

Abstract
Purpose of Review
Advances in understanding the genetic and molecular basis of innate immune system activation and function have supported the hypothesis that type I interferons (IFN-I), essential mediators of anti-viral host defense, are central contributors to the pathogenesis of systemic lupus erythematosus (SLE). This review addresses recent data that support the rationale for therapeutic targeting of the IFN-I pathway in SLE.
Recent Findings
New insights into mechanisms of cell-intrinsic innate immune system activation, driven by endogenous virus-like nucleic acids and potentially modified by environmental stressors, provide a model for induction of IFN-I that may precede clinically apparent autoimmunity in patients with lupus. Further amplification of IFN-α production, induced by nucleic acid-containing immune complexes that activate endosomal Toll-like receptors, augments and sustains immune system activation, autoimmunity and tissue damage.
Summary
As demonstrated in murine studies of persistent virus infection accompanied by sustained production of IFN-I, blockade of the IFN-I pathway may reverse the immune dysregulation and tissue damage that are essential features of the immunopathogenesis of SLE. Recent research progress has identified numerous therapeutic targets, and specific candidate therapeutics relevant to the IFN-I pathway are under investigation.
Keywords: Systemic lupus erythematosus, type I interferons, interferon-α, Toll-like receptors, cytoplasmic sensors, autoimmunity, long interspersed nuclear elements
Introduction
Evidence supporting a central and essential role for the type I interferons (IFN-I) in the pathogenesis of systemic lupus erythematosus (SLE) and other systemic autoimmune diseases has consistently grown over the past decade, building on observations described more than thirty-five years ago. Initially viewed as only one of a multitude of immune system alterations that characterize patients with SLE, elevated circulating type I interferon activity, and particularly high levels of IFN-α, along with evidence of a broad signature of gene products that are regulated by IFN-I, are now recognized as factors that reflect many of the genetic variations associated with a diagnosis of SLE and contribute to autoimmunity and tissue damage. In light of its role in disease, based on data from both murine and human systems, IFN-I is considered as a rational therapeutic target, with drug development efforts taking several distinct approaches. This review will summarize many of the important research observations published in recent months.
Genetic Associations
Genome-wide association studies have identified a long list of nucleotide variations that are associated with a diagnosis of SLE [1]. Meaningful insights into the significance of particular genetic associations have been informed by clinical and serologic phenotyping of patients, allowing analysis of sequence variants that are associated with presence of particular autoantibody specificities or clinical manifestations of disease. With regard to IFN-I, variants in genes encoding components of the endosomal Toll-like receptor (TLR) pathways and the signaling components downstream of the IFN-I receptor, IFNAR, have been striking [2]. Strong associations of gene variants related to the TLR pathway, particularly IRF5, with SLE in those patients with autoantibody specificities targeting RNA-binding proteins, such as Ro, have supported evidence from studies of patient cells ex vivo, indicating an important relationship between those autoantibodies and an IFN-I signature. The IRF5 risk haplotype is associated with anti-Ro antibodies in asymptomatic individuals, and more importantly, in those who later progress to SLE [3].
Recent genetic data have extended the pathways implicated in IFN-I production to those TLR-independent pathways that are involved in control of nucleic acid integrity and recognition of nucleic acids by cytoplasmic sensors. Rare mutations in several genes responsible for modifying cellular nucleic acids have been associated with Aicardi-Goutieres syndrome (AGS), characterized by high levels of IFN-I, autoantibodies, neurologic disease and skin rash [4, 5]*. So far, the genes implicated include TREX1; SAMHD1; RNASEH2A, B and C; and ADAR1, with recent studies documenting activation of the IFN-I pathway in patients with mutations in ADAR1, responsible for editing of noncoding RNA, and in 90% of AGS patients overall [4, 6]. Variations in these same genes have also been associated with some cases of SLE, although the specific mutations or common variants tend to be distinct from those that account for AGS [7].
Interest in epigenetics and publication of multiple studies from members of the ENCODE (Encyclopedia of DNA Elements) consortium, focused on identification of genomic sites of active transcriptional activity, were followed by recent publication of two studies documenting genome-wide hypomethylation of IFN-I-regulated genes in CD4+ T cells [8, 9]*. Among all hypomethylated genes identified, most are regulated by IFN-I, with the level of methylation independent of circulating levels of IFN-I. These observations suggested to the authors that epigenetic modification of the genome persists even when IFN-I levels are not abundant.
Subtypes of Type I Interferons
IFN-α comprises 13 subtypes encoded by distinct genes on chromosome 9p, and their protein products represent the majority of the IFN-I detected in lupus sera. However, analysis of gene transcripts preferentially regulated by IFN-α in comparison to those preferentially induced by other IFN-I's suggested potential roles for IFN-β or IFN-ω as inducers of the IFN signature in lupus patients [10]. Additional data demonstrated presence of IFN-I in lupus sera that could be inhibited by antibodies specific for IFN-β or IFN-ω [11]. Recent reports from extensive bioinformatics analyses of gene expression signatures present in lupus peripheral blood suggest that in addition to IFN-α, which generates a relatively stable pattern of IFN-I regulated gene transcripts over time, other gene clusters may reflect induction by IFN-β or IFN-γ and present a more variable pattern in longitudinal samples [12-14]*. This impressive analysis by Chaussabel and his colleagues raises important questions regarding the contributions of various cell populations and their triggers to the overall immune dysregulation observed in patients, and supports the need for more extensive studies of drivers of IFN-β production. Although both IFN-α and IFN-β utilize the same receptor to initiate downstream signaling and transcription of IFN-I-induced genes, recent data suggest that IFN-β can activate IFN-regulated gene transcription independently of the IFNAR2 receptor component and induce a somewhat distinct panel of target genes than those regulated by IFN-α [15]. A study of IFN-induced gene transcripts in tissue from patients with Sjogren's syndrome or dermatomyositis defined probes of IFN-γ effects that were distinguishable from IFN-α induction [16]. It will be beneficial to extend studies that distinguish signaling and gene regulation by IFN-α vs. IFN-β in order to inform therapeutic strategies targeting these pathways.
