

Abstract
Objective
To explore the relationship between DNA polymerase β (pol β) overexpression and benzo[a]pyrene (BaP) carcinogenesis.
Methods
Firstly, mouse embryonic fibroblasts that express wild-type level of DNA polymerase β (pol β cell) and high level of pol β (pol β oe cell) were treated by various concentrations of BaP to determine genetic instability induced by BaP under differential expression levels of pol β. Secondly, malignant transformation of pol β cells by low concentration of BaP (20 μM) was determined by soft agar colony formation assay and transformation focus assay. Thirdly, the mRNA and protein levels of BaP-transformed pol β cells (named pol β-T cells) was measured by reverse transcriptase-polymerase chain reaction (RT-PCR) and western blot, and the genetic instability of these cells were examined by HPRT gene mutation assay and random amplified polymorphic DNA (RAPD) assay.
Results
Pol β cells were successfully transformed into malignant pol β-T cells by an exposure to low concentration of BaP for 6 months. Pol β-T cells exhibited increased levels of pol β gene expression, HPRT gene mutation frequency and polymorphisms of RAPD products that were comparable to those of pol β oe cells.
Conclusion
Pol β overexpression and its-associated genetic instability may play a key role in BaP carcinogenesis.
Keywords: DNA polymerase β, benzo[a]pyrene, malignant transformation, base excision repair, genetic instability, carcinogenesis
Introduction
Benzo[a]pyrene (BaP), the most frequently studied polycyclic aromatic hydrocarbons (PAHs) family member, is a ubiquitous environmental pollutant which results from both natural and man-made sources. The linkage between BaP exposure and human cancers in lung, skin, and liver was initially established in early 1800s. An epidemiological study conducted by Percivall Pott in 1775 demonstrated a correlation between a high incidence of testis carcinoma, also known as chimney sweeper cancer, in chimney sweeper population in London and elevated concentrations of gas tar in their occupational environment, among which BaP was one of the major components. In 1915, Yamagiwa and Ichikawa successfully established a PAHs-induced skin cancer model in rabbits by repeatedly exposing the animals to coal tar through rabbit ears. Later studies have demonstrated that BaP can induce carcinogenic transformation of normal cells resulting in cellular malignancy by inducing a series of carcinogenic phenotypes that mainly include gene mutations and chromosomal aberrations, i.e. genetic instability. These can be mediated by BaP-induced BaP-DNA adducts and oxidative DNA damages (Nkrumah-Elie, et al., 2012). In vivo studies have also demonstrated that BaP-induced mutagenesis is associated with the development of cancers including colonic adenocarcinoma, lung cancer and hepatoma in mice (Chen, et al., 2011; Hakura, et al., 2011; Labib, et al., 2012). As a result, the International Agency for Research on Cancer (IARC) has upgraded benzo[a]pyrene from possible carcinogenic (Group II B) to identified carcinogenic to humans (Group I). However, the mechanisms by which BaP induces cancer remain poorly understood and need to be elucidated.
DNA damage resulting from endogenous and exogenous sources can be efficiently removed by robust DNA repair mechanisms that include base excision repair (BER), nucleotide excision repair (NER), and other DNA repair pathways. The damage repair pathways prevent genetic instability that can be lead to tumorigenesis or programmed cell death. DNA polymerase β (pol β) is a key enzyme in BER that excises the 5′-terminal deoxyribose phosphate (5′dRP) resulting from 5′-incision of an abasic site by apurinic/apyrimidinic (AP) endonuclease 1 (APE1). Pol β then catalyzes a temple-directed nucleotidyl-transfer reaction to fill in a single-nucleotide gaps resulting from its dRP lyase activity (Liu and Wilson, 2012). Compared with DNA replication polymerase, pol β is structurally simple and has relatively lower fidelity. Thus, excessive amount of pol β may facilitate error-prone synthesis during DNA damage repair leading to genetic instability.
Genetic instability plays an important role in the initiation, promotion and progression of neoplasm in humans caused by environmental carcinogens. Thus, environmental factors that promote genetic instability may induce tumorigenesis (Dizdaroglu, 2012). Early studies showed that BaP carcinogenesis is mainly associated with deficiency of NER pathway. However, another study has also demonstrated that PAHs is capable of inducing oxidative DNA damage that is mainly repaired by BER pathway in vitro and in vivo. In addition, it has been suggested that the BER pathway is involved in BaP carcinogenesis (Kundu, et al., 2007). Cellular co-exposure of BaP and quinhydrone can result in production of BaP-quinones (BPQs) that lead to generation of reactive oxygen species (ROS) such as hydrogen peroxide and hydroxyl radicals that can ultimately cause oxidative DNA damages (Kundu, et al., 2007). Recent studies have also demonstrated that BaP can promote lipid peroxidation in a dose-dependently manner and significantly enhance the production of ROS (Song, et al., 2011) that react with guanines and adenines to create oxidized DNA bases (Herbstman, et al., 2012).
Although pol β plays a vital role in repairing oxidative DNA damage induced by BaP, more and more evidences have suggested that the relatively low fidelity of the polymerase β may also be involved in the BaP carcinogenesis by performing error-prone DNA synthesis (Chary, et al., 2012). In this study, we explored a potential role of pol β overexpression in mediating BaP-induced cellular malignant transformation through genetic instability. We found that chronic exposure of wild-type pol β cells to low dose of BaP led to transformation of the cells into pol β-T cells that bear malignant phenotypes. Surprisingly, the pol β-T cells exhibited high levels of expression of pol β, gene mutation frequency and genetic instability that were comparable with pol β cells. These results suggest that BaP induced pol β overexpression resulting in genetic instability that ultimately causes cellular malignant transformation. This study has advanced our understanding about the roles of pol β overexpression and genetic instability in BaP carcinogenesis.
Material and Methods
2.1 Cell lines and culture conditions
Wild-type mouse embryonic fibroblasts (named pol β cells in this study) and mouse embryonic fibroblasts that expressed high level of pol β (named pol β oe cells in this study) were generous gifts from Dr. Samuel H. Wilson, National Institute of Environmental Health Sciences (NIEHS)/National Institutes of Health, NC, USA. Pol β protein and mRNA level in pol β oe cells is 2.11-fold and 2.20-fold of those of pol β cells (Luo, et al., 2012). Cells were cultured in 5% CO2 at 37 °C in Dulbecco’s modified eagle’s medium (DMEM) (Gibco, USA) supplemented with 10% fetal bovine serum (Fumeng Inc., China), 100 U/mL penicillin and 100 μg/mL streptomycin (Gibco, USA).
