP was cloned into pDONR221 attP4r-attP3r. Terminator sequences were amplified from T. pseudonana genomic DNA and cloned into pDONR221 attP3-attP2. The respective entry vectors were cloned into the destination vector pMHL_71 to create transformation vectors (see Table S2 in the supplemental material). The expression vectors were comprised of 429-, 875-, and 810-bp-long 5′ flanking regions incorporating the promoter sequences of SIT1 (Thaps3_ 268895), SIT2 (Thaps3_41392), and SIT3 (Thaps3_35133), respectively, whereas the 3′ flanking region incorporating the respective terminator sequences were 500 bp in all three vectors. Each transformation vector was cotransformed with pMHL_9 expressing the nat1 gene (received from N. Kroger, Germany) under the control of the acetyl coenzyme A carboxylase promoter, which confers resistance to the antibiotic nourseothricin.
(iii) RNAi vector.
The RNAi vector (see Fig. S4 in the supplemental material) was constructed by cloning a full-length nat1 gene with the stop codon followed by the first exon and intron of the SIT1 gene fragment in the sense strand and only the first exon in the antisense orientation. The nat1 gene was PCR amplified with primers GW-35/GW-36, and the entry vector pMHL_103 was generated by BP recombination using the donor vector pDONR221-P1P4. For cloning of a sense fragment, a 346-bp-long fragment of the SIT1 gene, including the 222-bp-long first exon and the 124-bp-long intron, which forms a hairpin loop in the RNA transcript, was PCR amplified using primers GW-19/GW-20, and the entry vector pMHL_104 was generated by BP recombination using the donor vector pDONR221-P5P2. For the antisense fragment, the first exon was amplified in the antisense orientation using primers GW-21/GW-22, and the entry vector pMHL_105 was generated by BP recombination using the donor vector pDONR221-P31P2. Entry vectors pMHL_103, pMHL_104, and pMHL_105 were cloned into the destination vector pMHL_78 by an LR recombination, and the expression vector pMHL_106, which was later transformed into diatom cells, was generated.
(iv) Antisense RNA expression vectors.
Antisense RNA expression vectors (see Fig. S4 in the supplemental material) were constructed by cloning a full-length nat1 gene with the stop codon followed by a SIT1 gene fragment in the antisense orientation to express antisense RNA. The nat1 gene was PCR amplified with primers GW-25/GW-26, and the entry vector pMHL_107 was generated by BP recombination using the donor vector pDONR221-P1P5r. For cloning of a DNA fragment encoding an antisense RNA, an 820-bp-long fragment of the SIT1 gene, including the 710-bp-long 3′ half of the gene and the 110-bp-long 3′ untranslated region (UTR), was PCR amplified using primers GW-13/GW-14, and entry vector pMHL_99 was generated by BP recombination using the donor vector pDONR221-P5P2. Entry vectors pMHL_107 and pMHL_99 were cloned into the destination vector pMHL_78 by LR recombination, and the expression vector pMHL_108 was generated.
Diatom transformation.
Expression vectors were cloned into T. pseudonana by microparticle bombardment using a Bio-Rad Biolistic PDS-1000/He particle delivery system (27, 28). Briefly, exponentially grown cells were harvested, and 1 × 108 cells were plated in a 5-cm-diameter circle in the middle of an NEPC agar plate lacking antibiotics. A nuclear transformation was then performed by bombarding plasmid DNA-coated tungsten beads (diameter, 1.1 μm; M-17; Bio-Rad) at 1,350 lb/in2 under vacuum at a distance of 8 cm onto cells plated on the agar plate (1.5% Bacto agar). The plates were bombarded twice to obtain a higher transformation efficiency. Immediately after bombardment, the cells were overlaid with 10 ml of NEPC medium and placed under light for 24 h. The cells were then plated on NEPC agar containing 100 μg/ml nourseothricin (clonNAT; Werner BioAgents, Germany). Resistant colonies were then picked and transferred into each well of 24-well plates containing 2 ml of NEPC medium with nourseothricin. Positive clones were then confirmed by PCR using nourseothricin-specific primers, as described below.
Genomic DNA extraction and PCR.
After 1 week, algal cultures grown in 24-well plates were centrifuged (10,000 × g, 2 min), washed once with phosphate-buffered saline (PBS), and resuspended in 25 μl of PBS. The cells were then subjected to three cycles of freeze-thawing by placing them in dry ice for 5 min and heating them for 2 min at 70°C. The lysate was then heated for 5 min at 95°C to denature DNases. After centrifugation, the supernatants containing genomic DNA were stored at −20°C until further use. Several antibiotic-resistant clones were screened by growing them in 24-well plates containing antibiotics, followed by confirmation by PCR.
RNA extraction and quantitative reverse transcription-PCR (RT-PCR).
Cells were harvested by centrifugation, and total RNA was extracted using TRIzol reagent (29). RNA was treated with RNase-free DNase, followed by purification and concentration using an RNeasy Plus minikit (Qiagen). cDNA was then synthesized from equal amounts of purified total RNA for each sample using SuperScript III reverse transcriptase (Life Technologies). Quantitative PCR was performed with equal amounts of cDNA using a LightCycler (v2.0) apparatus and a FastStart DNA MasterPlus Sybr green I kit (Roche Applied Science). Primers for quantitative PCR were designed using the Integrated DNA Technologies real-time primer design tool. Standards for real-time PCR were dilutions of T. pseudonana genomic DNA. Normalization was done using the TATA box binding protein (Thaps3_264095) as a housekeeping gene (30). Dilutions of cDNA were done to ensure that amplifications were in the linear range. The antisense clones were verified by antisense quantitative PCR (31, 32). Expression of the RNAi and antisense RNA transcripts was verified by PCR amplification with primers RT19/RT20 and RT25/RT26, respectively (see Table S1 in the supplemental material). The effect of antisense RNA and RNAi was analyzed by quantitative RT-PCR with two primer sets, RT30/RT31 and RT30/RT32, for SIT1 and SIT2, respectively (see Table S1 in the supplemental material), and mRNA levels were calculated as their means.
Western blotting.
Cells were resuspended in 2% SDS extraction buffer, pH 6.8, and frozen and thawed three times in dry ice (5 min) followed by 70°C water (2 min) and were then heated for 5 min at 95°C. The lysate was centrifuged, and the concentration of the supernatant containing the total protein extract was measured using a Bio-Rad DC protein assay kit before loading an equal amount of protein into each well containing NuPAGE Novex 4 to 12% bis-Tris gel (Life Technologies). Polyvinylidene difluoride membranes blotted with proteins were incubated with anti-SIT antibody (21) or antitubulin antibody (Santa Cruz Biotechnology), followed by horseradish peroxidase-conjugated antirabbit secondary antibodies (Pierce). The blots were then processed using a SuperSignal West Pico kit (Pierce) and exposed on X-ray film. Densitometry was performed using Bio-Rad Image Lab (v4.1) software.
Silicon uptake assay.
The silicomolybdate assay was used to measure the silicic acid concentration in the growth medium (8, 33). Cells were harvested by centrifugation at 10,000 × g for 10 min. A portion of the supernatants was collected, and the silicic acid concentration was quantified. The amount of silicic acid that disappeared from the medium was converted into silicic acid uptake in terms of the number of fmol per cell (8).
Silica incorporation assay.
Cells at logarithmic growth phase grown in NEPC medium [100 μM Si(OH)4] were harvested and incubated in silicic acid-free medium for 24 h. One hundred nanograms · milliliter−1 of PDMPO [2-(4-pyridyl)-5-((4-(2-dimethylaminoethylamino-carbamoyl)methoxy)phenyl)oxazole; LysoSensor YellowBlue DND-160; Life Technologies] was added, and after 5 min sodium silicate was added to achieve a 20 μM Si(OH)4 concentration. Cell samples were taken at various time points. Incorporation of newly formed silica was analyzed by virtue of PDMPO fluorescence (34, 35) using an Influx sorting flow cytometer (excitation, 488 nm; emission, 530 nm; 40-nm band pass; BD Biosciences). The flow cytometer data were analyzed using FlowJo (v7.6.5) software.
ImageStream flow cytometry.
Cells transformed with an eGFP tag were analyzed with an ImageStream imaging flow cytometer (X-100; Amnis Corp.) using 100-mW 488-nm laser excitation. At least 20,000 fluorescent as well as bright-field and side scatter cell images per sample were collected at ×40 magnification. Data were then analyzed using image analysis software (IDEAS, v5; Amnis Corp.).
Neutral lipid quantification.
The effect of downregulation of silicon transporters on neutral lipid accumulation was analyzed using a green lipophilic fluorescent dye, BODIPY 493/503 (4,4-difluoro-3a,4a diaza-s-indacene; Life Technologies), as a proxy for the neutral lipid (36). Exponentially grown cells were subcultured in NEPC medium with 100 μM Si and grown for 24 h to deplete Si from the medium. Thereafter, 20 μM Si was added to the cultures and samples were taken and stored at −20°C. Cell pellets were incubated with 2.56 μg/ml BODIPY for 15 min at room temperature and analyzed in the Influx flow cytometer (excitation, 488 nm; emission, 530 nm; 40-nm band pass). The data were subsequently analyzed using FlowJo (v7.6.4) software.