Cell Types Producing IFN-I, Including Neutrophils
Plasmacytoid dendritic cells (pDC) are poised to produce high levels of IFN-α, perhaps based on their constitutive high expression of IRF7, and account for much of the IFN-I produced in lupus patients, a conclusion supported by the ablation of the IFN signature and induction of pDC death in patients treated with high dose glucocorticoid therapy, as well as mouse studies [17]. However, recent data suggest contributions of other cell types, with interest in those cells that might be active producers of IFN-I either early in the preclinical phase of disease or as mediators of flare in those with established disease [18, 19]*. A study of TREX1-deficient mice, associated with increased levels of IFN-I, localized the initiation of disease to nonhematopoietic cells, those that might be more likely to produce IFN-β, with the participation of immune system cells following later [18]*.
Neutrophils have gained increased attention as inducers of IFN-I based on their production of pro-inflammatory defensins, their capacity to generate neutrophil extracellular traps (NETs) and extrude potentially stimulatory mitochondrial DNA [20-22]. Neutrophils may also produce IFN-I, with a recent study documenting TLR9-independent induction of IFN-α secretion induced by free chromatin [22-24]*. A study of SLE bone marrow cells demonstrated production of IFN-I by neutrophils, with an associated local IFN signature and alterations in B cell development [24]*. It should be noted that a study of lupus mice deficient in NADPH oxidase, required for NETosis, showed increased rather than decreased lupus disease [25]. Although the interpretation of those data questioned a role for in vivo NETosis in SLE, the developing data suggest that neutrophils can contribute to disease through additional mechanisms.
Mechanisms of Induction and Amplification of IFN-I
Important recent advances from studies addressing mechanisms of innate immune system activation, whether induced by viral infection or endogenous triggers, have extended the investigation of disease mechanisms in lupus beyond the activation of endosomal TLRs to address TLR-independent nucleic acid sensing pathways and their downstream consequences [26-28]*.
Toll-like receptor-independent induction
As noted above, insights from studies of AGS patients pointed attention to the intracytoplasmic sensors of RNA and DNA [29]. RIG-I and MDA5 are cytoplasmic receptors for RNA and trigger activation of TBK1 and IRF3, ultimately driving transcription of IFN-β and other proinflammatory cytokines through a process dependent on MAVS, an adaptor that associates with mitochondria, facilitating productive signaling [30]. Feedback regulation of this pathway can be mediated by IFN-I-dependent induction of a helicase, DDX24, that binds RNA and inhibits effective signaling [31]. Additional sensors of RNA and DNA have now been defined, with cyclic-GMP-AMP synthase (cGAS) holding particular interest [32-36]. Recognition of cytosolic double-stranded (ds)DNA by cGAS induces production of cGAMP and activates the stimulator of IFN genes (STING). Civril et al. pointed out the structural similarity between cGAS and 2′-5′ oligoandenylate synthase (OAS1), a cytosolic sensor of dsRNA and an IFN-I-regulated gene [36]. cGAMP binding to STING activates IRF3 and induces transcription of IFN-β but can also induce anti-viral genes independent of IFN-I [37]*. New information is also suggesting that IRF5 is involved in IFN-induced gene transcription mediated by the TLR-independent pathway in some cell types [38]. It is likely that additional sensors of intracellular nucleic acids and components of their downstream signaling pathways will be defined.
Induction of innate immune system activation and particularly IFN-I production by intracellular nucleic acids raises important questions regarding the nature of the endogenous RNAs and DNAs that are most effective in driving these pathways. The concept that endogenous nucleic acids might promote an anti-virus-like host response suggests novel mechanisms of induction of altered immune regulation and ultimately autoimmunity and autoimmune disease. Elucidation of those endogenous triggers is a particularly promising area of current and future research (Figure 1). Research characterizing the functions of proteins encoded by the AGS-associated genes, TREX1, SAMHD1, and RNASEH2A, B and C, has drawn particular attention to virus-like sequences derived from host genomic elements and suggests the intriguing hypothesis that self-nucleic acids, like exogenous microbes, can trigger common cytoplasmic sensors and IFN-I if they are not efficiently degraded or regulated [39-41]. Among the attractive genomic candidates that might generate nucleic acid stimuli of innate immune system activation is the non-long terminal repeat transposon long interspersed nuclear element-1 (LINE-1), which is present in the human genome in multiple copies and has the potential to transcribe an mRNA encoding an RNA-binding protein and a reverse transcriptase [42]. The TREX1 gene product can metabolize reverse-transcribed DNA, such as that encoded by LINE-1 [41]. SAMHD1 encodes an enzyme that removes phosphates from dNTPs, reducing available nucleotides for reverse transcription, and can act as a LINE-1 restriction factor [43-45]. SAMHD1 deficiency can trigger a DNA damage response and IFN-I production [45]. Members of the RNASEH2 family remove ribonucleotides from RNA-DNA hybrids. Innate immune activation, IFN-I production and AGS syndrome have also been associated with altered ADAR1 expression or function [6]. ADAR1 encodes an RNA-specific adenosine deaminase that converts adenosine to inosine in dsRNAs, particularly those enriched in Alu sequences, and typically functions as a suppressor of the cell intrinsic IFN-I response. In all cases, impaired regulation of genome integrity and quality control of nucleic acids can allow activation of intrinsic sensors that trigger transcription of IFN-I.
