
Fig. 2.
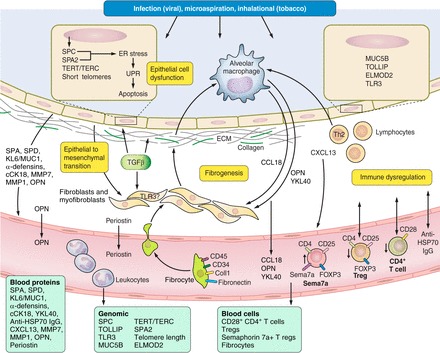
Core mechanisms and candidate molecular biomarkers for idiopathic pulmonary fibrosis (see text and Table 1). Pictured is the alveolar space (top, light blue), epithelial cell layer (tan), interstitial space (white), and capillary (bottom, red). Injury to the alveolar epithelium from a variety of inciting factors (dark blue box) leads to epithelial cell dysfunction, fibrogenesis/extracellular matrix (ECM) deposition, and immune dysregulation. Proposed molecular biomarkers of these processes include blood proteins, genomic markers, and blood cells (see light green boxes). cCK18, cleaved cytokeratin 18; CXCL, C-X-C chemokine ligand; ELMOD2, ELMO containing domain 2 gene mutations; EMT, epithelial-to-mesenchymal transition; ER, endoplasmic reticulum; HSP, heat shock protein; MMP, matrix metalloproteinase; KL6/MUC1, Krebs von den Lungen 6/mucin 1; MUC5B, mucin 5B promoter polymorphism; OPN, osteopontin; SPA, surfactant protein A; SPA2, surfactant protein A2 gene mutations; SPC, surfactant protein C gene mutations; SPD, surfactant protein D; TERT/TERC, telomerase gene mutations; TGF, transforming growth factor; TLR3, Toll-like receptor 3 gene mutation; TOLLIP, Toll-interacting protein gene mutations; Treg, regulatory T cell; UPR, unfolded protein response.