2.2 Malignant transformation of pol β cells
BaP stock solution (20 mM) was made by dissolving 25.2 mg BaP (Sigma, USA) into 5.0 mL dimethyl sulphoxide (DMSO) and was kept in darkness at 4 °C. The working solution of BaP at concentrations of 1.25, 5.00 and 20.00 μM was made by serial dilution of stock solution with DMEM. In this study, cells treated with 0.1% (v/v) DMSO alone served as solvent control. Rat liver S9 mixture was made via induction by combination of Phenobarbital (Sigma, USA) and β-Naphthoflavone (Sigma, USA) according to the guidelines approved by local Ethics Committee of Sichuan University. All experimental procedures and animal use were conducted in accordance with the Guide to the Care and Use of Laboratory Animals of Sichuan University. Malignantly transformed pol β cells (named pol β-T cells) were obtained by exposing cells to low concentrations of BaP and S9 mixture simultaneously. Pol β cells were seeded into 25 mL culture flask at a density of 1×106 cells in triplicates. Cells were treated with 1.25, 5.00 and 20.00 μM BaP for 48 hours. Subsequently, BaP was removed, and cells were washed by phosphate buffered saline (PBS) for three times and resupplied with fresh culture medium. Cells were continuously subjected to BaP treatment every ten days that lasted for six months. The malignant phenotypes of pol β-T cells were then characterized using the transforming focus assay and soft agar colony formation assay.
2.3 Characterization of malignant transformation of pol β cells
2.3.1 Transforming focus assay
Pol β cells treated with BaP were examined under microscope (Leica, Germany) for identification of transformation focus according to the standards of malignant transformation focus described previously by Ohno et al. (Ohno, et al., 2012). At the end of six month of BaP treatment, type III transforming focus were observed in 1.25, 5.00 and 20.00 μM BaP treatment groups. The colonies from 20.00 μM BaP treatment group that matched the standards of type III transforming focus were trypsinized by 0.25% EDTA-trypsin and isolated for soft agar colony formation assay and subsequent studies.
2.3.2 Soft agar colony formation assay
Soft agar cloning assay was performed in 6-well plates pre-filled with 2.00 mL 0.5% normal melting point agar (NMPA). Cells obtained from type III transforming focus were diluted using DMEM. Two milliliter of 0.3% low melting point agar (LMPA) was made through the mixture of 1.88 mL cell suspensions which contains two thousand cells and 0.12 mL 5% LMPA. Then, the mixture was spread into 6-well plates pre-coated with 0.5% NMPA. Cells were then cultured in 5% CO2, 37 °C incubator for two weeks. Two milliliters DMEM were added onto 6-well plates every other day to keep agar moist. On the 14th day after cell seeding, the plates were examined under microscope (Leica, Germany). Colonies that contained more than fifty cells or the diameter of colonies was larger than 75 μm (200×) were scored. Pol β cells treated with 0.1% (v/v) DMSO alone served as solvent control. The soft agar colony efficiency was calculated by the following equation: soft agar colony efficiency = (number of soft agar colonies/2000) × 100%.
2.4 HPRT gene mutation assay
Initially colony formation assay was performed to determine the viability of cells treated by BaP, and cloning efficiency (CE) was obtained. Two hundred cells were seeded into 24-well plates after trypsinization. BaP treated cells were cultured in 37 °C, 5% CO2 incubator for 14 days with culture medium replaced for every three days. Subsequently, cells were washed by PBS for three times, fixed by methanol for 20 min and stained by 10% Giemsa for 15 min. Plates were then washed by PBS and air-dried at room temperature. Colonies containing more than fifty cells were counted twice under microscope and the CE and inhibition rate of colony formation were calculated according to these equations: CE = (number of colonies/200) × 100%; Inhibition rate of colony formation = [1-(number of colonies of treated group/number of colonies of control group)] × 100%.
Since 6-thioguanine (6-TG) resistant mutant phenotype can only be manifested by a lack of HPRT gene activity in mammalian cells, a gene mutation detection system which combines HPRT gene deficiency and 6-TG resistant has been widely used to examine the mutagenic activity of various identified or potential mutagens on mammalian cells. Four milliliters of cell suspensions with 1×106 cells were seeded into 100 mL culture flasks overnight. Cells were then supplied with 5.00 mL of culture medium containing 40 μM of 6-TG (Sigma, USA) and incubated for 72 h. Cells were then supplied with fresh culture medium and grew for two weeks allowing colonies to form. Cell culture medium was replaced every three days. At the end of the experiment, cells were washed by PBS (pH 7.4) for three times, fixed by methanol for 20 min and stained by 10% Giemsa for 15 min. The colonies which contain more than fifty cells were counted twice under microscope and the mutation frequency (MF) was calculated as following: MF = number of colonies/(1×106×cloning efficiency).
2.5 Random amplified polymorphic DNA analysis (RAPD analysis)
The RAPD analysis was performed as described by Mehrotra et al. (Mehrotra, et al., 2012). Briefly, genomic DNA was extracted and purified using commercial kits (Tiangen Inc., China) according to the manufacturer’s instructions and DNA concentrations were measured by the absorbance at 260 nm using BioSpec-mini DNA/RNA/Protein Analyzer (Shimadzu, Japan). Then, RAPD reaction was performed by conducting PCR reactions (25 μl) containing 1 μl template DNA, 2.5 nmol of dNTP mixture, 1.25 U Taq DNA polymerase (Sangon Inc., China) and 25 pmol of ten pairs of random primers (Table 1) using the program denaturing at 94 °C for 3 min, 1 cycle; denaturing at 94 °C for 30 sec, annealing at 32 °C for 40 sec and extension at 72 °C for 1 min, 40 cycles; extension at 72 °C for 4 min, 1 cycle. PCR products were subjected to 2% agarose gel electrophoresis and stained with Green View (BioRule, China). PCR products were visualized by a UV-transilluminator (Bio-Rad, USA). PCR amplification by each pair of primers was repeated three times. The polymorphisms of genomic DNA were determined by the diversity of PCR products.
Table 1.