Figure 1. Induction of type I interferon in lupus pathogenesis.
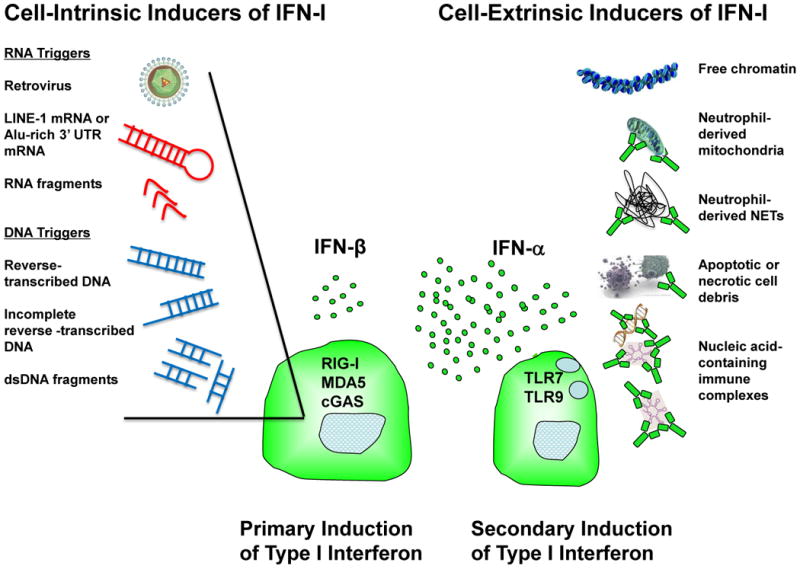
Recent data from studies of cytoplasmic sensors of RNA and DNA, including RIG-I, MDA5 and cGAS, suggest that in addition to exogenous microbes, endogenous triggers can activate signaling pathways that induce IFN-I, with IFN-β the dominant product. Stimulatory cytoplasmic RNA or DNA, in some cases enriched in endogenous retrotransposon sequences, may result from impaired regulation of genome integrity or altered nucleic acid degradation. Cell-intrinsic induction of IFN-I may represent a primary mechanism that primes the immune system for development of autoimmunity. Once autoantibodies have formed, nucleic acid-containing immune complexes, including those derived from products of apoptosis, necrosis, NETosis, or extrusion of mitochondria, can access endosomal TLR7 or TLR9 and induce high level production of IFN-α by plasmacytoid dendritic cells, amplifying and sustaining immune dysregulation and promoting inflammation and tissue damage. Free chromatin might be a cell-extrinsic inducer IFN-I production by neutrophils, contributing to lupus pathology.
A potential role for environmental factors to modify nucleic acid ligands of intracellular sensors is suggested by recent data indicating that oxidative modifications to DNA, resulting in 8-hydroxyguanosine (8-OHG), decreased degradation of that DNA by the TREX1 gene product, 3′ repair exonuclease I [46]*. 8-OHG was detected in skin of lupus patients that had been exposed to UV light and co-localized with IFN-I. These results suggest a potential mechanism that might incorporate a well-documented environmental trigger of lupus flare, IV light, with augmented intracellular sensing of DNA to generate pathogenic levels of IFN-I.
Toll-like receptor-dependent amplification
While new information regarding TLR-independent pathways driving IFN-I production suggests mechanisms that might act early in the pre-autoimmune phase of lupus, activation of endosomal TLRs, particularly TLR7, remains a highly significant pathway for amplification of the IFN-I response and lupus disease once autoantibodies have been produced [47]. It is well established that immune complexes carrying potentially stimulatory nucleic acids and their associated proteins access the endosomal compartments after binding the Fc receptor FCGR2A, and TLR7 activation is closely associated with presence of autoantibody specificities targeting RNA-associated proteins in lupus patients [48]. New data are extending understanding of the requirements for effective downstream signaling after those immune complexes access the TLR. Autophagy, a pathway that mediates isolation and digestion of intracellular pathogens, as well as targeting their components to the TLR compartments, has been implicated in IFN-I production in response to DNA immune complexes. In a process that requires ATG5 (with polymorphisms associated with SLE), activated PI3K, and microtubule associated protein (MAP) 1A/1B-light chain 3 (LC3), the autophagy pathway and TLR9 collaborate to promote IRF7-dependent IFN-I production induced by DNA-containing immune complexes [49]. These requirements are distinct from those that induce TNF through the NF-kB pathway downstream of TLR9. New data characterizing the potential role of TLR8 in autoimmune and inflammatory disease indicate distinct outcomes of TLR7 and TLR8 activation, with TLR8 promoting inflammatory arthritis [50].