RESULTS
Regulation of SIT mRNA in response to Si availability.
We analyzed SIT transcript levels during both Si starvation and replenishment. Microarray data showed that during Si starvation, SIT1 and SIT2 mRNAs were upregulated from the 4th hour (Fig. 1A). The SIT1 transcript was more highly induced (4.6 log2-fold change) than the SIT2 transcript (2.8 log2-fold change), and recent RNA-seq analysis indicated that the absolute amounts of SIT1 transcripts were on the order of 3- to 4-fold higher than those of SIT2 (S. R. Smith, C. Glé, R. M. Abbriano, J. C. Traller, E. Trentacoste, A. Davis, O. Cook, M. Vernet, A. E. Allen, and M. Hildebrand, submitted for publication). In contrast, both SIT1 and SIT2 mRNAs were downregulated upon addition of Si after 24 h starvation, with a slight increase being found at 4 h during the S phase of the cell cycle (Fig. 1B). SIT3 mRNA showed minimal responses to either the presence or the absence of Si, with a slight downregulation occurring at 4 h during Si starvation (Fig. 1A) and a slight downregulation occurring in the 2nd hour upon addition of Si (Fig. 1B).
FIG 1.
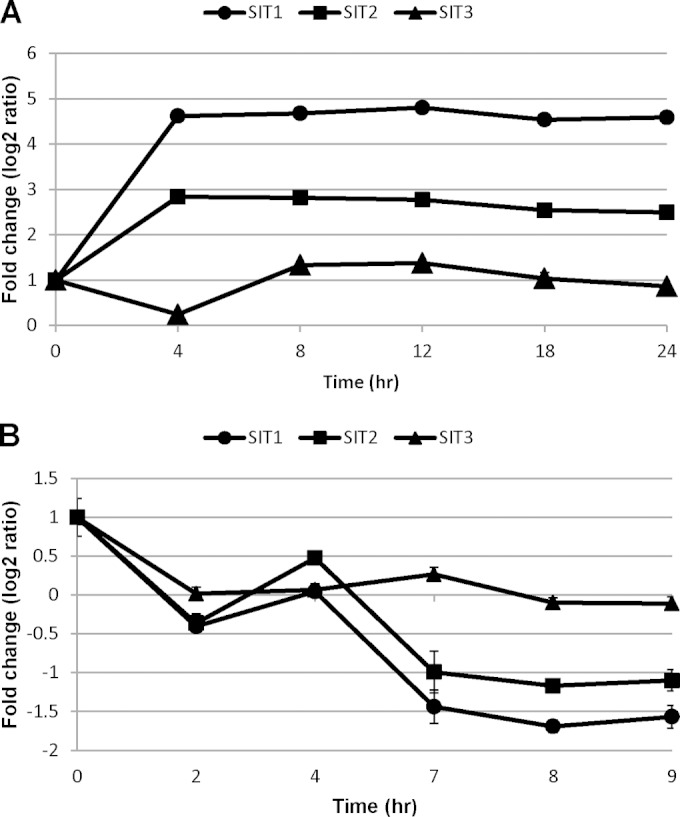
Patterns of T. pseudonana silicon transporter transcript expression under Si-replete conditions (42), deposited in NCBI's Gene Expression Omnibus and accessible through GEO Series accession number GSE37081, and Si-deplete conditions (Smith et al., submitted). The transcript levels were analyzed by probing microarrays with mRNA from Si-starved cultures (A) and an Si-replete synchronous culture (B). Each Affymetrix GeneChip whole-genome tiling array contained a total of 524,909 sense-strand probes (average, 16 probes per gene) and was probed with wild-type T. pseudonana cDNA. Bars indicate standard deviations from two microarrays on which an equivalent of 32 probes hybridized with cDNA from biological duplicates (42).
Molecular cloning, localization, and expression analysis of the SIT1 and SIT2 genes in T. pseudonana.
To determine the subcellular localization of the silicon transporters, we transformed T. pseudonana with T. pseudonana TpSIT1, TpSIT2, and TpSIT3 genes with eGFP fused to the C terminus under the transcriptional control of the respective native promoters (see Fig. S1 in the supplemental material). Epifluorescence microscopy of transgenic cells expressing SIT1 and SIT2 in Si-depleted cultures showed bright eGFP fluorescence in the plasma membrane under Si starvation for 24 h (Fig. 2). This is consistent with their primary involvement in silicic acid uptake into the cell. The interface regions between two dividing daughter cells had double the fluorescence of the rest of the plasma membrane (Fig. 2A). A faint circular eGFP label was often detected in the endoplasmic reticulum in G1 phase (Fig. 2B) and subsequently incorporated into the new plasma membrane. On the other hand, SIT3 was not localized specifically in the plasma membrane under either Si-replete or -deplete conditions, but we observed a single bright area within the cell, plus diffuse fluorescence in what appeared to be intracellular membranes, irrespective of the presence or absence of Si. Transgenic lines expressing TpSIT3-eGFP were unstable, and in spite of repeated attempts, no data other than initial localization data were obtained for these lines. Overall, the data suggest that SIT1 and SIT2 have differential contributions in Si transport into the cell, whereas there is little to no contribution of SIT3 in external Si uptake.
FIG 2.
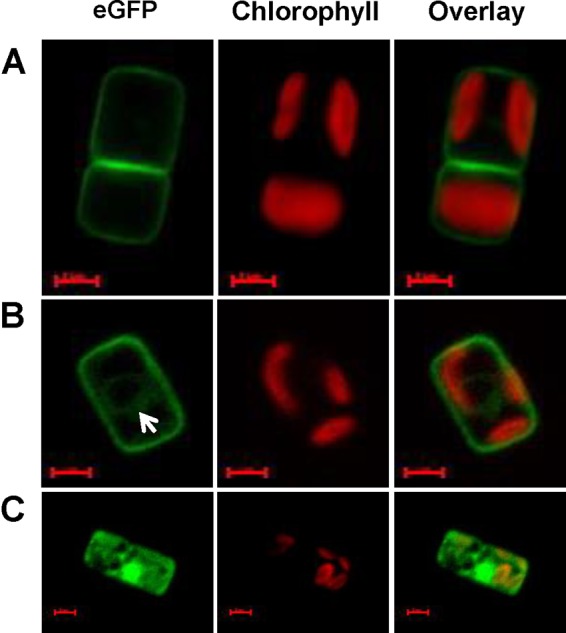
High-resolution images of transgenic T. pseudonana cells expressing eGFP-tagged SIT1 (A), SIT2 (B), and SIT3 (C) under the transcriptional control of the respective native promoters in Si-starved cultures. Green, eGFP fluorescence; red, chlorophyll autofluorescence. Images were taken using a Zeiss Axio Observer.Z1 microscope with a Plan-Apochromat 63×/1.40 oil DIC M27 objective and a 38 HE filter set. Exposures times for eGFP were 307 ms (A), 300 ms (B), and 425 ms (C). The white arrow in panel B indicates the faint circular eGFP label often detected in both SIT1 and SIT2. SIT3 localization is not specific to the plasma membrane. Bars = 2 μm.
Protein response to Si depletion.
We next monitored the relative abundance of SITs using eGFP fluorescence. To determine the cell-to-cell consistency in response, we quantified not only the fluorescence intensity but also the number of cells expressing eGFP-tagged SITs in Si-replete or -depleted medium using an imaging flow cytometer. Only a small percentage of the population (0.11 to 0.25%) exhibited fluorescence in the Si-replete culture, whereas more than 90% of the population expressed SIT1 and SIT2 after 12 h of Si starvation (see Fig. S2 in the supplemental material). The eGFP intensity of SIT1 was higher than that of SIT2 at all time points (Fig. 3A), reflecting a higher number of SIT1 molecules. The protein expression data, however, did not strictly correlate with mRNA expression levels. Although both were rapidly induced by 4 h, mRNA transcript levels remained constant after 4 h (Fig. 1A), but the SIT1 and SIT2 protein levels continuously increased (Fig. 3A), indicating an increasing number of transporters per cell. In addition, SIT levels continued to increase even until 48 h in some healthy cultures and decreased by 72 h (data not shown).
FIG 3.