Sequences of ten pairs of random primers for amplifying RAPD fragments
| Primer code | Sequence (5′–3′) | Tm (°C) | GC (%) |
|---|---|---|---|
| P1 | AAC GGT CAC G | 32 | 60 |
| P2 | TGG GCA TCT G | 32 | 60 |
| P3 | GGT CTG AAC C | 32 | 60 |
| P4 | ACG GTC TTG G | 32 | 60 |
| P5 | CCG GCT ACG G | 36 | 80 |
| P6 | AGC TGC CGG G | 36 | 80 |
| P7 | AAG GCT AGC G | 32 | 60 |
| P8 | AGG CAT TCC C | 32 | 60 |
| P9 | CGG CCC CTG T | 36 | 80 |
| P10 | CAG GCC CTT C | 34 | 70 |
2.6 Reverse transcription-polymerase chain reaction (RT-PCR)
The total RNA of 1×106 pol β or pol β-T cells was isolated using RNA extraction commercial kit (Geneaid, Inc., Taiwan). The amount of RNA was measured with BioSpec-mini DNA/RNA/Protein Analyzer (Shimadzu, Japan). Total RNA was transcribed into cDNA using a reverse-transcription kit (Toyobo Co., Japan) according to the protocols provided by the manufacturer. The primers for amplification of pol β and glyceraldehydes-3-phosphate dehydrogenase (G3PDH) were designed by Primer 5.0 and synthesized by GenScript (GenScript Inc., Nanjing, Jiangsu). The sequence of the primers are: pol β forward primer, 5′-AAC GAA TTG GGC TGA AAT-3′, and pol β reverse primer, 5′-TAG CGC CAC TGG ATG TAA-3′; G3PDH forward primer, 5′-ACC ACA GTC CAT GCC ATC AC-3′, and G3PDH reverse primer, 5′-TCC ACC ACC CTG TTG CTG TA-3′. PCR was performed with a program: denaturing at 94 °C for 3 min, 1 cycle; denaturing at 94 °C for 30 sec, annealing at 48 °C for 40 sec, extension at 72 °C for 1 min, 30 cycles; extension at 72 °C for 4 min, 1 cycle. The PCR products of pol β (574 bp) and G3PDH (450 bp) were separated by 2% agarose gel electrophoresis and stained by Green View (BioRule Inc., China). PCR products were visualized by a UV-transilluminator (Bio-Rad, USA) and quantified by Image J.
2.7 Western blot
Cells were washed by pre-cold PBS for three times and collected by trypsinization. Cellular proteins were extracted by commercial kit (Jiancheng Inc., China) according to the manufacturer’s instruction. The protein concentrations were determined using Coomassie brilliant blue protein kit (Jiancheng Inc., China). Thirty micrograms of total protein were subjected to 12% sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE). One gel was stained by Coomassie brilliant blue, and the other one was subjected to protein electro-transfer to a polyvinylidene difluoride (PVDF) membrane (Hybond Inc., USA) at a constant 200 mA electric current for 1 h. The PVDF membrane was subsequently blocked in 5% non-fat milk for 2 h. The membrane was then incubated with either a rabbit polyclonal anti-pol β (from Dr. Samuel H. Wilson, NIEHS/NIH, USA) or a rabbit polyclonal anti-β-actin (Bioss Inc., China) at a dilution of 1: 1000 in 5% non-fat milk at 4 °C overnight. The membrane were then incubated with horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (Zhongshan Goledn Bridge Inc., China) at a dilution of 1: 5000 in 5% non fat milk at room temperature for 2 h. After three washes, the membrane was incubated with enhanced chemiluminescence (ECL) substrate (Pierce, USA) and exposed to X-ray films (Kodak, USA). The quantitative analysis of western blot data was performed by Image J.
2.8 Statistical analysis
All experiments were performed for three times independently with four parallel experiments conducted for each concentration (n=12). Data were analyzed by SPSS 17.0 (IBM, USA) and illustrated as mean ± standard deviation. The statistical differences of proteins and mRNA levels between different groups were examined by student’s t-test. The significant differences of HPRT gene mutation frequencies and soft agar colony efficiency between different groups were examined by Poisson u test. The statistical differences of cloning efficiency and number of colonies among multiple groups were examined by one-way ANOVA or student’s t-test. A P value less than 0.05 indicated a significant difference.
Results
3.1 The cloning efficiency and inhibition rate of colony formation of pol β and pol β oe cells treated by BaP
The effect of BaP on cell viability was determined by measuring the cloning efficiency and inhibition rate of colony formation of pol β and pol β oe cells treated by BaP. The results were listed in Table 2. The cloning efficiency of pol β and pol β oe cells decreased with the increased concentrations of BaP, whereas the inhibition rate of colony formation of the cells increased with increased concentrations of BaP with a dose dependency. When pol β and pol β oe cells were treated with 5.00 and 20.00 μM BaP, their cloning efficiencies were significantly lower than untreated cells (P<0.05). These indicated that BaP decreased cell viability of both pol β and pol β oe cells. Interestingly, the cloning efficiency of pol β oe cells treated by 20.00 μM BaP was higher than that of pol β cells (P<0.05) indicating that overexpression of pol β prevented BaP-induced colony forming inhibition.
Table 2.
Effect of BaP on colony formation of pol β and pol β oe cells (n=12)
| Cell lines | BaP (μM) | Number of colonies | Cloning Efficiency (%) | Inhibition rate of colony formation (%) |
|---|---|---|---|---|
| pol β cells | 0a | 42.66±2.34 | 21.33±0.62 | — |
| 1.25 | 40.66±3.92 | 20.33±0.96 | 4.69 | |
| 5.00 | 35.42±2.16 | 17.71±0.74b | 16.97 | |
| 20.00 | 30.94±2.48 | 15.47±0.78b | 27.47 | |
| pol β oe cells | 0a | 43.34±3.45 | 21.67±1.76 | — |
| 1.25 | 42.18±1.98 | 21.09±0.83 | 2.68 | |
| 5.00 | 39.74±3.25 | 19.87±1.22c | 8.31 | |
| 20.00 | 37.10±1.33 | 18.55±0.59b,c | 14.40 |
denotes that pol β cells or pol β oe cells treated by 0.1% (v/v) DMSO served as solvent control;
denotes P<0.05 when compared with solvent control within the same cell lines;
denotes P<0.05 when compared with the corresponding BaP treatment of pol β cells.