Impact on Immune Function and Tissue Damage
The IFN-I signature provides a window into mechanisms of induction and amplification of autoimmunity and disease in lupus, but also reflects expression of gene products in multiple cell types with important impact on immune function. Of interest, a recent study of isolated mononuclear cell (T cell and monocyte) populations compared gene expression in a small number of SLE patients to healthy donors who had received a live attenuated yellow fever vaccine 7 days prior to blood collection [51]. Level of expression of most of the typical IFN-I regulated gene transcripts was stronger in the lupus cells than in those from subjects injected with live virus. In addition, the lupus cells demonstrated increased expression of pattern recognition receptors from both TLR-dependent and TLR-independent pathways. The authors suggested that their data were consistent with a sustained anti-microbial innate immune response. In that regard, two informative publications from studies of virus-infected mice provide an excellent model for predicting the impact of a sustained anti-virus like state characterized by persistent elevated levels of IFN-I, as is observed in patients with SLE [52, 53]*. Mice with persistent infection with lymphocytic choriomeningitis virus show sustained immune activation, impaired regulation of immune reactivity and lymphoid tissue disorganization. In the human system, IFN-α is expressed in untreated patients infected with HIV-1 and is associated with lymphopenia and lymphocyte activation [54]. It is likely that many of the similar defects or alterations in immune function in SLE cells that have been observed over decades of study can be attributed to the effects of IFN-I. For example, a recent study showed that IFN-α decreased the suppressive effect of apoptotic cells on dendritic cell function, a well described property of apoptotic cells [55]. A recent review describes many of the immunomodulatory actions of IFN-I that may be relevant to SLE [56].
In addition to promoting immune system activation and impaired regulatory mechanisms, the sustained production of IFN-I documented in many lupus patients contributes to tissue and organ inflammation and damage. Recent data demonstrate damage to renal podocytes and inhibition of differentiation of podocyte precursors to mature cells by IFN-I, with IFN-β responsible for inducing podocyte death [57]. Extensive data focused on mechanisms of the accelerated atherosclerosis characteristic of SLE document at least several effects of IFN-I, including reduction of endothelial cell precursors and propensity of low-density granulocytes to undergo NETosis with the potential to generate vascular damage [58]. Activated platelets can express CD154/CD40 ligand which when bound to its CD40 receptor on pDCs can induce IFN-I production [59]. The IFN-I signature and platelet aggregation are associated with vascular events, and Kaplan's group has shown an association of IFN-I with decreased endothelial function measured by flow mediated dilatation, increased carotid intimal medial thickness and severity of coronary calcification in lupus patients [60].
Interferon-Targeted Therapeutics
In view of the increasingly strong case for IFN-I playing a central pathogenic role in SLE, IFN-α and other IFN-I pathway components are recognized as rational therapeutic targets. Agents that directly bind and inhibit interaction of IFN-α with its receptor, IFNAR, are most advanced in clinical development. At least three monoclonal anti-IFN-α antibodies are in development, with two advancing through phase II studies [61-63]. Published studies to date indicate variable inhibition of the IFN-I signature in patients treated with these antibodies, with a sense that the signature is more refractory to inhibition in those with more active disease [62, 64]. Encouraging progress is suggested by the recent announcement that a phase IIb study of sifalimumab (MEDI-545) met its primary endpoint in patients with moderate or severe lupus (http://www.astrazeneca.com/Media/Press-releases/Article/20140512--astrazeneca-announces-medimmunes-mavrilimumab-sifalimumab-met-primary-endpoints-Phase-IIb-studies).
In addition to targeting IFN-I directly, antibodies specific for a chain of IFNAR and inhibitors of the endosomal TLRs are in development [65-67]. An interesting approach to generating endogenous neutralizing anti-IFN-α antibodies utilizes administration of a so-called IFN-α-kinoid, composed of IFN-α coupled to a classic T cell-dependent antigen, keyhole limpet hemocyanin [68]. Degradation of the stimulatory TLR7 ligands in RNA-containing immune complexes using RNase is another therapeutic concept in development [69]. Rheumatologists treating lupus patients should be gratified to recognize that treatment with hydroxychloroquine, extensively used as background therapy in patients with SLE, inhibits production of IFN-α by pDCs stimulated ex vivo with TLR9 and TLR7 agonists [70]. The current approaches to therapeutically target the IFN-I pathway in lupus have been summarized recently [71]*.
