

Abstract
Membrane-type 1 matrix metalloproteinase (MT1-MMP, MMP-14), a transmembrane proteinase with an extracellular catalytic domain and a short cytoplasmic tail, degrades extracellular matrix components and controls diverse cell functions through proteolytic and non-proteolytic interactions with extracellular, intracellular and transmembrane proteins. Here we show that in tumor cells MT1-MMP downregulates fibroblast growth factor-2 (FGF-2) signaling by reducing the amount of FGF-2 bound to the cell surface with high and low affinity. FGF-2 induces weaker activation of ERK1/2 MAP kinase in MT1-MMP expressing cells than in cells devoid of MT1-MMP. This effect is abolished in cells that express proteolytically inactive MT1-MMP but persists in cells expressing MT1-MMP mutants devoid of hemopexin-like or cytoplasmic domain, showing that FGF-2 signaling is downregulated by MT1-MMP proteolytic activity. MT1-MMP expression results in downregulation of FGFR-1 and -4, and in decreased amount of cell surface-associated FGF-2. In addition, MT1-MMP strongly reduces the amount of FGF-2 bound to the cell surface with low affinity. Because FGF-2 association with low-affinity binding sites is a prerequisite for binding to its high-affinity receptors, downregulation of low-affinity binding to the cell surface results in decreased FGF-2 signaling. Consistent with this conclusion, FGF-2 induction of tumor cell migration and invasion in vitro is stronger in cells devoid of MT1-MMP than in MT1-MMP expressing cells. Thus, MT1-MMP controls FGF-2 signaling by a proteolytic mechanism that decreases the cell’s biological response to FGF-2.
Keywords: Membrane-type-1 matrix metalloproteinase (MT1-MMP), fibroblast growth factor (FGF), fibroblast growth factor receptor (FGFR), proteoglycans, receptors, MAP kinases (MAPKs), ERK1/2, cell migration
INTRODUCTION
Membrane-type 1 matrix metalloproteinase (MT1-MMP, MMP-14), the prototypical member of the MT-MMP subclass of matrix metalloproteinases (MMP), is a cell-membrane-bound proteinase with an extracellular catalytic site and a 20-amino acid cytoplasmic tail (Deryugina and Quigley, 2006). MT1-MMP degrades a variety of extracellular matrix (ECM) components including fibrillar collagen, and activates the proenzyme forms of MMP-2 and MMP-13. These features have implicated MT1-MMP as an important component of the proteolytic mechanisms of a variety of physiological and pathological processes, including tumor invasion, metastasis and angiogenesis (Hotary, et al., 2006, Sabeh, et al., 2004). The analysis of the phenotype of mice genetically deficient in MT1-MMP (MT1-MMP−/−) has shown important roles of this enzyme in development, connective tissue metabolism and angiogenesis. The genetic deficiency of MT1-MMP in the mouse causes a variety of severe abnormalities in postnatal development, including dwarfism, musculoskeletal defects, lipodystrophy, defective angiogenesis and vascular invasion of the skeleton, defective pulmonary alveoli septation and submandibular gland branching. MT1-MMP−/− mice die between 3 weeks and 3 months of age of an unknown cause(s) (Atkinson, et al., 2005, Chun, et al., 2006, Galvez, et al., 2005, Holmbeck, et al., 1999, Holmbeck, et al., 1999, Ohtake, et al., 2006, Zhou, et al., 2000). Because of the important role of MT1-MMP in ECM degradation, it has been proposed that the phenotype of MT1-MMP−/− mice results from defective collagen turnover (Holmbeck, et al., 1999).