3.2 Overexpression of pol β increased BaP-induced HPRT gene mutation frequency
To determine the mutagenic effect of BaP under various levels of pol β, we examined the BaP-induced HPRT gene mutation frequency of pol β and pol β oe cells. The results showed that the number of mutant colony and mutation frequency of pol β and pol β oe cells increased with increasing concentration of BaP (Table 3). The mutation frequencies of the cells treated with 1.25, 5.00 and 20.00 μM BaP were significantly higher than that of control cells (P<0.05). In addition, the mutation frequency of pol β oe cells was significantly higher than that of pol β cells (P<0.05) at 5.00 and 20.00 μM BaP tested concentrations suggesting that high HPRT gene mutation frequency induced by BaP was associated with pol β overexpression.
Table 3.
Mutation frequency of HPRT gene induced by BaP in pol β and pol β oe cells (n=12)
| Cell lines | BaP (μM) | Number of mutant colony | Mutation frequency (×10−6) |
|---|---|---|---|
| Pol β cells | 0a | 8.08±1.16 | 6.25 |
| 1.25 | 15.00±2.04b | 12.29b | |
| 5.00 | 20.75±2.34b | 19.76b | |
| 20.00 | 24.00±2.41b | 24.78b | |
| Pol β oe cells | 0a | 8.00±1.81 | 6.15 |
| 1.25 | 17.00±2.37b | 13.43b | |
| 5.00 | 32.58±2.84b,c | 27.68b,c | |
| 20.00 | 43.25±3.60b,c | 38.63b,c |
denotes that pol β cells or pol β oe cells treated by 0.1% (v/v) DMSO served as solvent control;
denotes P<0.05 when compared with solvent control within the same cell lines;
denotes P<0.05 when compared with the corresponding BaP treatment of pol β cells.
3.3 Overexpression of pol β increased BaP-induced polymorphism of RAPD fragments
Random amplified polymorphic DNA (RAPD) analysis was performed to examine the BaP-induced genetic instability under various expression levels of pol β. The RAPD fragments amplified by the random primer P3, P4, P6, P8, P9 and P10 exhibited the same size patterns for both untreated and BaP-treated cells. However, different size patterns of the RAPD products amplified by the random primer P1, P2, P5 and P7 were identified (Table 4 and Figure 1). Specifically, loss or addition of the RAPD products amplified by primer P1, P2 and P5 were detected in all BaP-treated cells compared with untreated cells (Figure 1: Primer 1, lanes 1 and 2, lanes 3 and 4, lanes 5 and 6; Primer 2, lanes 3 and 4, lanes 5 and 6; Primer 5, lanes 3 and 4, lanes 5 and 6). BaP-treated pol β oe cells exhibited more polymorphic changes in the sizes of the RAPD products amplified by the primers (Figure 1, Primer 7, lanes 5 and 6). This indicated that pol β overexpression facilitated BaP-induced genetic instability.
Table 4.
Polymorphism of RAPD fragments induced by BaP in pol β and pol β oe cells
| Cell lines | Random Primers
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| P1 | P2 | P3 | P4 | P5 | P6 | P7 | P8 | P9 | P10 | |
|
| ||||||||||
| pol β cells | a, b | - | - | - | - | - | - | - | - | - |
| pol β oe cells | a | a | - | - | a | - | c | - | - | - |
“a” denotes missing band(s); “b” denotes increasing band(s); “c” denotes shifting band(s); and “-” denotes monomorphic band pattern.
Figure 1. PCR products of pol β and pol β oe cells amplified with random primers.

Random Primer P1 and Random Primer P7: M, 50 bp DNA ladder marker; Lane 1 and 2, pol β cells exposed to 0 (0.1% v/v DMSO) and 20 μM BaP, respectively; Lane 3 and 4, pol β cells exposed to 0 (0.1% v/v DMSO) and 20 μM BaP, respectively; Lane 5 and 6, pol β oe cells exposed to 0 (0.1% v/v DMSO) and 20.00 μM BaP, respectively.
Random Primer P2 and Random Primer P5: M, 50 bp DNA ladder marker; Lane 1 and 2, pol β cells exposed to 0 (0.1% v/v DMSO) and 20.00 μM BaP, respectively; Lane 3 and 4, pol β oe cells exposed to 0 (0.1% v/v DMSO) and 20 μM BaP, respectively; Lane 5 and 6, pol β oe cells exposed to 0 (0.1% v/v DMSO) and 20.00 μM BaP, respectively.
3.4 BaP-transformed pol β cells (pol β-T cells) exhibited anchorage independent growth
To find out if a chronic cellular exposure to BaP can lead to transformation of wild-type pol β cells into malignant cells, we examined the anchorage independent growth of pol β cells that were chronically treated by a low concentration of BaP (20 μM). The results showed that BaP-treated pol β cells formed a large number of colonies on soft agar, whereas untreated pol β cells formed a few colonies (Figure 2 and Table 5). The colony efficiency of pol β-T cells treated by 5.00 and 20.00 μM BaP was 11-fold and 16-fold of that of untreated cells, respectively (Table 5). With increasing concentrations of BaP, the colony efficiency of pol β-T cells was significantly increased (P<0.05). These indicated that chronic exposure to BaP induced the transformation of pol β cells into pol β-T cells with malignant phenotypes in a dose-dependent manner.
Figure 2. Morphological alterations of cells grown in soft agar (100×).
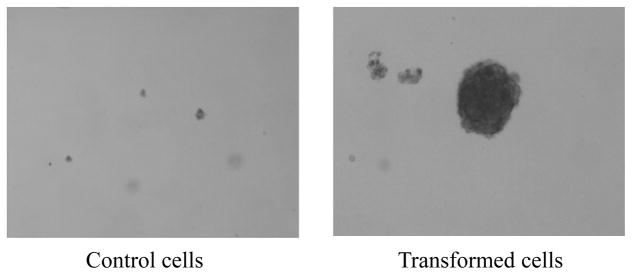
Control cells: 0 μM BaP (0.1% v/v DMSO) treated pol β cells served as solvent control; Transformed cells: pol β cells treated with long time and low concentration BaP and S9 mixture coexposure.
Table 5.