Conclusions
IFN-I is increasingly recognized as a central pathogenic mediator in patients with SLE. Characterization of cytoplasmic sensors of nucleic acids and genetic data identifying mutations in regulators of genome integrity and nucleic acid degradation support the hypothesis that cell-intrinsic production of IFN-I driven by endogenous nucleic acids is an early event that can prime the immune system for development of autoimmunity and immune dysregulation. With more sophisticated analysis of complex gene expression profiles, it is now recognized that IFN-β, in addition to IFN-α, could be an important early innate immune system product that promotes cell priming and autoimmunity. In that regard, data demonstrating a transient increase of IFN-I-induced gene transcripts in children genetically predisposed to type I diabetes but prior to the development of autoantibodies support IFN-I pathway activation as a primary event in some autoimmune diseases [72]. Abundant data support an important role for the endosomal TLRs in driving sustained production of IFN-α, with TLR7 and immune complexes containing its RNA ligands representing the system most responsible for persistent IFN-α production, immune dysregulation and tissue damage. Although pDCs are the most active producers of IFN-α, neutrophils are gaining attention as both drivers and potential producers of IFN-I. The evolution of research that has defined the genetic basis of increased IFN-I production, the molecular mechanisms of its induction, its impact on autoimmunity and immune regulation and ultimately tissue damage together make a strong case for continued development of therapies that target the relevant mechanisms and mediators.
Key Points.
Characterization of the altered function of enzymes associated with the Aicardi-Goutieres syndrome, along with advances in studies of the innate immune response to virus infection, draw attention to the potential role of cytoplasmic receptors for RNA and DNA and their nucleic acid triggers in cell-intrinsic production of IFN-I.
Virus-like sequences in the human genome are attractive candidates for triggering of innate immune responses in SLE.
Environmental stressors, including UV light, may impair regulation of genome integrity and nucleic acid quality, contributing to innate immune system activation.
Activation of endosomal TLRs with nucleic acid-containing immune complexes provides a strong and sustained stimulus for IFN-α production.
The IFN-I pathway is a rational target for therapeutic modulation in SLE.
Footnotes
Conflicts of Interest: Dr. Crow has consulted for Bristol Myers-Squibb, Eisai, EMD Merck Serono, GSK, Lilly, Takeda and UCB and she has received research grants from Novo Nordisk and Alliance for Lupus Research-Pfizer Centers for Therapeutic Innovation.
Reference Section
- 1.Rullo OJ, Tsao BP. Recent insights into the genetic basis of systemic lupus erythematosus. Annals of the rheumatic diseases. 2013;72(Suppl 2):ii56–61. doi: 10.1136/annrheumdis-2012-202351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ghodke-Puranik Y, Niewold TB. Genetics of the type I interferon pathway in systemic lupus erythematosus. Int J Clin Rheumtol. 2013;8 doi: 10.2217/ijr.13.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cherian TS, Kariuki SN, Franek BS, et al. Brief Report: IRF5 systemic lupus erythematosus risk haplotype is associated with asymptomatic serologic autoimmunity and progression to clinical autoimmunity in mothers of children with neonatal lupus. Arthritis and rheumatism. 2012;64:3383–7. doi: 10.1002/art.34571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rice GI, Forte GM, Szynkiewicz M, et al. Assessment of interferon-related biomarkers in Aicardi-Goutieres syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: a case-control study. Lancet neurology. 2013;12:1159–69. doi: 10.1016/S1474-4422(13)70258-8. This large collaborative study documents expression of an IFN-I signature in nearly all patients bearing a mutation in one of the genes associated with AGS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee-Kirsch MA, Wolf C, Gunther C. Aicardi-Goutieres syndrome: a model disease for systemic autoimmunity. Clinical and experimental immunology. 2014;175:17–24. doi: 10.1111/cei.12160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rice GI, Kasher PR, Forte GM, et al. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nature genetics. 2012;44:1243–8. doi: 10.1038/ng.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Orebaugh CD, Fye JM, Harvey S, et al. The TREX1 C-terminal region controls cellular localization through ubiquitination. The Journal of biological chemistry. 2013;288:28881–92. doi: 10.1074/jbc.M113.503391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coit P, Jeffries M, Altorok N, et al. Genome-wide DNA methylation study suggests epigenetic accessibility and transcriptional poising of interferon-regulated genes in naive CD4+ T cells from lupus patients. Journal of autoimmunity. 2013;43:78–84. doi: 10.1016/j.jaut.2013.04.003. This study, along with reference 9, characterizes genome methylation in samples from patients with SLE and demonstrates that IFN-I regulated genes comprise the majority of those hypomethylated. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Absher DM, Li X, Waite LL, et al. Genome-wide DNA methylation analysis of systemic lupus erythematosus reveals persistent hypomethylation of interferon genes and compositional changes to CD4+ T-cell populations. PLoS genetics. 2013;9:e1003678. doi: 10.1371/journal.pgen.1003678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crow MK, Kirou KA, Wohlgemuth J. Microarray analysis of interferon-regulated genes in SLE. Autoimmunity. 2003;36:481–90. doi: 10.1080/08916930310001625952. [DOI] [PubMed] [Google Scholar]