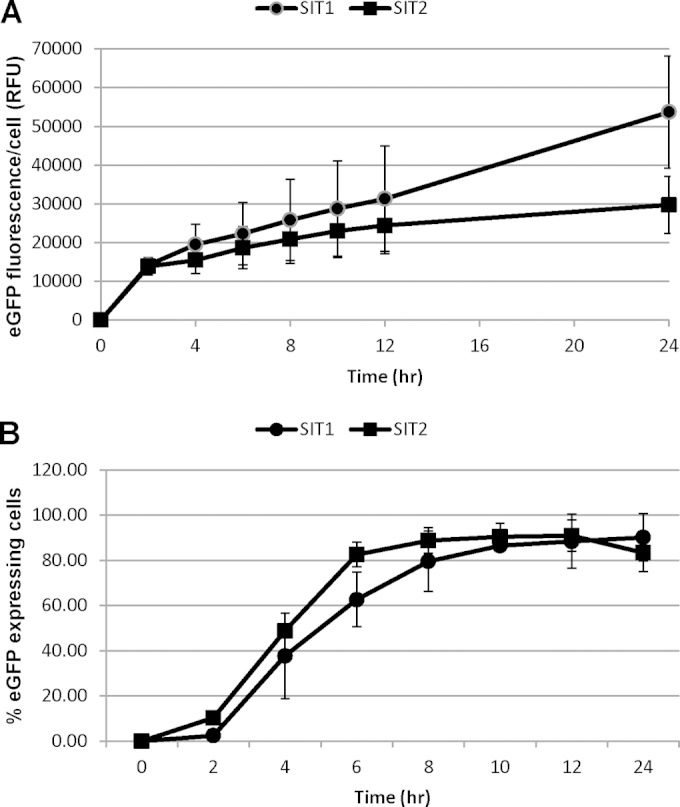
Time course expression analysis of eGFP-tagged SIT1 and SIT2 under the control of the respective native promoters in Si-starved T. pseudonana cells. Exponentially grown cells were transferred to silicon-deprived medium. Expression levels were then followed by measuring eGFP fluorescence by imaging flow cytometry (488-nm laser, 100 mW) of the 20,000 cell images collected. (A) Normalized mean fluorescence intensity per cell. RFU, relative fluorescence intensity. (B) Percentage of cells expressing SITs. Bars represent standard deviations (n = 3).
Protein response to Si replenishment at different concentrations and correlation of SIT level to Si incorporation and cell cycle S phase.
We next analyzed changes in SIT1 and SIT2 protein abundance upon addition of different concentrations of Si to Si-starved cultures. It was previously shown that the SITs are predominantly needed for Si transport at an Si concentration of ≤30 μM, but at higher concentrations, diffusion of Si predominates (10). Exponentially grown cells were incubated in silicon-free medium for 24 h and made replete with 20, 30, 50, and 200 μM silicic acid, and eGFP fluorescence was monitored using imaging flow cytometry. Initially, cells were brightly fluorescent after 24 h of silicon starvation. The fluorescence levels then decreased in the presence of silicic acid, but the extent of the decrease was concentration dependent (Fig. 4). In the case of SIT1, the fluorescence level decreased similarly under all conditions until 3 h, and then the response differentiated, with an increase in levels occurring at the two lowest silicic acid concentrations and a relatively sharp decrease occurring by 4 h for the two higher concentrations. The responses to these two higher concentrations were further differentiated by an eventual increase for 50 μM silicic acid but a decrease followed by a leveling off for 200 μM. In general, the reduction in protein levels related inversely to the Si concentration added, but additional features were present. Unlike the results for SIT1, SIT2 levels dipped by 2 h for all Si concentrations. Thereafter, expression continuously increased in the presence of 20 μM Si, whereas in the presence of other concentrations, the level of protein increased, decreased, and then increased in a concentration-dependent manner. As described previously (8), after addition of 200 μM Si into cultures starved for 24 h, releasing Si-starved diatoms from Si arrest, a graph of 1/SIT1 level (derived from Fig. 4A) against the level of rhodamine 123 (a fluorescent dye for staining newly deposited silica)-stained silica incorporation showed a perfect but inverse relation between the level of Si incorporation and the SIT1 level (Fig. 5A and B), consistent with the SIT1 amount being primarily regulated by silica incorporation. Interestingly, the change in the pattern of the SIT2 level (derived from Fig. 4B) coincided with the S-phase peak of the cell cycle, as described previously (8) (Fig. 5C). We repeatedly observed a brief increase in the SIT2 level and mRNA transcript level (1 log2-fold) during the S phase of the cell cycle (Fig. 1B). On the other hand, the SIT1 mRNA level was upregulated by 0.6 log2-fold during this time, but an increase in protein level was not observed.
FIG 4.
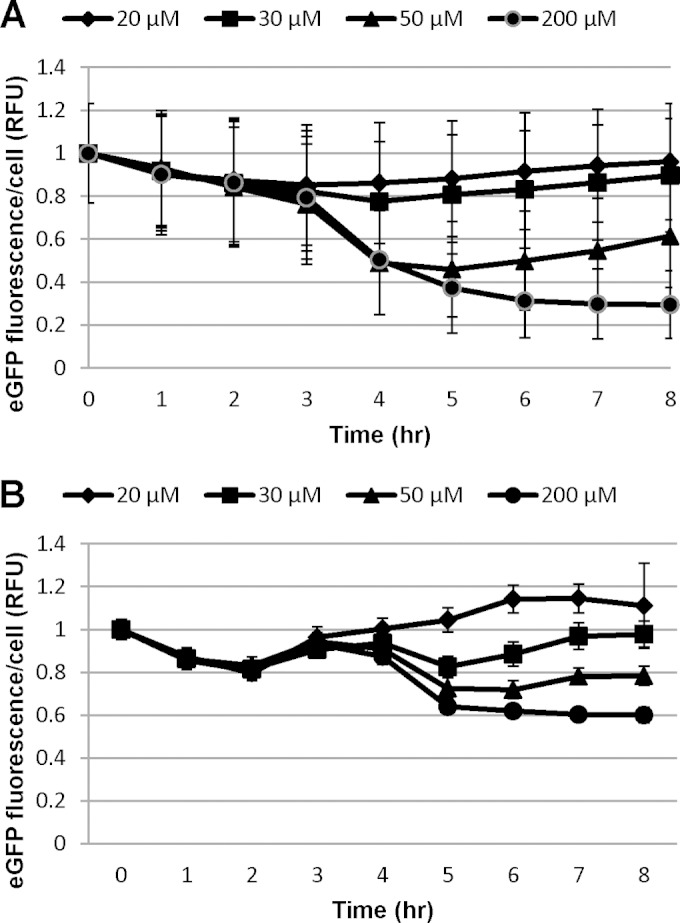
Expression of eGFP-tagged SIT1 (A) and SIT2 (B) in the presence of various concentrations of silicic acid added to 24-h silicon-starved cultures. Expression was monitored by measuring the eGFP fluorescence, using an imaging flow cytometer, of the 20,000 cell images collected. Bars represent standard deviations (n = 3).
FIG 5.
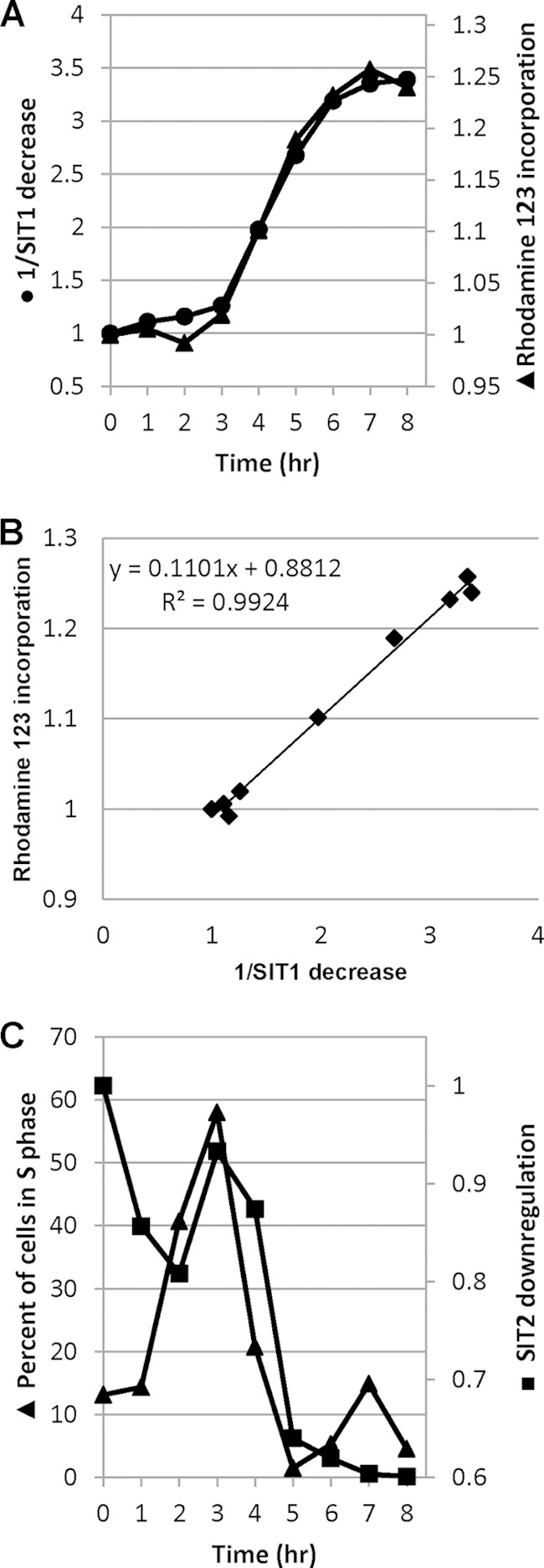
Correlation between SIT expression and cellular processes. (A) Inverse of the SIT1 level (derived from the results obtained with 200 μM Si, shown in Fig. 4A) versus incorporation of a fluorescent silica tracer, rhodamine 123, as described previously (8), in the cell walls of T. pseudonana; (B) correlation between 1/SIT1 level and Si incorporation; (C) SIT2 expression level relative to percentage of cells in S phase (8).