Colony formation of pol β-T cells in soft agar (n=12)
| BaP (μM) | Number of colony formation | Cloning Efficiency (‰) |
|---|---|---|
| 0a | 5.08±1.51 | 0.83±0.12 |
| 1.25 | 24.92±2.84b | 4.17±0.46b |
| 5.00 | 54.33±3.58b | 9.00±1.83b |
| 20.00 | 80.00±2.04b | 13.33±1.41b |
denotes that pol β cells treated by 0.1% (v/v) DMSO served as solvent control;
denotes P<0.05 when compared with solvent control.
3.5 Malignant pol β-T cells exhibited high level of pol β
To further determine if malignant transformation of pol β cells induced by BaP may result from the alteration of pol β expression level, we examined pol β mRNA and protein levels in pol β-T cells and found that the cells exhibited significantly 2-3-fold increase of pol β mRNA and protein levels (Figure 3A lane 2 and Figure 4A right lane, Figure 3B and 4B) compared with pol β cells (Figure 3A lane 1 and Figure 4A left lane, Figure 3B and 4B) (P<0.05). The difference in pol β expression level was not caused by the variations in the amount of loaded samples in our experiments because no significant difference was detected in the mRNA and protein levels of internal controls, i.e. G3PDH mRNA and β-actin protein in the experiments. Thus, the results indicated that chronic exposure to BaP induced pol β overexpression in pol β-T cells.
Figure 3. Pol β mRNA level in pol β cells and pol β-T cells.

M: 100 bp DNA ladder marker; Lane1: pol β cells; Lane2: pol β-T cells.
Quantification of the bands intensity of PCR products was analyzed by Image J. Integral optical density value (IOV) = average optical density value of bands × area of bands. Relative optical density value (rIOV) = IOVpol β/IOVG3PDH.
Figure 4. Pol β protein level in pol β cells and pol β-T cells.
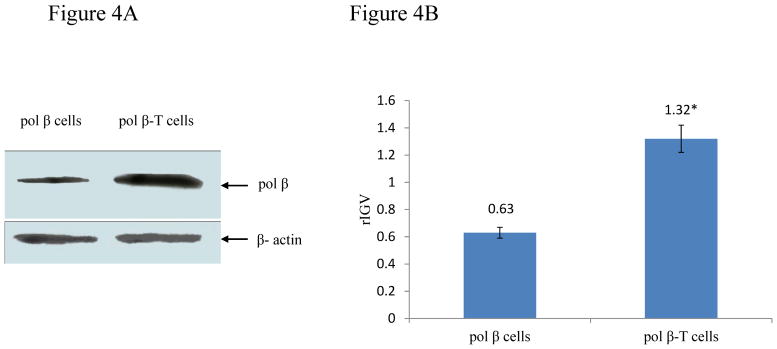
Quantitative analysis was performed by Image J. Integral grayscale value (IGV) = average grayscale value of protein bands × area of protein bands. Relative integral grayscale value (rIGV) = IGVpol β/IGVβ-actin.
3.6 Pol β-T cells exhibited an enhanced HPRT gene mutation frequency and abnormal colony formation
To further characterize the genetic instability of pol β-T cells, we examined the HPRT gene mutation frequency and colony formation of the cells. We found that the colony formation efficiency of pol β-T cells was comparable with that of pol β oe cells (Table 5 and Table 2). Furthermore, we observed that pol β-T cells formed multi-layer and pycnomorphous colonies that represent a feature of unlimited growth (Figure 5, right panel), whereas pol β cells formed colonies with monolayer that attached to the bottom of culture dishes that represent a feature of regulated growth (Figure 5, left panel) indicating malignancy of the BaP-transformed pol β-T cells. Moreover, we discovered that pol β-T cells exhibited significantly increased levels of cloning efficiency (1.8-fold) and mutation frequency (8.5-fold) compared with pol β cells when treated by 20 μM BaP (P<0.05) (Table 6). The results demonstrated that BaP induced malignant transformation and increased gene mutation frequency in pol β-T cells.
Figure 5.
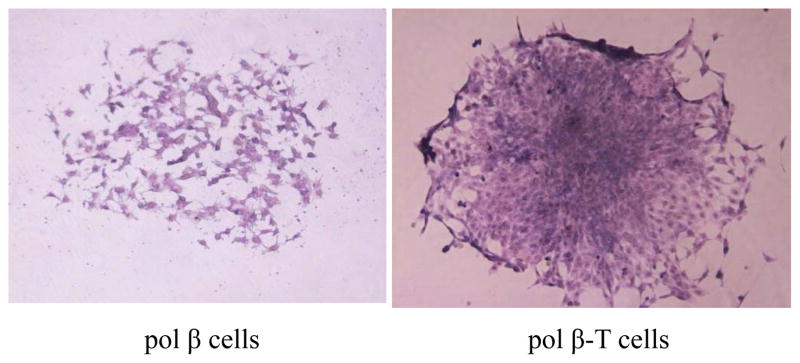
Morphology of mutant colony of pol β cells and pol β-T cells (100×)
Table 6.
Mutation frequency of HPRT gene in pol β-T and pol β cells (n=12)
| Cell lines | Number of mutant colony | Cloning Efficiency (%) | Mutation Frequency (×10−6) |
|---|---|---|---|
| pol β cells | 7.08±1.83 | 21.36±0.92 | 5.46 |
| pol β-T cells | 105.67±7.09a | 38.21±1.36a | 46.23a |
denotes P<0.05 when compared with pol β cells
3.7 Pol β-T cells exhibited high level of polymorphism of RAPD fragments
To characterize the genetic instability of pol β-T cells, the polymorphism of RAPD fragments was studied. Loss of some fragments in pol β-T cells was detected when RAPD fragments were amplified by the random primer P1, P2 and P5 (Table 7 and Figure 6). Additional fragments in pol β-T cells were detected when RAPD fragments were amplified by random primer P1, P4 and P6 (Table 7 and Figure 6). These indicate that pol β-T cells exhibited genetic instability, which may be attributed to the malignancy of pol β-T cells induced by chronic BaP exposure.
Table 7.
Polymorphism of RAPD fragments in pol β-T cells
| Cell line | Random Primers | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| P1 | P2 | P3 | P4 | P5 | P6 | P7 | P8 | P9 | P10 | |
| pol β-T cells | a, b | a | - | b, c | a | b | - | - | - | - |
“a” denotes missing band(s); “b” denotes increasing band(s); “c” denotes band(s) intensity change; and “-” denotes monomorphic band pattern.
Figure 6. PCR products amplified with random primers P1, P2, P4, P5 and P6 in pol β cells and pol β-T cells.