- 11.Yao Y, Higgs BW, Morehouse C, et al. Development of Potential Pharmacodynamic and Diagnostic Markers for Anti-IFN-alpha Monoclonal Antibody Trials in Systemic Lupus Erythematosus. Human genomics and proteomics : HGP. 2009:2009. doi: 10.4061/2009/374312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bloom CI, Graham CM, Berry MP, et al. Transcriptional blood signatures distinguish pulmonary tuberculosis, pulmonary sarcoidosis, pneumonias and lung cancers. PloS one. 2013;8:e70630. doi: 10.1371/journal.pone.0070630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chiche L, Jourde-Chiche N, Pascual V, Chaussabel D. Current perspectives on systems immunology approaches to rheumatic diseases. Arthritis and rheumatism. 2013;65:1407–17. doi: 10.1002/art.37909. [DOI] [PubMed] [Google Scholar]
- 14.Chiche L, Jourde-Chiche N, Whalen E, et al. Modular transcriptional repertoire analyses of adults with systemic lupus erythematosus reveal distinct type I and type II interferon signatures. Arthritis Rheumatol. 2014 doi: 10.1002/art.38628. This recently published study describes a beautiful analysis of longitudinal gene expression data from study of lupus patient peripheral blood Gene transcript clusters defined by statistical analysis were related to published databases and to clinical disease activity in the patient donors The authors suggest that in addition to a gene transcript signature induced by IFN-α, other gene transcript clusters that are more variable over time may represent genes regulated by IFN-β or IFN-γ. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Weerd NA, Vivian JP, Nguyen TK, et al. Structural basis of a unique interferon-beta signaling axis mediated via the receptor IFNAR1. Nature immunology. 2013;14:901–7. doi: 10.1038/ni.2667. [DOI] [PubMed] [Google Scholar]
- 16.Hall JC, Casciola-Rosen L, Berger AE, et al. Precise probes of type II interferon activity define the origin of interferon signatures in target tissues in rheumatic diseases. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:17609–14. doi: 10.1073/pnas.1209724109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baccala R, Gonzalez-Quintial R, Blasius AL, et al. Essential requirement for IRF8 and SLC15A4 implicates plasmacytoid dendritic cells in the pathogenesis of lupus. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:2940–5. doi: 10.1073/pnas.1222798110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gall A, Treuting P, Elkon KB, et al. Autoimmunity initiates in nonhematopoietic cells and progresses via lymphocytes in an interferon-dependent autoimmune disease. Immunity. 2012;36:120–31. doi: 10.1016/j.immuni.2011.11.018. This study, using the TREX1-1 deficient mouse, strongly suggests that IFN-I production initiates in non-hematopoietic cells and is followed by immune activation, induction of autoimmunity and tissue damage. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaplan MJ. Role of neutrophils in systemic autoimmune diseases. Arthritis research & therapy. 2013;15:219. doi: 10.1186/ar4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garcia-Romo GS, Caielli S, Vega B, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Science translational medicine. 2011;3:73ra20. doi: 10.1126/scitranslmed.3001201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lande R, Ganguly D, Facchinetti V, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Science translational medicine. 2011;3:73ra19. doi: 10.1126/scitranslmed.3001180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ries M, Schuster P, Thomann S, et al. Identification of novel oligonucleotides from mitochondrial DNA that spontaneously induce plasmacytoid dendritic cell activation. Journal of leukocyte biology. 2013;94:123–35. doi: 10.1189/jlb.0612278. [DOI] [PubMed] [Google Scholar]
- 23.Denny MF, Yalavarthi S, Zhao W, et al. A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. Journal of immunology. 2010;184:3284–97. doi: 10.4049/jimmunol.0902199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Palanichamy A, Bauer JW, Yalavarthi S, et al. Neutrophil-mediated IFN activation in the bone marrow alters B cell development in human and murine systemic lupus erythematosus. Journal of immunology. 2014;192:906–18. doi: 10.4049/jimmunol.1302112. These authors succeeded in obtaining bone marrow samples from patients with SLE and documented production of IFN-I by bone marrow neutrophils along with a local IFN-I signature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Campbell AM, Kashgarian M, Shlomchik MJ. NADPH oxidase inhibits the pathogenesis of systemic lupus erythematosus. Science translational medicine. 2012;4:157ra141. doi: 10.1126/scitranslmed.3004801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shrivastav M, Niewold TB. Nucleic Acid Sensors and Type I Interferon Production in Systemic Lupus Erythematosus. Frontiers in immunology. 2013;4:319. doi: 10.3389/fimmu.2013.00319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hasan M, Yan N. Safeguard against DNA sensing: the role of TREX1 in HIV-1 infection and autoimmune diseases. Front Microbiol. 2014;5:193. doi: 10.3389/fmicb.2014.00193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu J, Chen ZJ. Innate immune sensing and signaling of cytosolic nucleic acids. Annual review of immunology. 2014;32:461–88. doi: 10.1146/annurev-immunol-032713-120156. This recent review provides a comprehensive description of current understanding of the TLR-independent mechanisms of innate immune system activation. [DOI] [PubMed] [Google Scholar]