However, increasing evidence shows that MT1-MMP is a multifunctional protein that, in addition to remodeling the ECM, controls intracellular signaling by a variety of different mechanisms. MT1-MMP interaction with platelet-derived growth factor receptor-beta (PDGFRβ) and LDL receptor-related protein-1 (LRP1) controls PDGF-B induction of mitogenic signaling and differentiation in vascular smooth muscle cells (Lehti, et al., 2009, Lehti, et al., 2005). MT1-MMP controls fibroblast growth factor-2 (FGF-2) signaling by several mechanisms. It forms a complex with FGF receptor (FGFR)-4 (Sugiyama, et al., 2010), and potentiates FGF-2 induction of corneal angiogenesis by modulating FGF-2-activated intracellular signaling pathways (Onguchi, et al., 2009). In calvarial osteoblasts MT1-MMP upregulates FGF signaling by shedding ADAM-9, which in turn cleaves FGF receptor-2 (FGFR-2) (Chan, et al., 2012).
FGF-2 is the prototype member of a family of heparin-binding proteins with growth factor or hormone activity (Beenken and Mohammadi, 2009, Brooks, et al., 2012, Presta, et al., 2005). Formerly known as basic FGF (bFGF), FGF-2 is a ubiquitous growth factor implicated in many physiological and pathological processes, such as cell survival, proliferation, migration, growth and differentiation (Presta, et al., 2005). The biological effects of FGF-2 are mediated by four high-affinity tyrosine kinase receptors (FGFR-1 to -4) and low-affinity receptors, consisting of the heparan sulfate (HS) and heparan sulfate proteoglycans (HSPG) present in the ECM or bound to the membrane of essentially all cell types. FGF-2 binding of HSPG provides a fine control of the bioavailability and interaction of this growth factor with cells (Schlessinger, et al., 2000). Cell-associated HSPG are required for FGF-2 binding to, and activation of FGFRs, which involves the dimerization of a ternary complex consisting of two molecules of FGF-2, two FGFRs and two heparan sulfate chains (Plotnikov, et al., 1999, Schlessinger, et al., 2000). HSPG on the cell membrane and in the ECM also act as a high-capacity reservoir for FGF-2. By virtue of its binding to HSPG FGF-2 is highly concentrated in insoluble phase on the cell surface and ECM, and can thus provide prolonged stimulation to the cell (Flaumenhaft, et al., 1989, Moscatelli, 1992). ECM-degrading proteinases including plasminogen activators/plasmin and MMPs have been implicated in the mobilization of FGF-2 from the insoluble phase, necessary for FGFR activation (Flaumenhaft, et al., 1992, Rifkin, et al., 1990).
Here we report that MT1-MMP lowers the amount of cell surface-associated FGF-2 and downregulates FGF-2 activation of intracellular signaling. This effect results in a reduced biological response of MT1-MMP expressing cells to FGF-2.
MATERIALS AND METHODS
Materials
Rabbit anti-human MT1-MMP antibody (hinge region), Ilomastat (GM6001), and PVDF membranes were purchased from Millipore (Billerica, MA); mouse anti-human phosphop44/42 MAPK (ERK1/2) (Thr202/Tyr204) from Cell Signaling Technology (Danver, MA); mouse anti-human CD138 clone MI15 antibody, biocoat matrigel invasion chamber and matrigel basement membrane matrix from BD Biosciences (San Jose, CA), doxycycline, 1,10-phenantroline, bovine serum albumin (BSA), and mouse anti-human tubulin antibody from Sigma-Aldrich (St. Louis, MO); donkey anti-mouse IgG antibody, donkey IgG anti-rabbit antibody, and streptavidin (all conjugated with horseradish peroxidase) from Jackson ImmunoResearch Laboratories (West Grove, PA); sulfosuccinimidyl-6-[biotin-amido]hexanoate (sulfo-NHS-LCbiotin), bicinchoninic acid (BCA) protein assay kit, and SuperSignal West Pico chemiluminescent substrate from Thermo Scientific (Rockford, IL); heparin-Sepharose beads from Reprokine Research Immunity (Valley Cottage, NY); protein A/G PLUS-agarose immunoprecipitation reagent, rabbit anti-human ERK2, rabbit anti-human FGFR-1, and rabbit anti-human FGFR-4 antibodies from Santa Cruz Biotechnology (Santa Cruz, CA); rabbit anti-human FGFR-2 antibody from Abcam (Cambridge, MA); mouse anti-human ADAM-9 antibody from R&D Systems (Minneapolis, MN); recombinant human basic FGF (155) from Akron Biotech (Boca Raton, FL); GoTaq DNA Polymerase from Promega (Madison, WI); Lipofectamine 2000 Reagent, TRIzol Reagent, SuperScript II Reverse Transcriptase, Alexa Fluor 488 goat anti-mouse antibody, Alexa Fluor 432 goat anti-mouse antibody and DAPI from Invitrogen (Grand Island, NY); MT1-MMP siRNA (siGENOME SMART) and control siRNA pools from Dharmacon (GE Healthcare Life Sciences, Piscataway, NJ); complete mini protease inhibitor cocktail, PhosSTOP phosphatase inhibitor cocktail, and DNAse I from Roche (Indianapolis, IN); Dulbecco’s modified Eagle’s medium (DMEM), DMEM/F-12 Ham’s medium, fetal bovine serum (FBS), L-glutamine, penicillin, and streptomycin from CellGro (Manassas, MA). Mouse anti-human FGF-2 monoclonal antibody (354FI) was a generous gift from Texas Bio-Technologies (Houston, TX).
Cells and culture media
Human MCF-7 breast adenocarcinoma cells and MCF-7 cells stably transfected with MT1-MMP cDNA under control by the tetracycline resistance transactivator in the Tet-Off conformation have been described (D’Alessio, et al., 2008). The human pediatric glioma cell lines: SF188, KNS42, UW479, Res259 and Res186 (Bax, et al., 2009) were kindly provided by Dr. C. Jones (The Institute of Cancer Research, Sutton, UK). Transfected and nontransfected MCF-7 cells were grown in DMEM supplemented with 10% (v/v) FBS, 2 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. The glioma cells were grown in DMEM/F12 Ham’s medium supplemented with FBS, L-glutamine and antibiotics as above.
FGF-2 treatment
Sub-confluent MT1-MMP Tet-Off MCF-7 cells grown for 24 h in medium containing 0.5% FBS with or without doxycycline (DOX; 1 μg/ml) were incubated with the indicated concentrations of FGF-2 for 15 min. FGF-2 was added to the cultures in a volume of 1 μl in medium without changing the medium in the cell cultures. An equivalent volume of medium without FGF-2 was added as a control. Where indicated, Ilomastat (50 μM final concentration) was added to the medium 15 min before FGF-2 addition.
Transient transfections
The plasmids encoding wild-type MT1-MMP, the cytoplasmic tail deleted (Δcyt) and the proteolytically inactive (E240A) MT1-MMP mutants have been described (D’Alessio et al., 2008). The plasmid encoding the MT1-MMP mutant lacking the hemopexin domain (Δpex) was a kind gift from Dr. J. Cao (Stony Brook University School of Medicine) (Zarrabi, et al., 2011). The constructs (3 μg) were transiently transfected into sub-confluent MCF-7 cells in 6-well plates using 7.5 μl of Lipofectamine according to the manufacturer’s instructions. The MT1-MMP siRNAs pool or control scrambled siRNAs (5 nM) were transiently transfected into sub-confluent Res186 cells in 6-well plates using 5 μl of Lipofectamine according to the manufacturer’s instructions. Twenty-four hours after transfection, the cells were incubated with medium containing 0.5% FBS for additional 24 h, and immediately used for the experiments.