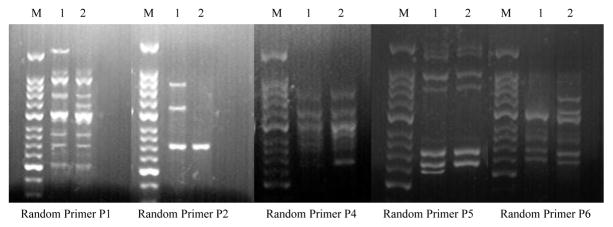
M: 50 bp DNA ladder marker; Lane 1: pol β cells; Lane 2: pol β-T cells.
Discussion
Initiation, promotion and progression of BaP carcinogenesis are multi-stage processes, and their underlying mechanisms remain to be elucidated. Here we hypothesize that BaP may induce cell malignant transformation by altering cellular pol β expression level. To test this hypothesis we have established a cell malignant model (pol β-T cells) created by chronic exposure to low level of BaP. We found that malignant pol β-T cells exhibited high levels of pol β expression, HPRT gene mutation frequency, and polymorphism of RAPD fragments compared with wild-type pol β cells. This indicates that BaP-induced carcinogenesis may be mediated through pol β overexpression, gene mutation and genetic instability. The discovery is consistent with previous findings showing that an alkylating DNA damaging agent methyl methanesulfonate (MMS) can also induce high levels of pol β expression in human embryonic lung fibroblast (HLF) and gene mutation frequency (Du LT, et al., 2006). A correlation among high levels of pol β expression, gene mutation and genetic instability in pol β-T cells suggests that BaP-induced overexpression of pol β can cause high frequency of gene mutation and promote genetic instability possibly through pol β relatively low fidelity DNA synthesis as well as its nucleotide misinsertion in bypassing a template BaP adducted-dA or dG (Chary, et al., 2012).
In this study, for the first time, we successfully established a BaP-induced malignant mouse embryonic fibroblast model by co-exposing BaP and S9 mixture to wild-type pol β cells. We showed that this model exhibited malignant phenotypes and genetic instability demonstrating that our cellular transformation procedure was effective and practical. Because BaP is an indirect carcinogen, and its carcinogenic effects are mediated through metabolic activation, our procedure has paved a way for establishing a cell model for other indirect carcinogens as well as for establishing an effective in vitro system for screening potential indirect carcinogens as recommended by the International Agency for Research on Cancer (IARC). Furthermore, our BaP-transformed cell model provides a useful system to study the mechanisms of BaP carcinogenesis.
Pol β is a critical enzyme in BER pathway. It has a dRP lyase activity that removes 5′-terminal deoxyribose phosphate (5′-dRP) and a gap-filling synthesis activity that fills in a single-nucleotide gap generated from removal of a damaged base by a DNA glycosylase and 5′-incision of an abasic site by APE1 (Liu and Wilson, 2012). Pol β is also actively involved in DNA replication, translesion DNA synthesis and many other biological processes (Lang, et al., 2007; Nemec, et al., 2012). Thus, pol β plays a critical role in maintaining genome integrity and stability as well as a role in causing genomic instability. Pol β functional deficiency can increase frequency of sister chromatid exchange (SCE) and the rate of Rad51 transformation foci as well as spontaneous mutation frequency in human cell line BL2 (Pascucci, et al., 2005; Poltoratsky, et al., 2007). On the other hand, pol β overexpression can induce genetic instability that further leads to the cellular malignant transformation. This include the formation of aneuploid, abnormal location of γ-tubulin, a key component of mitosis, deficiency in mitotic division check points and others (Bergoglio, et al., 2001). In addition, increased pol β expression level can enhance cellular microsatellite instability which in turn accelerates cellular malignant proliferation and transformation (Yamada and Farber, 2002; Al, 2007). Pol β expression can be stimulated by alkylating DNA damaging agent N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) through activation of the cAMP-PKA-CREB signal transduction pathway. Thus it is possible that BaP induces pol β overexpression through the same signaling pathway. Our discovery of BaP-induced cellular overexpression of pol β and gene mutation in mouse embryonic fibroblasts is also consistent with previous findings showing that pol β overexpression in Chinese hamster ovary (CHO) cells increased gene mutation frequency by 4- to 12-fold compared with CHO cells that express wild-type level of pol β (Canitrot, et al., 2000; Frechet, et al., 2001, 2002). This is because pol β lacks a proof-reading 3′–5′ exonuclease activity, thus this confers its relatively low fidelity in DNA synthesis compared with replication DNA polymerases. Excessive amount of pol β may compete and substitute high fidelity DNA polymerases during DNA repair, thereby increasing the probability of nucleotide misinsertions, gene mutation and cancer susceptibility (El-Andaloussi, et al., 2006). Overexpression of pol β may also promote translesion DNA synthesis and error-prone DNA synthesis during repair of alkylated DNA base lesions (Bambara and Huang, 1995). It has been found that high level of pol β expression can promote DNA translesion synthesis in bypassing an O6-methylguanine (O6mG) by preferentially inserting a thymine resulting in a guanine to adenine transition mutation during the repair of alkylated DNA base lesions (Singh, et al., 1996; Wood and Shivji, 1997). Our previous study also suggests that overexpression of pol β induces base loss and/or base substitutions during BER that further lead to increased gene mutation frequency in cells (Luo, et al., 2012).
Our results suggest a mechanism by which BaP causes malignant transformation of pol β cells and carcinogenesis by promoting gene mutation and genetic instability through increasing cellular pol β expression level. This has been supported by previous studies. Differentiated liver cells with high pol β expression exhibited stronger proliferative capacity with accumulation of base loss, misinsertion and/or base substitution than embryonic liver cells with wild-type level of pol β expression (Liang, et al., 2007). It’s also reported that increased pol β activity resulting from enhanced pol β mRNA and protein levels in various types of cancer tissues was remarkably higher than normal tissues (Albertella, et al., 2005). The levels pol β mRNA and protein were found to be increased in one third of neoplasm tissues and cancer cell lines including colon cancer, breast cancer, ovary cancer and leukaemia cell lines (Tan, et al., 2005). The pol β level in breast adenocarcinoma tissue was 286-fold of that in the adjacent normal tissues (Albertella, et al., 2005). Overexpression of pol β has also been found to mediate tumorigenesis possibly by promoting mutator phenotypes resulting from pol β mutant proteins that have been identified in neurospongioma, esophageal carcinoma, nasopharyngeal carcinoma, gastric cancer, prostate cancer and cervical cancer by epidemiological studies (Zhao, et al., 2012). C127 mouse cells that expressed high levels of wild-type pol β protein exhibited no malignant phenotypes. However, C127 cells that overexpressed a mutant pol β protein with deficiency in dRP lyase exhibited malignant transformation features (Lang, et al., 2007). This mutant pol β along with the others, i.e. pol β E295K, K289M and I260M mutants identified in gastric cancer, colon cancer and prostate cancer can compete with wild-type pol β to interfere base lesion repair conferring a dominant-negative effect on BER leading to cellular malignant transformation phenotypes such as loss of anchorage-dependent growth and focus formation and ultimately tumorigenesis.