Effect of RNAi- and antisense RNA-mediated downregulation of SIT1 and SIT2.
We employed antisense RNA and RNAi to knock down silicon transporter genes in T. pseudonana. We chose SIT1 and SIT2 as the targets for knockdown because of their implied major role in silicic acid transport (10, 21), which includes a role in plasma membrane localization and mRNA and protein changes in relation to silicon status; in contrast, SIT3 did not share these features (Fig. 1 and 2) (21). Because of sequence similarity (see Fig. S3 in the supplemental material), one construct could be used to knock down both genes simultaneously, which, according to our hypothesis, would have a substantial effect on silicic acid transport at low extracellular concentrations. Separate knockdowns of SIT1 and SIT2 could not be pursued with the vector construction design used in this study.
Wild-type T. pseudonana cells were transformed with plasmid vectors expressing TpSIT antisense RNA or RNAi constructs (see Fig. S4 in the supplemental material). Several antibiotic resistance clones were analyzed for the effect of SIT downregulation by Western blotting and real-time quantitative PCR. Two pairs of quantitative RT-PCR primers specific for either TpSIT1 or TpSIT2 were used, and mRNA levels were calculated as their means. RNAi clones R-13, R-19, R-20, and R-21 had a lower level of SIT mRNA than the wild type, whereas clone R-14 had a higher level (Fig. 6A and B). Clone R-13 showed the greatest effect of RNAi on the mRNA level (a 95% decrease) relative to that for the wild type; clones R-19 to R-22 showed reductions of 50 to 75%. Antisense clone A-2 showed the greatest effect of antisense RNA on the mRNA level (80% decrease), followed by clones A-7 and A-8 (15% and 10% reductions, respectively). On average, the effect of knockdown was greater for the RNAi clones (50% reduction) than the antisense clones (35% reduction) (Fig. 6B).
FIG 6.

RNAi and antisense RNA knockdowns of SITs. (A) Quantitative RT-PCR evaluation of the SIT mRNA level in RNAi and antisense RNA transgenic lines. TATA box binding protein mRNA was used for normalization. A and R, antisense RNA and RNAi clones, respectively. Bars represent the standard errors from quantitative RT-PCRs performed with primers specific for SIT1 and SIT2. (B) Average levels of SIT mRNA determined from the results presented in panel A. (C) (Top) Western blot of clones in which SIT was knocked down by antisense RNA and RNAi probed with anti-SIT antiserum; (bottom) Western blot of α-tubulin antibodies used to indicate the amount of protein loaded. (D) Densitometry data normalizing the SIT level to the α-tubulin level. WT, wild type.
Both RNAi and antisense clones were subjected to Western blot analysis using anti-SIT antibodies (21). The antiserum recognizes all three SITs (21); thus, a decrease is representative of a decrease in total SIT levels. Immunoblots were quantified, and the amounts were normalized against the amount for the loading control, α-tubulin. All the transgenic strains showed a decreased amount of SIT compared with that of the wild type (Fig. 6C and D). Overall, the amount of SIT protein in all the RNAi clones was reduced by 80% compared with that in the wild type, whereas the decrease in SIT protein level in the antisense clones was 55% (Fig. 6D), a trend which is consistent with the overall mRNA downregulation (Fig. 6B).
We observed that both the RNAi- and antisense RNA-mediated knockdown methods reduced the protein abundance, but the results did not always correlate with the changes in transcript levels. Clone R-14 had a higher mRNA level, despite a lower protein level, for example, than the wild type. Similarly, mRNA levels in clones A-7 and A-8 were not much lower than the level in the wild type, but the protein levels were reduced (Fig. 6B).
Effect of SIT downregulation on Si transport and incorporation.
After establishing the effect of antisense RNA and RNAi on SIT mRNA and protein levels, we investigated the effect of the knockdowns on silicon transport and cell wall silica incorporation. Following preliminary Si uptake experiments with RNAi and antisense RNA clones in the presence of 10, 20, and 200 μM Si(OH)4, we chose 20 μM silicate for use in further experiments, in which the involvement of SITs in the transportation of silicic acid was clearly observed. The net transport of silicic acid was measured by the disappearance of silicic acid from the medium (8). The three antisense and three RNAi clones examined had reduced silicic acid transport compared with that of the wild type (Fig. 7). While all silicic acid was taken up by the wild type in 2 h, three RNAi clones took one extra hour to take up the same quantity of silicic acid (Fig. 7A). Similar results were obtained with the antisense RNA knockdowns (Fig. 7B).
FIG 7.
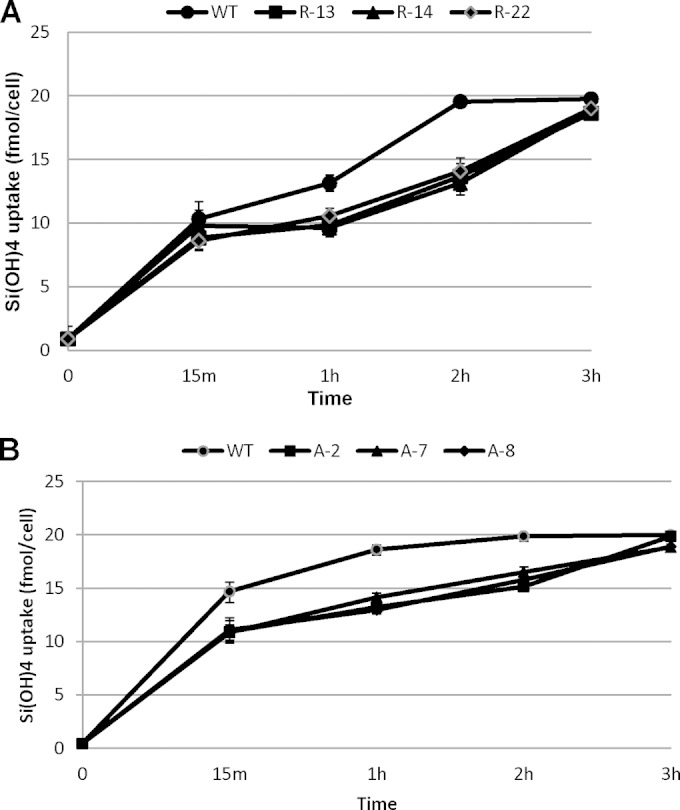
Effect of SIT downregulation on silicon uptake in T. pseudonana. Cultures starved overnight were made replete with 20 μM Si(OH)4, and silicic acid uptake was measured by monitoring the disappearance of Si(OH)4 from the medium. Bars represent standard deviations (n = 3).
We analyzed the effect of SIT downregulation on silica incorporation in the cell wall by virtue of PDMPO labeling (34, 35). In the presence of 20 μM Si(OH)4, all the antisense and RNAi clones showed lower levels of silica incorporation than the wild type (Fig. 8A and B). Control experiments with a subset of clones showed no difference in incorporation between the wild type and knockdown clones in the presence of 200 μM silicate (Fig. 8C). The relative extent of uptake and incorporation differed substantially when the wild type and the knockdown lines were compared. Comparing the uptake by the wild type with the average uptake by all three clones at 3 h indicated that the clones took up 97% of the silicic acid that the wild type did but incorporated only 62% of the amount of silica (Fig. 7 and 8).
FIG 8.