Reverse Transcriptase Polymerase Chain Reaction
Total RNA was extracted with the TRIzol Reagent including DNase I treatment. Yield and purity were characterized by Nanodrop spectrophotometry. One microgram of total RNA was reverse-transcribed using 500 ng of random primers and 200 U of SuperScript Reverse Transcriptase. All the protocols were performed following the manufacturer’s instructions. For amplification of FGFR-1, -2, -3, and -4 cDNA GoTaq polymerase, 5 μmoles of forward and reverse primers, and the following conditions were used: denaturation at 95° C for 10 min, followed by 28 cycles of denaturation at 95° C for 30 sec, annealing at 58° C for 30 sec, and elongation at 72° C for 30 sec. GAPDH was amplified as a loading control under the same conditions. The following primers were designed with Primer3 (v. 0.4.0) using default settings. Because different FGFR isoforms are generated by alternative splicing, Fast DB software was first used to identify the exons shared by all FGFR variants, and primers were subsequently designed with Primer3: FGFR-1_FOR 5′ – ACCACCGACAAAGAGATGGA – 3′; FGFR-1_REV 5′ – GCCCCTGTGCAATAGATGAT – 3′; FGFR-2_FOR 5′ – TCTAAAGGCAACCTCCGAGA; FGFR-2_REV 5′ – CTCTGGCGAGTCCAAAGTCT – 3′; FGFR-3_FOR 5′ – CCACTGTCTGGGTCAAGGAT – 3′; FGFR-3_REV 5′ – CCAGCAGCTTCTTGTCCATC – 3′; FGFR-4_FOR 5′ – TCATCAACCTGCTTGGTGTC – 3′; FGFR-4_REV 5′ – CGGGACTCCAGATACTGCAT – 3′; GAPDH_FOR 5′ – AACATCATCCCTGCCTCTAC – 3′; GAPDH_REV 5′ – CCCTGTTGCTGTAGCCAAAT – 3′
Western blotting
Cells were washed with ice-cold PBS and lysed in RIPA buffer (150 mM NaCl, 1% Igepal, 0.5% sodium deoxycholate, 0.1% SDS in 50 mM Tris-HCl, pH 8.0) containing protease (Complete) and phosphatase (PhosSTOP) inhibitors. The lysates were sonicated and centrifuged (14,000 rpm for 15 min at 4° C in an Eppendorf centrifuge). Cell extract protein (20-40 μg) was electrophoresed in SDS/10% or 12% polyacrylamide gels, and analyzed by Western blotting with the indicated antibodies as described (D’Alessio, et al., 2008). For analysis of FGF-2 in cell-conditioned medium or washing buffers heparin-Sepharose beads (20 μl) equilibrated with serum-free medium were incubated with 200 μl of the sample for 2 h at 4° C in an end-over-end mixer. Following centrifugation, the pelleted beads were resuspended in reducing sample buffer, boiled at 95° C for 5 min, and loaded onto a SDS/12% polyacrylamide gel. In most experiments the membranes were stripped of the antibodies by incubation in a mild stripping buffer (20 mM Glycine, 0.1% SDS, 1% Tween 20, pH 2.2) for 30 min at room temperature with gentle agitation, re-blocked and re-probed with other antibodies.
Densitometry
Quantitative analysis of Western blot bands was performed with ImageJ 10.2 software (National Institutes of Health). Data are shown as the ratio between the readings of the sample and that of the corresponding loading control, unless indicated otherwise.
Gelatin zymography analysis of MMP-2 activation
Because MCF-7 cells do not express MMP-2 (Rozanov, et al., 2001), cells transfected with MT1-MMP or control empty vector were incubated for 2 h in serum-free medium conditioned by human umbilical vein endothelial (HUVE) cells, which secrete proMMP-2 and no MMP-9 (Shamamian, et al., 2001). The conditioned medium was then analyzed by gelatin zymography as described (Mazzieri, et al., 1997).