- 29.Barbalat R, Ewald SE, Mouchess ML, Barton GM. Nucleic acid recognition by the innate immune system. Annual review of immunology. 2011;29:185–214. doi: 10.1146/annurev-immunol-031210-101340. [DOI] [PubMed] [Google Scholar]
- 30.Crampton SP, Bolland S. Spontaneous activation of RNA-sensing pathways in autoimmune disease. Current opinion in immunology. 2013;25:712–9. doi: 10.1016/j.coi.2013.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma Z, Moore R, Xu X, Barber GN. DDX24 Negatively Regulates Cytosolic RNA-Mediated Innate Immune Signaling. PLoS pathogens. 2013;9:e1003721. doi: 10.1371/journal.ppat.1003721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun L, Wu J, Du F, et al. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–91. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu J, Sun L, Chen X, et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. 2013;339:826–30. doi: 10.1126/science.1229963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Diner EJ, Burdette DL, Wilson SC, et al. The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell reports. 2013;3:1355–61. doi: 10.1016/j.celrep.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ablasser A, Goldeck M, Cavlar T, et al. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature. 2013;498:380–4. doi: 10.1038/nature12306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Civril F, Deimling T, de Oliveira Mann CC, et al. Structural mechanism of cytosolic DNA sensing by cGAS. Nature. 2013;498:332–7. doi: 10.1038/nature12305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barber GN. STING-dependent cytosolic DNA sensing pathways. Trends in immunology. 2013 doi: 10.1016/j.it.2013.10.010. A nice overall review of the cGAS-STING pathway of innate immune activation. [DOI] [PubMed] [Google Scholar]
- 38.Lazear HM, Lancaster A, Wilkins C, et al. IRF-3, IRF-5, and IRF-7 coordinately regulate the type I IFN response in myeloid dendritic cells downstream of MAVS signaling. PLoS pathogens. 2013;9:e1003118. doi: 10.1371/journal.ppat.1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stetson DB, Ko JS, Heidmann T, Medzhitov R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell. 2008;134:587–98. doi: 10.1016/j.cell.2008.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stetson DB. Endogenous retroelements and autoimmune disease. Current opinion in immunology. 2012;24:692–7. doi: 10.1016/j.coi.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Volkman HE, Stetson DB. The enemy within: endogenous retroelements and autoimmune disease. Nature immunology. 2014;15:415–22. doi: 10.1038/ni.2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Crow MK. Long interspersed nuclear elements (LINE-1): potential triggers of systemic autoimmune disease. Autoimmunity. 2010;43:7–16. doi: 10.3109/08916930903374865. [DOI] [PubMed] [Google Scholar]
- 43.Behrendt R, Schumann T, Gerbaulet A, et al. Mouse SAMHD1 has antiretroviral activity and suppresses a spontaneous cell-intrinsic antiviral response. Cell reports. 2013;4:689–96. doi: 10.1016/j.celrep.2013.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao K, Du J, Han X, et al. Modulation of LINE-1 and Alu/SVA retrotransposition by Aicardi-Goutieres syndrome-related SAMHD1. Cell reports. 2013;4:1108–15. doi: 10.1016/j.celrep.2013.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kretschmer S, Wolf C, Konig N, et al. SAMHD1 prevents autoimmunity by maintaining genome stability. Annals of the rheumatic diseases. 2014 doi: 10.1136/annrheumdis-2013-204845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gehrke N, Mertens C, Zillinger T, et al. Oxidative damage of DNA confers resistance to cytosolic nuclease TREX1 degradation and potentiates STING-dependent immune sensing. Immunity. 2013;39:482–95. doi: 10.1016/j.immuni.2013.08.004. These authors provide an example of a mechanism by which environmental stressors might modify endogenous regulatory mechanisms and promote inappropriate activation of the innate immune response and induction of IFN-I. [DOI] [PubMed] [Google Scholar]
- 47.Kono DH, Baccala R, Theofilopoulos AN. TLRs and interferons: a central paradigm in autoimmunity. Current opinion in immunology. 2013;25:720–7. doi: 10.1016/j.coi.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kirou KA, Lee C, George S, et al. Activation of the interferon-alpha pathway identifies a subgroup of systemic lupus erythematosus patients with distinct serologic features and active disease. Arthritis and rheumatism. 2005;52:1491–503. doi: 10.1002/art.21031. [DOI] [PubMed] [Google Scholar]
- 49.Henault J, Martinez J, Riggs JM, et al. Noncanonical autophagy is required for type I interferon secretion in response to DNA-immune complexes. Immunity. 2012;37:986–97. doi: 10.1016/j.immuni.2012.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guiducci C, Gong M, Cepika AM, et al. RNA recognition by human TLR8 can lead to autoimmune inflammation. The Journal of experimental medicine. 2013;210:2903–19. doi: 10.1084/jem.20131044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kyogoku C, Smiljanovic B, Grun JR, et al. Cell-specific type I IFN signatures in autoimmunity and viral infection: what makes the difference? PloS one. 2013;8:e83776. doi: 10.1371/journal.pone.0083776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wilson EB, Yamada DH, Elsaesser H, et al. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science. 2013;340:202–7. doi: 10.1126/science.1235208. This paper, along with reference 53, describe the impact of short term vs. sustained virus infection in a murine system and demonstrate the pathologic effects of prolonged IFN-I production. Blockade of the IFN-I pathway reverses many of the pathologic manifestations of sustained IFN-I production, supporting the rationale for therapeutic targeting of the IFN-I pathway in patients with SLE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Teijaro JR, Ng C, Lee AM, et al. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science. 2013;340:207–11. doi: 10.1126/science.1235214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hardy GA, Sieg S, Rodriguez B, et al. Interferon-alpha is the primary plasma type-I IFN in HIV-1 infection and correlates with immune activation and disease markers. PloS one. 2013;8:e56527. doi: 10.1371/journal.pone.0056527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Abeler-Dorner L, Rieger CC, Berger B, et al. Interferon-alpha abrogates the suppressive effect of apoptotic cells on dendritic cells in an in vitro model of systemic lupus erythematosus pathogenesis. The Journal of rheumatology. 2013;40:1683–96. doi: 10.3899/jrheum.121299. [DOI] [PubMed] [Google Scholar]
- 56.Gonzalez-Navajas JM, Lee J, David M, Raz E. Immunomodulatory functions of type I interferons. Nature reviews Immunology. 2012;12:125–35. doi: 10.1038/nri3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Migliorini A, Angelotti ML, Mulay SR, et al. The antiviral cytokines IFN-alpha and IFN-beta modulate parietal epithelial cells and promote podocyte loss: implications for IFN toxicity, viral glomerulonephritis, and glomerular regeneration. The American journal of pathology. 2013;183:431–40. doi: 10.1016/j.ajpath.2013.04.017. [DOI] [PubMed] [Google Scholar]
- 58.Knight JS, Kaplan MJ. Cardiovascular disease in lupus: insights and updates. Current opinion in rheumatology. 2013;25:597–605. doi: 10.1097/BOR.0b013e328363eba3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Duffau P, Seneschal J, Nicco C, et al. Platelet CD154 potentiates interferon-alpha secretion by plasmacytoid dendritic cells in systemic lupus erythematosus. Science translational medicine. 2010;2:47ra63. doi: 10.1126/scitranslmed.3001001. [DOI] [PubMed] [Google Scholar]
- 60.Somers EC, Zhao W, Lewis EE, et al. Type I interferons are associated with subclinical markers of cardiovascular disease in a cohort of systemic lupus erythematosus patients. PloS one. 2012;7:e37000. doi: 10.1371/journal.pone.0037000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stohl W. Future prospects in biologic therapy for systemic lupus erythematosus. Nature reviews Rheumatology. 2013;9:705–20. doi: 10.1038/nrrheum.2013.136. [DOI] [PubMed] [Google Scholar]
- 62.Merrill JT, Wallace DJ, Petri M, et al. Safety profile and clinical activity of sifalimumab, a fully human anti-interferon alpha monoclonal antibody, in systemic lupus erythematosus: a phase I, multicentre, double-blind randomised study. Annals of the rheumatic diseases. 2011;70:1905–13. doi: 10.1136/ard.2010.144485. [DOI] [PubMed] [Google Scholar]
- 63.McBride JM, Jiang J, Abbas AR, et al. Safety and pharmacodynamics of rontalizumab in patients with systemic lupus erythematosus: results of a phase I, placebo-controlled, double-blind, dose-escalation study. Arthritis and rheumatism. 2012;64:3666–76. doi: 10.1002/art.34632. [DOI] [PubMed] [Google Scholar]
- 64.Yao Y, Richman L, Higgs BW, et al. Neutralization of interferon-alpha/beta-inducible genes and downstream effect in a phase I trial of an anti-interferon-alpha monoclonal antibody in systemic lupus erythematosus. Arthritis and rheumatism. 2009;60:1785–96. doi: 10.1002/art.24557. [DOI] [PubMed] [Google Scholar]
- 65.Wang B, Higgs BW, Chang L, et al. Pharmacogenomics and translational simulations to bridge indications for an anti-interferon-alpha receptor antibody. Clinical pharmacology and therapeutics. 2013;93:483–92. doi: 10.1038/clpt.2013.35. [DOI] [PubMed] [Google Scholar]
- 66.Barrat FJ, Coffman RL. Development of TLR inhibitors for the treatment of autoimmune diseases. Immunological reviews. 2008;223:271–83. doi: 10.1111/j.1600-065X.2008.00630.x. [DOI] [PubMed] [Google Scholar]
- 67.Kandimalla ER, Bhagat L, Wang D, et al. Design, synthesis and biological evaluation of novel antagonist compounds of Toll-like receptors 7, 8 and 9. Nucleic acids research. 2013;41:3947–61. doi: 10.1093/nar/gkt078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lauwerys BR, Hachulla E, Spertini F, et al. Down-regulation of interferon signature in systemic lupus erythematosus patients by active immunization with interferon alpha-kinoid. Arthritis and rheumatism. 2013;65:447–56. doi: 10.1002/art.37785. [DOI] [PubMed] [Google Scholar]
- 69.Sun X, Wiedeman A, Agrawal N, et al. Increased ribonuclease expression reduces inflammation and prolongs survival in TLR7 transgenic mice. Journal of immunology. 2013;190:2536–43. doi: 10.4049/jimmunol.1202689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sacre K, Criswell LA, McCune JM. Hydroxychloroquine is associated with impaired interferon-alpha and tumor necrosis factor-alpha production by plasmacytoid dendritic cells in systemic lupus erythematosus. Arthritis research & therapy. 2012;14:R155. doi: 10.1186/ar3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kirou KA, Gkrouzman E. Anti-interferon alpha treatment in SLE. Clinical immunology. 2013;148:303–12. doi: 10.1016/j.clim.2013.02.013. This review summarizes many of the therapeutic approaches in development for modulation of the IFN-I pathway and treatment of patients with SLE. [DOI] [PubMed] [Google Scholar]
- 72.Ferreira RC, Guo H, Coulson RM, et al. A type I interferon transcriptional signature precedes autoimmunity in children genetically at-risk of type 1 diabetes. Diabetes. 2014 doi: 10.2337/db13-1777. [DOI] [PMC free article] [PubMed] [Google Scholar]