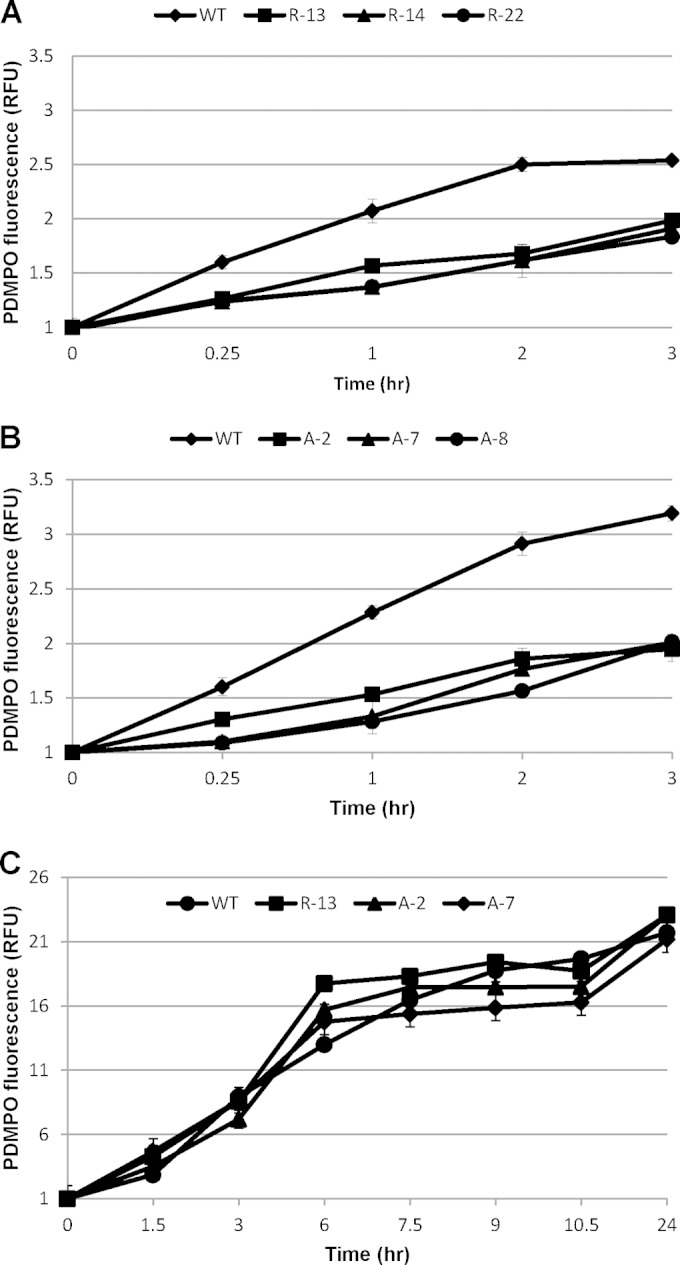
Effect of SIT downregulation on silica incorporation in T. pseudonana frustules after cultures starved overnight were made replete with 20 μM Si(OH)4 (A and B) and 200 μM Si(OH)4 (C). Newly synthesized cell wall silica was measured by virtue of PDMPO labeling in a flow cytometer. Bars represent standard deviations (n = 3).
Effect of knockdowns on TAG accumulation.
Silicon starvation is an established means to induce the accumulation of triacylglycerol (TAG) in diatoms (37, 38). Obtaining conditions where the amount of Si in the growth medium is minimal in an open outdoor cultivation system, as is anticipated for use for large-scale biofuels production, may be problematic, due to wind-blown dust and other factors. We tested whether the SIT knockdowns could induce TAG more rapidly than the wild type by effectively inducing starvation under conditions in which silicon was still present in small amounts in the medium (Fig. 9). In this experiment, cultures were grown with 100 μM Si(OH)4 for 24 h, and after harvesting, Si was added at 20 μM and then TAG accumulation was followed by flow cytometry using BODIPY staining (39). According to the data in Fig. 7, all the silicic acid should have been depleted from the medium by the 4-h time point, yet all knockdown clones accumulated TAG more rapidly and to higher levels than the wild type starting at that time. There was no increase in lipid accumulation in the wild type by 4 h, whereas increases of 1.2- to 2.2-fold were observed in the RNAi clones and increases of 1.2- to 1.5-fold were observed in the antisense clones (Fig. 9). A 1.5-fold increase in the amount of neutral lipid in the wild type was observed only by 12 h.
FIG 9.

Effect of SIT downregulation on neutral lipid accumulation. Exponential-phase cultures were grown overnight in the presence of 100 μM Si(OH)4, harvested, and resuspended in 20 μM Si(OH)4. Samples were then incubated with BODIPY, and fluorescence was monitored by flow cytometry. Bars represent standard deviations (n = 3).
DISCUSSION
Here we explored the roles of individual SITs in Si transport in T. pseudonana by eGFP tagging and RNAi- and antisense RNA-mediated knockdown and the effect of knockdown on TAG accumulation. Previous work relied on bulk analyses in which mRNA levels for the different SITs could be distinguished but protein levels could not (21). The development of new genetic manipulation tools enabled examination of the SITs individually at the protein level, and further distinction of their roles was documented. We were able to examine transcript level changes for all three SITs and protein-level changes for SIT1 and SIT2. The knockdown approaches used in this study focused on the SITs predominantly involved in extracellular uptake. Because of extensive sequence similarity and the particular knockdown approaches that we chose to use, we could not distinguish between the roles of SIT1 and SIT2 (both were knocked down), but the results from this study suggest that developing the means to knock them down individually could be fruitful.
It was likely that at least a subset of the SITs in T. pseudonana would be localized in the plasma membrane, which is the most logical site for silicon transport from the external milieu into the cell. Here we demonstrated that eGFP-tagged SIT1 and SIT2 were indeed localized in the plasma membrane (Fig. 2), consistent with a major role in silicon transport into the cell. Plasma membranes were strongly fluorescent under Si-starved conditions, whereas some weak labeling of what appeared to be the endoplasmic reticulum was also detected. On the other hand, SIT3 under the control of either the native promoter (Fig. 2) or a more highly expressed constitutive fcp promoter was not localized to the plasma membrane (data not shown). This may be due to the fact that SIT3 contains a type II signal anchor peptide that usually remains uncleaved (40), whereas SIT1 and SIT2 have type I signal peptides (see Table S3 in the supplemental material). Target prediction with pTARGET predicted the plasma membrane for SIT1 and SIT2 but the peroxisome for SIT3 (see Table S3 in the supplemental material). SIT3 did not appear to have a precise intracellular location but exhibited general labeling in intracellular membrane systems which have features of the endoplasmic reticulum, but it may also be more broadly distributed. Further characterization of the location of SIT3 was not possible because the transgenic lines were consistently unstable and eGFP fluorescence was rapidly lost. We can say with certainty only that the protein does not appear to be localized specifically to the plasma membrane, suggesting that SIT3 is not involved in Si uptake into the cell. Potentially, it may play an intracellular transport role or serve a regulatory function. We made other attempts to localize SIT3 by tagging eGFP at the N terminus under the control of the native promoter and terminator. However, we could not detect any fluorescence under either Si-replete or Si-starved conditions.
SIT mRNA levels showed a differential response to Si starvation and replenishment (Fig. 1), consistent with the SITs playing different functional roles. The induction of SIT1 and SIT2 mRNA is consistent with a starvation response in which transporter levels increase in an attempt to scavenge whatever substrate is available (29, 41). Downregulation of the SITs after silicic acid addition is consistent with their minor role in uptake under conditions where Si diffusion into the cell would predominate (10). An interesting response seen here and previously (21) is the induction of mRNA during S phase, a time when Si is not needed for cell wall synthesis. This is consistent with a response to populate new cells with the transporters and is not related to a cellular need for Si, as no silica structures are being made at that time.
Previous work indicated a disconnection between SIT transcript and overall protein levels (21), but in this study, SIT1 and SIT2 levels could be distinctly and directly monitored by eGFP tagging. The lack of appreciable SIT during growth in the presence of 200 μM exogenous Si (Fig. 3, 0 h) is consistent with a minor role for the SITs in the presence of high Si concentrations that are sufficient to meet cellular Si demand entirely through diffusion. A very small percentage of cells in the population expressed SIT1 and SIT2 at 0 h, and not all cells expressed SIT1 and SIT2 by the time of maximal induction (Fig. 3). This indicates a very tight control of expression, with some variability occurring, possibly due to epigenetic variations or differences in silicon status in individual cells.
The response of eGFP-tagged SIT1 and SIT2 to silicon replenishment indicated distinct roles of SIT1 and SIT2. The response for SIT1 was consistent with a need for SITs under conditions with small amounts of Si but not under those with larger amounts of Si. In the presence of 20 and 30 μM Si, cultures would proceed through the girdle band formation stage of cell wall synthesis (8) but then become depleted of silicon, resulting in an increase in SIT1 levels (Fig. 4A). The dose dependency of the response is evident because SIT1 increased sooner in the presence of 20 μM Si than in the presence of 30 μM Si. In the presence of 50 μM Si, enough silicon was present to at least initiate valve synthesis at between 3 and 5 h (8), which was manifested as a more rapid decrease in SIT1 levels, after which silicon became limiting, resulting in a subsequent increase in SIT1 levels. In the presence of 200 μM Si, cells would complete valve synthesis and would never be limited for silicon, and SIT1 levels did not increase. The pattern of decrease in the amount of SIT1 at higher concentrations was distinctive and corresponded exactly with previously determined periods of silicic acid requirements for cell wall formation (8). The decrease in the amount of SIT1 during valve formation, when the cell requires more silicic acid, is counterintuitive. This demonstrates that the cell is actively decreasing the SIT1 level in inverse correspondence with Si needs. This indicates a negative regulatory coupling between SIT1 levels and cell wall synthesis. One possible explanation is that the cell is responding to the rapid influx of Si and turns down SIT1 as a response to total influx (or the buildup of an internal pool). An important concept in this hypothesis, consistent with the findings of a previous analysis (8), is that incorporation and not uptake is controlling the process: the cell is drawing in silicic acid when needed and not pumping it in to drive silicification. Another contribution to silicon homeostasis could be Si efflux via efflux transporter proteins. We identified a putative efflux protein via transcriptomics of T. pseudonana, but we have not yet analyzed the role of this protein (42).