In this study, we employed RAPD technique to explore genetic instability induced by BaP in pol β cells and pol β-T cells by determining the polymorphism of RAPD fragments. This technique allows the amplification of genome DNA through PCR with a series of random primers for detecting genetic instability resulting from base loss, insertion and deletions and transposition. With optimized genomic DNA concentration and annealing temperature, we successfully identified a significant increase in polymorphic variations of RAPD fragments in pol β cells and pol β-T cells with several random primers. This demonstrates that RAPD technique can be used to as an efficient and sensitive approach for detecting BaP-induced genetic instability in mouse embryonic fibroblasts.
Conclusion
In this study, we have discovered that BaP can induce malignant transformation of wild-type pol β cells through metabolic activation by promoting cellular pol β overexpression. We further identified a correlation between pol β overexpression and high levels of HPRT gene mutation frequency and polymorphism of RAPD fragments in the BaP-transformed pol β-T cells. The results suggest that BaP-induced carcinogenesis is mediated by cellular high level of pol β expression that further results in gene mutation and genetic instability. Thus, our study reveals an importance of DNA polymerase β overexpression in promoting xenobiotic carcinogenesis. Our BaP-induced malignant cell model allows us to further study the roles of other DNA repair pathways in BaP carcinogenesis by using small molecule inhibitors of DNA polymerase β in the future.
Acknowledgments
This research was supported by the grant No. 30872079 and 81172632 from the National Natural Science Foundation of China to Zunzhen Zhang and partially supported by National Institutes of Health grant ES017476 to Yuan Liu.
Footnotes
Conflict of interest statement:
The authors declare no potential conflict of interest relevant to this article.
References
- Al MAA. Elevated expression of DNA polymerase II increases spontaneous mutagenesis in Escherichia coli. Mutat Res. 2007;625:29–39. doi: 10.1016/j.mrfmmm.2007.05.002. [DOI] [PubMed] [Google Scholar]
- Albertella MR, Lau A, O’Connor MJ. The overexpression of specialized DNA polymerases in cancer. DNA Repair (Amst) 2005;4:583–593. doi: 10.1016/j.dnarep.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Bambara RA, Huang L. Reconstitution of mammalian DNA replication. Prog Nucleic Acid Res Mol Biol. 1995;51:93–122. doi: 10.1016/s0079-6603(08)60877-6. [DOI] [PubMed] [Google Scholar]
- Bergoglio V, Canitrot Y, Hogarth L, Minto L, Howell SB, Cazaux C, Hoffmann JS. Enhanced expression and activity of DNA polymerase beta in human ovarian tumor cells: impact on sensitivity towards antitumor agents. Oncogene. 2001;20:6181–6187. doi: 10.1038/sj.onc.1204743. [DOI] [PubMed] [Google Scholar]
- Canitrot Y, Hoffmann JS, Calsou P, Hayakawa H, Salles B, Cazaux C. Nucleotide excision repair DNA synthesis by excess DNA polymerase beta: a potential source of genetic instability in cancer cells. FASEB J. 2000;14:1765–1774. doi: 10.1096/fj.99-1063com. [DOI] [PubMed] [Google Scholar]
- Chary P, Beard WA, Wilson SH, Lloyd RS. DNA polymerase beta gap-filling translesion DNA synthesis. Chem Res Toxicol. 2012;25:2744–2754. doi: 10.1021/tx300368f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Zhang Y, Yang J, Jin M, Wang XW, Shen ZQ, Qiu Z, Zhao G, Wang J, Li JW. Estrogen promotes benzo[a]pyrene-induced lung carcinogenesis through oxidative stress damage and cytochrome c-mediated caspase-3 activation pathways in female mice. Cancer Lett. 2011;308:14–22. doi: 10.1016/j.canlet.2011.04.007. [DOI] [PubMed] [Google Scholar]
- Dizdaroglu M. Oxidatively induced DNA damage: mechanisms, repair and disease. Cancer Lett. 2012;327:26–47. doi: 10.1016/j.canlet.2012.01.016. [DOI] [PubMed] [Google Scholar]
- Du LT, Xu L, Yang XF, He Y, Wei Q, Zhuang ZX. Effects of overexpression of human pol-beta on cellular response to DNA damage. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi. 2006;24:88–91. [PubMed] [Google Scholar]
- El-Andaloussi N, Valovka T, Toueille M, Steinacher R, Focke F, Gehrig P, Covic M, Hassa PO, Schar P, Hubscher U, Hottiger MO. Arginine methylation regulates DNA polymerase beta. Mol Cell. 2006;22:51–62. doi: 10.1016/j.molcel.2006.02.013. [DOI] [PubMed] [Google Scholar]
- Frechet M, Canitrot Y, Bieth A, Dogliotti E, Cazaux C, Hoffmann JS. Deregulated DNA polymerase beta strengthens ionizing radiation-induced nucleotidic and chromosomal instabilities. Oncogene. 2002;21:2320–2327. doi: 10.1038/sj.onc.1205295. [DOI] [PubMed] [Google Scholar]
- Frechet M, Canitrot Y, Cazaux C, Hoffmann JS. DNA polymerase beta imbalance increases apoptosis and mutagenesis induced by oxidative stress. FEBS Lett. 2001;505:229–232. doi: 10.1016/s0014-5793(01)02834-4. [DOI] [PubMed] [Google Scholar]
- Hakura A, Seki Y, Sonoda J, Hosokawa S, Aoki T, Suganuma A, Kerns WD, Tsukidate K. Rapid induction of colonic adenocarcinoma in mice exposed to benzo[a]pyrene and dextran sulfate sodium. Food Chem Toxicol. 2011;49:2997–3001. doi: 10.1016/j.fct.2011.07.057. [DOI] [PubMed] [Google Scholar]