Biotinylation of cell surface-associated and soluble FGF-2
To label cell surface associated FGF-2 we used water-soluble, cell membrane-impermeable Sulfo-NHS-LC-biotin. Cell monolayers incubated with 30 ng/ml FGF-2 for 15 min were washed 3 times with ice-cold PBS, and incubated on ice for 30 min with 1 mM Sulfo-NHS-LC-biotin. Following 3 washings with 100 mM glycine in PBS to quench and remove excess biotin, the cells were lysed in RIPA buffer (50 mM Tris-HCl, 125 mM NaCl, 0.5% Igepal, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM DTT, pH 8.0) containing protease and phosphatase inhibitors. The cell lysate was sonicated, centrifuged (14,000 rpm for 15 min at 4° C in an Eppendorf centrifuge), and assayed for protein concentration. Anti-FGF-2 antibody (1 μg) or mouse non-immune IgG (1 μg) was added to cell extract protein (1 mg), and incubated with protein A/G PLUS-agarose beads (30 μl) overnight at 4° C. Following centrifugation at 2,000 rpm for 4 min at 4° C, the pelleted beads were washed 3 times with RIPA buffer and resuspended in 4X reducing sample buffer, loaded onto a SDS/12% polyacrylamide gel for Western blotting analysis, and the immunocomplexes were detected with peroxidase-conjugated streptavidin or FGF-2 antibody as indicated. To biotinylate soluble, purified FGF-2 10 mM Sulfo-NHS-LC-biotin was added to 100 μg/ml FGF-2 in 100 μl of PBS, and incubated on ice for 2 h. The biotinylated FGF-2 was analyzed by Western blotting with peroxidase-conjugated streptavidin without further purification.
Elution of FGF-2 from the cell surface
MT1-MMP Tet-Off MCF-7 cells grown for 24 h in medium containing 0.5% FBS with or without DOX (1 μg/ml) were incubated with 30 ng/ml of FGF-2 for 15 min. The cells were washed 3 times with PBS followed by one washing with 2 M NaCl in 20 mM HEPES, pH 7.5 or one washing with PBS as a control (Moscatelli, 1987). The cells were lysed with RIPA buffer. Cell lysates and the final PBS and 2 M NaCl washings (900 μl) were incubated with 50 μl of heparin-Sepharose overnight at 4° C in an end-over-end mixer. Following centrifugation at 2,000 rpm for 4 min at 4° C, the pelleted beads were resuspended in 4 X reducing sample buffer and loaded onto a SDS/12% polyacrylamide gel for Western blotting analysis.
In vitro scratch assay for cell migration
For MT1-MMP Tet-Off MCF-7 cell transfectants confluent monolayers of cells grown for 24 h in the presence or absence of DOX (1 μg/ml) in medium containing 0.5% FBS were scratched as described (Liang, et al., 2007). After photographing the cells (time 0), incubation was continued in the same media with or without addition of FGF-2 (10 ng/ml). The cultures were observed twice daily, and photographed after 42 h incubation, when wound closure was first observed. Wound areas were measured using Adobe Photoshop and photographs of multiple areas taken at 0 and 42 h. For Res186 glioma cells confluent monolayers were scratched; multiple areas of the wound were labeled on the bottom of the dish and photographed immediately (time 0). The same areas were photographed 24 h, 45 h and 69 h later. The area covered by cells on one side of the picture at time 0 was selected with Photoshop and moved onto the same side of the picture taken at 69 h, and the cells covering the wound area were counted manually. These cells represent the cells migrated from the edge of the wound into the wound area.
In vitro invasion assay
MT1-MMP Tet-Off MCF-7 cells grown for 24 h in serum-free medium with or without DOX (1 μg/ml) were placed into the upper chamber of a matrigel-coated insert (8-μm pore size). Serum-free medium with or without FGF-2 (10 ng/ml) was added to the lower chamber. After overnight incubation, the cells attached to the upper aspect of the membrane were removed by gently scraping with a Q-tip. The cells that had invaded through the membrane were stained with the Diff-Quick staining solutions, and counted using an inverted microscope.