SIT2 had a response distinct from that of SIT1 after replenishment with different Si concentrations. Similar to the findings for SIT1, a dose dependency was observed, but a distinctive increase occurred at 3 to 4 h (Fig. 4B). Comparison with previous data (8) indicates that the response correlates well with the percentage of cells in the S phase of the cell cycle, after an initial decline related to recovery from silicon starvation (Fig. 5). Two possible explanations for an increase in SIT2 during S phase, when silicic acid is not needed for cell wall component formation, are that (i) the cell is preloading silicic acid in anticipation of valve formation or (ii) the cell is populating new daughter cells with SITs. Because no appreciable increase in silicic acid intracellular pools occurs during S phase and prior to valve formation (8), the former hypothesis is not supported. This suggests that SIT2 could be replenishing transporter levels during the expansion of cellular membranes.
SIT-knockdown lines enabled us to test the effect of inhibiting transport on cellular processes. In order to select knockdown clones, we had to screen only a few transgenic clones from the agar plates and found that protein in most of them was downregulated, unlike the findings in previous knockdown studies (43). We attributed it to the efficiency of our vector design. A spliceable intron was included as a spacer in the RNAi vector to increase the frequency of silencing (44, 45). Moreover, we used a long gene fragment, which can facilitate efficient gene silencing by minimizing the variability in efficiency attributed to different sites of genes and the length of the short interfering RNA (siRNA) (46). In addition, a long double-stranded inverted repeat cleaved into a number of 19- to 21-bp-long siRNAs by RNase III (47) can contribute to gene silencing efficiency (48). In the case of antisense constructs, we included a 110-bp-long 3′ UTR, in addition to the coding region, which, upon entry into the nucleus, could contribute to increasing the gene silencing efficiency by masking the 3′ UTR from microRNA binding and by modulating polyadenylation and thus alter the function of the target RNA (49–53). On the other hand, stoichiometric binding of antisense RNA at the coding region of the target mRNA in the nucleus induces RNase H1-mediated degradation of the target mRNA, whereas in the cytoplasm it prevents translation into a full-length protein and the resulting improperly folded proteins are destined for destruction (53–55). We observed a range of levels of mRNA transcription and protein expression in knockdown lines (Fig. 6), which could be due to the randomness of transgene insertion in chromosomes, or the positional effect. The general effectiveness of knockdown is obvious from the average level of reduction of mRNA and protein (Fig. 6B and D), and thus, knockdown methods with both RNAi and antisense RNA were generally found to be effective when a complete knockout was not desirable. When required, an individual clone with the strongest knockdown effect can be selected by screening enough transgenic lines.
Initially, we tested the effect of knockdown of SITs in the presence of 200 μM Si(OH)4 and observed that silicic acid uptake and silica incorporation in all the clones were more or less equal to those in the wild type, consistent with diffusion being predominant. In the presence of 20 μM extracellular silicon (Fig. 7), the wild type depleted the medium of silicon by 2 h, whereas the transgenic clones lagged and took 1 h longer. Diffusion still contributes to overall uptake even at low Si concentrations, but it is clear that knockdown of the SITs has a significant effect. Silica incorporation not only was faster in the wild type than in the downregulated clones (Fig. 8) but also was more complete. This suggests a tighter coupling between the SITs and silica deposition than between the SITs and silicon uptake, which would be expected if diffusion contributes, and this finding is supported by several other pieces of evidence presented here (e.g., Fig. 2) and our hypothesis about coupling between SIT stability and cell wall synthesis. Silica incorporation occurs as a stepwise process during the first 0 to 3 h with the sequential formation of girdle bands (8); perhaps the difference in incorporation between the wild type and the knockdown lines resulted from fewer girdle bands being synthesized in the latter. A nonlinear correlation between the extent of knockdown and a change in Si uptake was observed (Fig. 6 and 7). This could be due to uptake involving both SIT-mediated and diffusional transport, which is especially exacerbated at low Si concentrations. In addition, there was not a linear relation between SIT protein and uptake activity, as has been demonstrated by kinetics measurements showing a sigmoidal curve indicative of a cooperative interaction between proteins involved in affecting transport (10).
An established effect of silicon starvation-induced cell cycle arrest in diatoms is the induction of TAG accumulation (37, 38). This is an attractive feature for the use of diatoms for biofuel production (56), because silicon limitation occurs more rapidly than nitrogen limitation (due to a lack of appreciable intracellular stores) and silicon limitation has fewer detrimental effects on cellular metabolism. The practical limitation of silicic acid in a large-scale open cultivation system may be challenging, given the possibility of undesired Si input from wind or other sources. If the cell could be engineered to sense silicon starvation before levels reach zero, then the induction of TAG accumulation could still occur. This was tested in SIT-knockdown transgenic lines (Fig. 9). The data indicate that even though extracellular Si should be depleted by 3 h in all lines (Fig. 7), there was a more rapid induction of TAG accumulation in the transgenic lines by 4 h. The average induction by all three transgenic lines at 4 h was greater than that by the wild type at 12 h. The differences between the transgenic lines and the wild type were consistent from 4 to 12 h, with the average difference being from 1.7- to 1.8-fold higher at each time point evaluated. Thus, the effect of SIT knockdown on lipid accumulation is most significant at the early hours of Si depletion, with little effect subsequently being detected.
Overall, the data presented here substantiate several features related to the presumed roles of SITs but also raise some unanticipated questions regarding their true functions. A general conclusion is that SIT1 and SIT2 play major roles in silicic acid uptake only at environmentally relevant extracellular Si concentrations, consistent with previous kinetic measurements (10), and this has now been substantiated in terms of the regulation of mRNA and protein. This was documented by the decrease in SIT levels with increasing amounts of added Si (Fig. 4) and the lack of an effect of knockdowns on Si uptake in the presence of 200 μM Si (Fig. 8C). The concept of SITs being important in Si uptake only in the presence of current environmentally relevant concentrations raises questions about the evolution of these transporters. At the time that diatoms evolved, Si levels in the oceans were high (57), and diffusion-mediated transport of silicon would have been predominant. As diatoms and other siliceous organisms reduced the ocean levels of Si, a need for transporter-mediated uptake would have arisen. This suggests that the SITs may not have been involved in the development of the silicification process, unless they were required for transport across intracellular membranes.
Unanticipated results relate to SIT1 and SIT2 expression patterns and the effect of knockdowns on silica incorporation and TAG accumulation. It may make logical sense to downregulate SIT1 in the presence of excess Si, but why would the cell correlate the downregulation so precisely in accordance with silica incorporation, especially since the cell does not need the SITs when exogenous Si passively diffuses into cells (Fig. 4A)? A hypothesis consistent with the data is that the cell is using the SITs to monitor whether sufficient silicic acid is present to complete the formation of a cell wall component, particularly the valve. Alternatively, another silicon-sensing protein upstream of silicon transport could be involved; however, the SITs are the only proteins yet identified to specifically interact with silicic acid (15), and there is precedence for transporters functioning as sensors (58, 59). The TAG accumulation results (Fig. 9) also support the suggestion that the SITs are directly involved in sensing. Knowing that sufficient Si was present would be essential to cell survival; an incomplete cell wall structure could compromise the integrity of the cell. Titration of the amount of SIT could be a mechanism for monitoring extracellular Si: higher SIT levels relate to lower Si levels. This concept may also relate to the TAG accumulation results. In Fig. 9, at 4 h, Si was depleted from all cultures, but the knockdown lines had accumulated TAG many hours before the wild type did. The difference between the wild type and the knockdowns is the lower abundance of the SITs in the latter. TAG accumulation in diatoms is associated with cell cycle arrest (56). If the cell uses SITs to sense whether enough Si is present to complete the cell wall and cell cycle, and knockdown lines have less SIT, then the mechanism accounting for Si levels could be affected, signaling, artificially, that the cell will not be able to progress through the cell cycle. In the wild type, the cell will also not be able to progress through the cell cycle, but the signal for this could be lacking. This suggests that the TAG accumulation response could be due to a signaling mechanism and not cell cycle arrest per se.