- Herbstman JB, Tang D, Zhu D, Qu L, Sjodin A, Li Z, Camann D, Perera FP. Prenatal exposure to polycyclic aromatic hydrocarbons, benzo[a]pyrene-DNA adducts, and genomic DNA methylation in cord blood. Environ Health Perspect. 2012;120:733–738. doi: 10.1289/ehp.1104056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundu CN, Balusu R, Jaiswal AS, Gairola CG, Narayan S. Cigarette smoke condensate-induced level of adenomatous polyposis coli blocks long-patch base excision repair in breast epithelial cells. Oncogene. 2007;26:1428–1438. doi: 10.1038/sj.onc.1209925. [DOI] [PubMed] [Google Scholar]
- Labib S, Yauk C, Williams A, Arlt VM, Phillips DH, White PA, Halappanavar S. Subchronic oral exposure to benzo(a)pyrene leads to distinct transcriptomic changes in the lungs that are related to carcinogenesis. Toxicol Sci. 2012;129:213–224. doi: 10.1093/toxsci/kfs177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang T, Dalal S, Chikova A, DiMaio D, Sweasy JB. The E295K DNA polymerase beta gastric cancer-associated variant interferes with base excision repair and induces cellular transformation. Mol Cell Biol. 2007;27:5587–5596. doi: 10.1128/MCB.01883-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang L, Deng L, Mendonca MS, Chen Y, Zheng B, Stambrook PJ, Shao C, Tischfield JA. X-rays induce distinct patterns of somatic mutation in fetal versus adult hematopoietic cells. DNA Repair (Amst) 2007;6:1380–1385. doi: 10.1016/j.dnarep.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wilson SH. DNA base excision repair: a mechanism of trinucleotide repeat expansion. Trends Biochem Sci. 2012;37(4):162–172. doi: 10.1016/j.tibs.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Q, Lai Y, Liu S, Wu M, Liu Y, Zhang Z. Deregulated expression of DNA polymerase beta is involved in the progression of genomic instability. Environ Mol Mutagen. 2012;53:325–333. doi: 10.1002/em.21697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrotra S, Khwaja O, Kukreja AK, Rahman L. ISSR and RAPD based evaluation of genetic stability of encapsulated micro shoots of Glycyrrhiza glabra following 6 months of storage. Mol Biotechnol. 2012;52:262–268. doi: 10.1007/s12033-011-9491-6. [DOI] [PubMed] [Google Scholar]
- Nemec AA, Donigan KA, Murphy DL, Jaeger J, Sweasy JB. Colon cancer-associated DNA polymerase beta variant induces genomic instability and cellular transformation. J Biol Chem. 2012;287:23840–23849. doi: 10.1074/jbc.M112.362111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nkrumah-Elie YM, Reuben JS, Hudson A, Taka E, Badisa R, Ardley T, Israel B, Sadrud-Din SY, Oriaku E, Darling-Reed SF. Diallyl trisulfide as an inhibitor of benzo(a)pyrene-induced precancerous carcinogenesis in MCF-10A cells. Food Chem Toxicol. 2012;50:2524–2530. doi: 10.1016/j.fct.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno E, Itoh A, Kawashima H, Ishikawa T, Matsubara H, Itoh Y, Nakamura Y, Hiramatsu T, Nakamura M, Miyahara R, Ohmiya N, Ishigami M, Katano Y, Goto H, Hirooka Y. Malignant transformation of branch duct-type intraductal papillary mucinous neoplasms of the pancreas based on contrast-enhanced endoscopic ultrasonography morphological changes: focus on malignant transformation of intraductal papillary mucinous neoplasm itself. Pancreas. 2012;41:855–862. doi: 10.1097/MPA.0b013e3182480c44. [DOI] [PubMed] [Google Scholar]
- Pascucci B, Russo MT, Crescenzi M, Bignami M, Dogliotti E. The accumulation of MMS-induced single strand breaks in G1 phase is recombinogenic in DNA polymerase beta defective mammalian cells. Nucleic Acids Res. 2005;33:280–288. doi: 10.1093/nar/gki168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poltoratsky V, Prasad R, Horton JK, Wilson SH. Down-regulation of DNA polymerase beta accompanies somatic hypermutation in human BL2 cell lines. DNA Repair (Amst) 2007;6:244–253. doi: 10.1016/j.dnarep.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J, Su L, Snow ET. Replication across O6-methylguanine by human DNA polymerase beta in vitro. Insights into the futile cytotoxic repair and mutagenesis of O6-methylguanine. J Biol Chem. 1996;271:28391–28398. doi: 10.1074/jbc.271.45.28391. [DOI] [PubMed] [Google Scholar]
- Song MK, Kim YJ, Song M, Choi HS, Park YK, Ryu JC. Polycyclic aromatic hydrocarbons induce migration in human hepatocellular carcinoma cells (HepG2) through reactive oxygen species-mediated p38 MAPK signal transduction. Cancer Sci. 2011;102:1636–1644. doi: 10.1111/j.1349-7006.2011.02000.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan XH, Zhao M, Pan KF, Dong Y, Dong B, Feng GJ, Jia G, Lu YY. Frequent mutation related with overexpression of DNA polymerase beta in primary tumors and precancerous lesions of human stomach. Cancer Lett. 2005;220:101–114. doi: 10.1016/j.canlet.2004.07.049. [DOI] [PubMed] [Google Scholar]
- Wood RD, Shivji MK. Which DNA polymerases are used for DNA-repair in eukaryotes. Carcinogenesis. 1997;18:605–610. doi: 10.1093/carcin/18.4.605. [DOI] [PubMed] [Google Scholar]
- Yamada NA, Farber RA. Induction of a low level of microsatellite instability by overexpression of DNA polymerase Beta. Cancer Res. 2002;62:6061–6064. [PubMed] [Google Scholar]
- Zhao W, Wu M, Yang M, Zhang ZZ. Mechanism of DNA polymerase beta hyper-expression in malignant transformation induced by benzo[a] pyrene. Sichuan Da Xue Xue Bao Yi Xue Ban. 2012;43:801–806. [PubMed] [Google Scholar]