Flow cytometry
Cells were harvested, washed with PBS and incubated for 30 min at 4° C with mouse anti- human CD138 (syndecan-1) antibody (clone MI15; 0.5 μg) in PBS, 2 mM EDTA, 0.05% BSA. Subsequently the cells were incubated for 20 min at 4° C with Alexa Fluor 532-conjugated goat anti-mouse antibody (0.5 μg) in the same buffer. Isotype control was included in the analysis. The cells were fixed in 1% PFA, and fluorescence was measured using a FACScalibur.
Immunofluorescence
Cells fixed in 4% PFA were blocked for 1 h at room temperature with 3% BSA in PBS and incubated for 1 h at 4° C with mouse anti-human CD138 antibody (clone MI15; 0.5 μg) in PBS 0.2% BSA. Following 1 h incubation at 4° C with Alexa Fluor 488-conjugated goat anti-mouse antibody in PBS 0.2% BSA, the cells were counterstained with DAPI to evidence the nuclei.
Statistical analysis
The data were analyzed using the Students’ t test. A p value ≤ 0.05 was considered as significant.
RESULTS
MT1-MMP expression downregulates ERK1/2 activation by FGF-2
To study the effect of MT1-MMP on FGF-2 signaling we used human MCF-7 mammary carcinoma cells stably transfected with MT1-MMP under control by the tetracycline resistance transactivator (Tet-Off) (D’Alessio, et al., 2008). Non-transfected MCF-7 cells do not express MT1-MMP, MMP-2 or FGF-2 (D’Alessio, et al., 2008, Rozanov, et al., 2001). Addition of doxycycline (DOX; 1 μg/ml) to the culture medium of Tet-Off MT1-MMP transfectants blocks MT1-MMP expression but has no effect on FGF-2 expression (Figs. 1 A, and 5). By semi-quantitative RT-PCR analysis our Tet-Off MT1-MMP transfectants show expression of FGFR-1, -2 and -4, and almost undetectable levels of FGFR-3 (Fig. 1 C).
Figure 1. MT1-MMP downregulates ERK1/2 activation by FGF-2.

Western blotting analysis of ERK1/2 activation (p-ERK1/2) and MT1-MMP expression. A. MT1-MMP Tet-Off MCF-7 cells grown for 24 h in the presence or absence of DOX (1 μg/ml) in medium containing 0.5% FBS were treated with 0.75 ng/ml of FGF-2 for 15 min. B. Densitometric analysis. C. RT-PCR analysis of FGFR expression in MT1-MMP Tet-Off MCF-7 cells. H2O and GAPDH are shown as negative and loading controls, respectively. D and E. Cells grown for 24 h in the presence or absence of DOX in medium containing 0.5% FBS were treated with increasing concentrations of FGF-2 for 15 min (D) or with 0.75 ng/ml FGF-2 for the indicated times (E). Tubulin (TUB) and ERK2 are shown as loading controls. Panel B and the lower panels of D and E show mean ± S.E. of densitometric readings normalized to the corresponding loading control; *, p ≤ 0.05; - MT1-MMP vs. the corresponding + MT1-MMP sample. The experiment shown in panel A was repeated multiple times with comparable results. The experiments shown in panels D and E were repeated three times with similar results
Figure 5. FGF-2 binding to cells inversely correlates with MT1-MMP expression.

A. MT1-MMP Tet-Off MCF-7 cells grown for 24 h in the presence or absence of DOX (1 μg/ml) in medium containing 0.5% FBS were incubated with FGF-2 (30 ng/ml) for the indicated time. ERK1/2 phosphorylation, MT1-MMP expression and cell-associated or soluble FGF-2 in the conditioned medium were characterized by Western blotting. Tubulin (TUB) is shown as a loading control. B. Densitometric analysis of ERK1/2 activation and cell extract-associated FGF-2. The histograms show mean ± S.E. of densitometric readings normalized to the corresponding loading control; *, p ≤ 0.05; - MT1-MMP vs. the corresponding + MT1-MMP sample. This experiment was repeated three times with similar results.