The ability to monitor individual SIT levels and to knock down the ability of the cell to use SIT-mediated uptake alters our concepts of the role of these proteins in unexpected ways. In addition to the SITs playing a major role in silicon uptake at environmentally relevant extracellular Si concentrations, which was previously documented in kinetic analyses (10), they also appear to play a more substantial regulatory role related to the extracellular availability of silicon. Considering that when diatoms arose evolutionarily, proteins to transport silicon into cells should not have been necessary, it would make sense that the SIT function evolved under conditions with lower levels of ambient environmental silicon to sense whether sufficient silicon was present to complete cell wall structures. The distinct protein-level response comparing SIT1 and SIT2 (Fig. 4) as well as the localization of SIT3 (Fig. 2) suggests further specialization of the roles of SITs. The development of more sophisticated manipulation approaches, such as the use of individual knockdowns for SIT1 and SIT2 or complete knockouts, will facilitate their further study.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by AFOSR MURI grant FA9550-10-1-0555 to M.H.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/EC.00209-14.
REFERENCES
- 1.Monroe JS, Wicander R. 2011. The changing earth: exploring geology and evolution, 6th ed Cengage Learning-Wadsworth, Belmont, CA. [Google Scholar]
- 2.Treguer PJ, De La Rocha CL. 2012. The world ocean silica cycle. Annu Rev Mar Sci 5:477–501. doi: 10.1146/annurev-marine-121211-172346. [DOI] [PubMed] [Google Scholar]
- 3.Wallace AF, Wang D, Hamm LM, Knoll AH, Dove PM. 2012. Eukaryotic skeletal formation. In Knoll AH, Canfield DE, Konhauser KO (ed), Fundamentals of geobiology. John Wiley & Sons, Ltd, Chichester, United Kingdom. doi: 10.1002/9781118280874.ch10. [DOI] [Google Scholar]
- 4.Nelson DM, Tréguer P, Brzezinski MA, Leynaert A, Quéguiner B. 1995. Production and dissolution of biogenic silica in the ocean: revised global estimates, comparison with regional data and relationship to biogenic sedimentation. Global Biogeochem Cycles 9:359–372. doi: 10.1029/95GB01070. [DOI] [Google Scholar]
- 5.Martin-Jezequel V, Hildebrand M, Brzezinski MA. 2000. Silicon metabolism in diatoms: implications for growth. J Phycol 36:821–840. doi: 10.1046/j.1529-8817.2000.00019.x. [DOI] [Google Scholar]
- 6.Skinner HCW, Jahren AH. 2003. Biomineralization, p 1–69. In Holland HD, Turekian KK (ed), Treatise on geochemistry. Pergamon, Oxford, United Kingdom. doi: 10.1016/B0-08-043751-6/08128-7. [DOI] [Google Scholar]
- 7.Brzezinski MA, Olson RJ, Chisholm SW. 1990. Silicon availability and cell-cycle progression in marine diatoms. Mar Ecol Prog Ser 67:83–96. doi: 10.3354/meps067083. [DOI] [Google Scholar]
- 8.Hildebrand M, Frigeri LG, Davis AK. 2007. Synchronized growth of Thalassiosira pseudonana (Bacillariophyceae) provides novel insights into cell-wall synthesis processes in relation to the cell cycle. J Phycol 43:730–740. doi: 10.1111/j.1529-8817.2007.00361.x. [DOI] [Google Scholar]
- 9.Treguer P, Nelson DM, Van Bennekom AJ, DeMaster DJ, Leynaert A, Queguiner B. 1995. The silica balance in the world ocean: a reestimate. Science 268:375–379. doi: 10.1126/science.268.5209.375. [DOI] [PubMed] [Google Scholar]
- 10.Thamatrakoln K, Hildebrand M. 2008. Silicon uptake in diatoms revisited: a model for saturable and nonsaturable uptake kinetics and the role of silicon transporters. Plant Physiol 146:1397–1407. doi: 10.1104/pp.107.107094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hildebrand M. 2008. Diatoms, biomineralization processes, and genomics. Chem Rev 108:4855–4874. doi: 10.1021/cr078253z. [DOI] [PubMed] [Google Scholar]
- 12.Hildebrand M, Dahlin K, Volcani BE. 1998. Characterization of a silicon transporter gene family in Cylindrotheca fusiformis: sequences, expression analysis, and identification of homologs in other diatoms. Mol Gen Genet 260:480–486. doi: 10.1007/s004380050920. [DOI] [PubMed] [Google Scholar]
- 13.Likhoshway YV, Masyukova YA, Sherbakova TA, Petrova DP, Grachev MA. 2006. Detection of the gene responsible for silicic acid transport in chrysophycean algae. Dokl Biol Sci 408:256–260. doi: 10.1134/S001249660603015X. [DOI] [PubMed] [Google Scholar]
- 14.Thamatrakoln K, Alverson AJ, Hildebrand M. 2006. Comparative sequence analysis of diatom silicon transporters: toward a mechanistic model of silicon transport. J Phycol 42:822–834. doi: 10.1111/j.1529-8817.2006.00233.x. [DOI] [Google Scholar]
- 15.Hildebrand M, Volcani BE, Gassmann W, Schroeder JI. 1997. A gene family of silicon transporters. Nature 385:688–689. doi: 10.1038/385688b0. [DOI] [PubMed] [Google Scholar]
- 16.Sherbakova T, Masyukova YA, Safonova T, Petrova D, Vereshagin A, Minaeva T, Adelshin R, Triboy T, Stonik I, Aizdaitcher N. 2005. Conserved motif CMLD in silicic acid transport proteins of diatoms. Mol Biol 39:269–280. doi: 10.1007/s11008-005-0038-4. [DOI] [PubMed] [Google Scholar]
- 17.Alverson AJ. 2007. Strong purifying selection in the silicon transporters of marine and freshwater diatoms. Limnol Oceanogr 52:1420–1429. doi: 10.4319/lo.2007.52.4.1420. [DOI] [Google Scholar]
- 18.Sapriel G, Quinet M, Heijde M, Jourdren L, Tanty V, Luo GZ, Le Crom S, Lopez PJ. 2009. Genome-wide transcriptome analyses of silicon metabolism in Phaeodactylum tricornutum reveal the multilevel regulation of silicic acid transporters. PLoS One 4:e7458. doi: 10.1371/journal.pone.0007458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bowler C, Allen AE, Badger JH, Grimwood J, Jabbari K, Kuo A, Maheswari U, Martens C, Maumus F, Otillar RP. 2008. The Phaeodactylum genome reveals the evolutionary history of diatom genomes. Nature 456:239–244. doi: 10.1038/nature07410. [DOI] [PubMed] [Google Scholar]
- 20.Armbrust EV, Berges JA, Bowler C, Green BR, Martinez D, Putnam NH, Zhou S, Allen AE, Apt KE, Bechner M, Brzezinski MA, Chaal BK, Chiovitti A, Davis AK, Demarest MS, Detter JC, Glavina T, Goodstein D, Hadi MZ, Hellsten U, Hildebrand M, Jenkins BD, Jurka J, Kapitonov VV, Kroger N, Lau WW, Lane TW, Larimer FW, Lippmeier JC, Lucas S, Medina M, Montsant A, Obornik M, Parker MS, Palenik B, Pazour GJ, Richardson PM, Rynearson TA, Saito MA, Schwartz DC, Thamatrakoln K, Valentin K, Vardi A, Wilkerson FP, Rokhsar DS. 2004. The genome of the diatom Thalassiosira pseudonana: ecology, evolution, and metabolism. Science 306:79–86. doi: 10.1126/science.1101156. [DOI] [PubMed] [Google Scholar]
- 21.Thamatrakoln K, Hildebrand M. 2007. Analysis of Thalassiosira pseudonana silicon transporters indicates distinct regulatory levels and transport activity through the cell cycle. Eukaryot Cell 6:271–279. doi: 10.1128/EC.00235-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vrieling EG, Sun Q, Tian M, Kooyman PJ, Gieskes WW, van Santen RA, Sommerdijk NA. 2007. Salinity-dependent diatom biosilicification implies an important role of external ionic strength. Proc Natl Acad Sci U S A 104:10441–10446. doi: 10.1073/pnas.0608980104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thamatrakoln K, Kustka AB. 2009. When to say when: can excessive drinking explain silicon uptake in diatoms? Bioessays 31:322–327. doi: 10.1002/bies.200800185. [DOI] [PubMed] [Google Scholar]
- 24.Brasser H, van der Strate H, Gieskes W, Krijger G, Vrieling E, Wolterbeek H. 2012. Compartmental analysis suggests macropinocytosis at the onset of diatom valve formation. Silicon 4:39–49. doi: 10.1007/s12633-010-9059-2. [DOI] [Google Scholar]
- 25.Curnow P, Senior L, Knight MJ, Thamatrakoln K, Hildebrand M, Booth PJ. 2012. Expression, purification, and reconstitution of a diatom silicon transporter. Biochemistry 51:3776–3785. doi: 10.1021/bi3000484. [DOI] [PubMed] [Google Scholar]