The mitogen-activated protein kinases (MAPK) are components of the signaling mechanism activated by FGF-2 (Klint, et al., 1995). Therefore, to analyze FGF-2 signaling we characterized the phosphorylation of the MAPK extracellular signal-regulated kinases 1 and 2 (ERK1/2). For this purpose, cells grown in the presence or absence of DOX - i.e. without or with expression of MT1-MMP, respectively - were treated with human recombinant FGF-2 (0.75 ng/ml) for 15 min, and ERK1/2 activation was analyzed by Western blotting with antibody to phosphorylated ERK1/2. As shown in Fig. 1 A and B, neither DOX nor FGF-2 affected ERK2 expression significantly; however, ERK1/2 phosphorylation by FGF-2 in MT1-MMP expressing cells was over three-fold lower than in cells with no MT1-MMP expression. Although this effect could be seen with FGF-2 concentrations ranging from 0.5 ng/ml to 30 ng/ml, it was most evident with a concentration of 0.75 ng/ml, and 15 min of incubation (Fig. 1 D and E). Therefore, we used these experimental conditions for subsequent experiments.
To investigate whether the effect of MT1-MMP on FGF-2 signaling requires the proteolytic activity of MT1-MMP or is mediated by a non-proteolytic mechanism, we transiently transfected MCF-7 cells with wt MT1-MMP, or with mutant MT1-MMP devoid of the hemopexin-like (PEX) or cytoplasmic domain (Δpex and Δcyt, respectively), or with a mutant devoid of proteolytic activity (E240A) (D’Alessio, et al., 2008). Cells transfected with the empty vector were used as controls. The transfected cells were treated with FGF-2, and ERK1/2 activation was analyzed by Western blotting (Fig. 2). As expected, cells transfected with the control empty vector, which expressed no MT1-MMP, showed a level of ERK1/2 activation in response to FGF-2 higher than cells transfected with wt MT1-MMP, and similar to cells expressing the proteolytically inactive MT1-MMP E240A mutant. Cells expressing Δpex or Δcyt MT1-MMP, which are both proteolytically active, showed levels of ERK1/2 activation similar to that of cells expressing wt MT1-MMP (Fig. 2 A) and lower than cells expressing catalytically inactive or no MT1-MMP. Consistent with these results, MT1-MMP-expressing cells pretreated with Ilomastat, an MMP inhibitor, showed a level of ERK1/2 activation in response to FGF-2 higher than cells treated with FGF-2 in the absence of Ilomastat (Fig. 2 B). Therefore, these results showed that the inhibitory effect of MT1-MMP on FGF-2 signaling requires the proteolytic activity of MT1-MMP.
Figure 2. The proteolytic activity of MT1-MMP is required for downregulation of ERK1/2 activation by FGF-2.

Western blotting analysis of ERK1/2 activation (p-ERK1/2) and MT1-MMP expression. A. MCF-7 cells transiently transfected with wt or mutant MT1-MMP cDNAs were grown for 24 h in medium containing 0.5% FBS and treated with FGF-2 (0.75 ng/ml) for 15 min. -, empty vector; wt, wild-type MT1-MMP; Δpex, Δcyt, MT1-MMP devoid of the hemopexin or the cytoplasmic domain, respectively; E240A, proteolytically inactive MT1-MMP mutant with ala substitution of glu 240 in the catalytic domain. Bottom panel: gelatin zymography. To characterize the proteolytic activity of the various forms of MT1-MMP the transfectants were analyzed for their capacity to activate proMMP-2 as described in Materials and Methods. B. MT1-MMP Tet-Off MCF-7 cell transfectants grown for 24 h in medium containing 0.5% FBS were treated with FGF-2 (0.75 ng/ml) for 15 min in the presence or absence of Ilomastat (50 μM), an MMP inhibitor. Tubulin (TUB) is shown as a loading control. The lower panels of A and B show mean ± S.E. of densitometric readings normalized to the corresponding loading control; *, p ≤ 0.05; control and E240A transfectants + FGF-2 vs. wt, Δpex and Δcyt transfectants + FGF-2. This experiment was repeated three times with similar results.