- 26.Poulsen N, Chesley PM, Kröger N. 2006. Molecular genetic manipulation of the diatom Thalassiosira pseudonana (Bacillariophyceae). J Phycol 42:1059–1065. doi: 10.1111/j.1529-8817.2006.00269.x. [DOI] [Google Scholar]
- 27.Poulsen N, Kröger N. 2005. A new molecular tool for transgenic diatoms. Control of mRNA and protein biosynthesis by an inducible promoter-terminator cassette. FEBS J 272:3413–3423. doi: 10.1111/j.1742-4658.2005.04760.x. [DOI] [PubMed] [Google Scholar]
- 28.Dunahay TG, Jarvis EE, Roessler PG. 1995. Genetic transformation of the diatoms Cyclotella cryptica and Navicula saprophila. J Phycol 31:1004–1012. doi: 10.1111/j.0022-3646.1995.01004.x. [DOI] [Google Scholar]
- 29.Hildebrand M, Dahlin K. 2000. Nitrate transporter genes from the diatom Cylindrotheca fusiformis (Bacillariophyceae): mRNA levels controlled by nitrogen source and by the cell cycle. J Phycol 36:702–713. doi: 10.1046/j.1529-8817.2000.99153.x. [DOI] [PubMed] [Google Scholar]
- 30.Huysman MJ, Martens C, Vandepoele K, Gillard J, Rayko E, Heijde M, Bowler C, Inze D, Van de Peer Y, De Veylder L, Vyverman W. 2010. Genome-wide analysis of the diatom cell cycle unveils a novel type of cyclins involved in environmental signaling. Genome Biol 11:R17. doi: 10.1186/gb-2010-11-2-r17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cummings M, McGurk C, Masters JR. 2003. Rapid identification of antisense mRNA-expressing clones using strand-specific RT-PCR. Antisense Nucleic Acid Drug Dev 13:115–117. doi: 10.1089/108729003321629656. [DOI] [PubMed] [Google Scholar]
- 32.Yeung MC, Lau AS. 2002. Detection of antisense RNA transcripts by anti-sense RT-PCR. Methods Mol Biol 193:341–346. doi: 10.1385/1-59259-283-X:341. [DOI] [PubMed] [Google Scholar]
- 33.Strickland J, Parsons T. 1968. A practical handbook of sea water analysis. Bull Fish Res Board Can 167:1–311. [Google Scholar]
- 34.Shimizu K, Del Amo Y, Brzezinski MA, Stucky GD, Morse DE. 2001. A novel fluorescent silica tracer for biological silicification studies. Chem Biol 8:1051–1060. doi: 10.1016/S1074-5521(01)00072-2. [DOI] [PubMed] [Google Scholar]
- 35.Tesson B, Hildebrand M. 2010. Dynamics of silica cell wall morphogenesis in the diatom Cyclotella cryptica: substructure formation and the role of microfilaments. J Struct Biol 169:62–74. doi: 10.1016/j.jsb.2009.08.013. [DOI] [PubMed] [Google Scholar]
- 36.Cooper MS, Hardin WR, Petersen TW, Cattolico RA. 2010. Visualizing “green oil” in live algal cells. J Biosci Bioeng 109:198–201. doi: 10.1016/j.jbiosc.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 37.Yu ET, Zendejas FJ, Lane PD, Gaucher S, Simmons BA, Lane TW. 2009. Triacylglycerol accumulation and profiling in the model diatoms Thalassiosira pseudonana and Phaeodactylum tricornutum (Baccilariophyceae) during starvation. J Appl Phycol 21:669–681. doi: 10.1007/s10811-008-9400-y. [DOI] [Google Scholar]
- 38.Roessler PG. 1988. Changes in the activities of various lipid and carbohydrate biosynthetic enzymes in the diatom Cyclotella cryptica in response to silicon deficiency. Arch Biochem Biophys 267:521–528. doi: 10.1016/0003-9861(88)90059-8. [DOI] [PubMed] [Google Scholar]
- 39.Traller JC, Hildebrand M. 2013. High throughput imaging to the diatom Cyclotella cryptica demonstrates substantial cell-to-cell variability in the rate and extent of triacylglycerol accumulation. Algal Res 2:244–252. doi: 10.1016/j.algal.2013.03.003. [DOI] [Google Scholar]
- 40.von Heijne G. 1988. Transcending the impenetrable: how proteins come to terms with membranes. Biochim Biophys Acta 947:307–333. [DOI] [PubMed] [Google Scholar]
- 41.Hildebrand M. 2005. Cloning and functional characterization of ammonium transporters from the marine diatom Cylindrotheca fusiformis (Bacillariophyceae). J Phycol 41:105–113. doi: 10.1111/j.1529-8817.2005.04108.x. [DOI] [Google Scholar]
- 42.Shrestha RP, Tesson B, Norden-Krichmar T, Federowicz S, Hildebrand M, Allen AE. 2012. Whole transcriptome analysis of the silicon response of the diatom Thalassiosira pseudonana. BMC Genomics 13:499. doi: 10.1186/1471-2164-13-499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.De Riso V, Raniello R, Maumus F, Rogato A, Bowler C, Falciatore A. 2009. Gene silencing in the marine diatom Phaeodactylum tricornutum. Nucleic Acids Res 37:e96. doi: 10.1093/nar/gkp448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith NA, Singh SP, Wang M-B, Stoutjesdijk PA, Green AG, Waterhouse PM. 2000. Gene expression: total silencing by intron-spliced hairpin RNAs. Nature 407:319–320. doi: 10.1038/35030305. [DOI] [PubMed] [Google Scholar]
- 45.Hellens RP, Allan AC, Friel EN, Bolitho K, Grafton K, Templeton MD, Karunairetnam S, Gleave AP, Laing WA. 2005. Transient expression vectors for functional genomics, quantification of promoter activity and RNA silencing in plants. Plant Methods 1:13. doi: 10.1186/1746-4811-1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scherczinger CA, Yates AA, Knecht DA. 1992. Variables affecting antisense RNA inhibition of gene expression. Ann N Y Acad Sci 660:45–56. doi: 10.1111/j.1749-6632.1992.tb21056.x. [DOI] [PubMed] [Google Scholar]
- 47.Elbashir SM, Lendeckel W, Tuschl T. 2001. RNA interference is mediated by 21-and 22-nucleotide RNAs. Genes Dev 15:188–200. doi: 10.1101/gad.862301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu ZH, Xiao HW, Zheng XM, Qiao XF, Wang HY. 2011. Silencing effect of shRNA expression vectors with stem length of 21, 27 or 29 bp. Afr J Biotechnol 10:1073–1080. [Google Scholar]
- 49.Gray NK, Wickens M. 1998. Control of translation initiation in animals. Annu Rev Cell Dev Biol 14:399–458. doi: 10.1146/annurev.cellbio.14.1.399. [DOI] [PubMed] [Google Scholar]
- 50.Mignone F, Gissi C, Liuni S, Pesole G. 2002. Untranslated regions of mRNAs. Genome Biol 3:1–10. doi: 10.1186/gb-2002-3-3-reviews0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Flynt AS, Lai EC. 2008. Biological principles of microRNA-mediated regulation: shared themes amid diversity. Nat Rev Genet 9:831–842. doi: 10.1038/nrg2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kobayashi M, Yamauchi Y, Yamaguchi K, Tanaka A. 1995. Transient expression assay for antisense RNAs using episomal replication of plasmids: effective reduction of retinoblastoma gene (Rb-1) product by its antisense RNA complementary to 3′-untranslated region. Antisense Res Dev 5:141–148. [DOI] [PubMed] [Google Scholar]
- 53.DeVos SL, Miller TM. 2013. Antisense oligonucleotides: treating neurodegeneration at the level of RNA. Neurotherapeutics 10:486–497. doi: 10.1007/s13311-013-0194-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McManus MT, Sharp PA. 2002. Gene silencing in mammals by small interfering RNAs. Nat Rev Genet 3:737–747. doi: 10.1038/nrg908. [DOI] [PubMed] [Google Scholar]
- 55.Dobson CM. 2004. Experimental investigation of protein folding and misfolding. Methods 34:4–14. doi: 10.1016/j.ymeth.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 56.Hildebrand M, Davis AK, Smith SR, Traller JC, Abbriano R. 2012. The place of diatoms in the biofuels industry. Biofuels 3:221–240. doi: 10.4155/bfs.11.157. [DOI] [Google Scholar]
- 57.Libes S. 2011. Introduction to marine biogeochemistry. Academic Press, New York, NY. [Google Scholar]
- 58.Krämer R. 2010. Sensory transport proteins. In Krämer R, Jung K (ed), Bacterial signaling, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany. doi: 10.1002/9783527629237.ch12.60. [DOI] [Google Scholar]
- 59.Ho C-H, Lin S-H, Hu H-C, Tsay Y-F. 2009. CHL1 functions as a nitrate sensor in plants. Cell 138:1184–1194. doi: 10.1016/j.cell.2009.07.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.