To investigate whether MT1-MMP inhibition of FGF-2 signaling is a unique feature of our MCF-7 cell transfectants, we characterized FGF-2 induction of ERK1/2 activation in a panel of human pediatric glioma cell lines. As shown in Fig. 3 A and B, these cells constitutively express diverse levels of MT1-MMP, as well as both low-molecular weight (18 kDa) and high-molecular weight FGF-2 (Florkiewicz and Sommer, 1989, Florkiewicz, et al., 1991, Prats, et al., 1989) (22-24 kDa; Fig. 3). By RT-PCR all these cell lines express FGFR-1; one cell line also expressing low levels of FGFR-2 and FGFR-3, and two cell lines very low FGFR-4 levels. Because of the relatively high levels of endogenous FGF-2 of some of these cells, we used a higher concentration of FGF-2 than in the previous experiments. Treatment of the cells with 10 ng/ml of FGF-2 induced strong activation of ERK1/2 in the three cell lines devoid of MT1-MMP (Fig. 3; SF188, KNS42 and Res259). In contrast, FGF-2 induced no or very low ERK1/2 activation in the two cell lines that express MT1-MMP (UW479 and Res186; the former cells express very low amounts of FGFR-1; however, the latter cells show a level of FGFR-1 expression comparable to the other cell lines).
Figure 3. Constitutive MT1-MMP expression inversely correlates with ERK1/2 activation by FGF-2 in pediatric glioma cells.
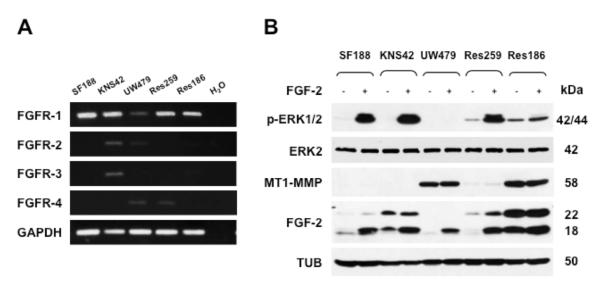
A. RT-PCR analysis of FGFR expression. H2O and GAPDH are shown as negative and loading controls, respectively. B. Western blotting analysis of ERK1/2 activation, MT1-MMP and FGF-2 expression in protein extracts of pediatric glioma cells grown for 24 h in medium containing 0.5% FBS and treated with FGF-2 (10 ng/ml for 15 min). The cells express both 18 kDa and 22 kDa FGF-2 (low- and high-molecular weight FGF-2). The molecular weight of recombinant FGF-2 is 18 kDa. Tubulin (TUB) and ERK2 are shown as loading controls. This experiment was repeated twice with comparable results.
To validate these results we characterized FGF-2 activation of ERK1/2 in cells in which the endogenous expression of MT1-MMP was downregulated by siRNA-mediated gene silencing. For this purpose Res186 glioma cells, which express endogenous MT1-MMP (Fig. 3), were transfected with MT1-MMP siRNA or control scrambled siRNA. Forty-eight hours later the cells were treated with FGF-2 (30 ng/ml) for 15 min, and ERK1/2 activation was analyzed by Western blotting. MT1-MMP expression was reduced by approximately 70% in cells transfected with MT1-MMP siRNA relative to the control siRNA transfectants (Fig. 4). Consistent with our previous result (Fig. 3 B) FGF-2 did not upregulate ERK1/2 activation in cells transfected with the control siRNA. Conversely, FGF-2 induced a 2-fold increase in the level of phosphorylated ERK1/2 in MT1-MMP siRNA transfected cells. Thus, these results showed that MT1-MMP expression downregulates FGF-2 activation of ERK1/2.
Figure 4. Silencing of MT1-MMP expression correlates with ERK1/2 activation by FGF-2 in Res186 pilocytic astrocytoma cells.
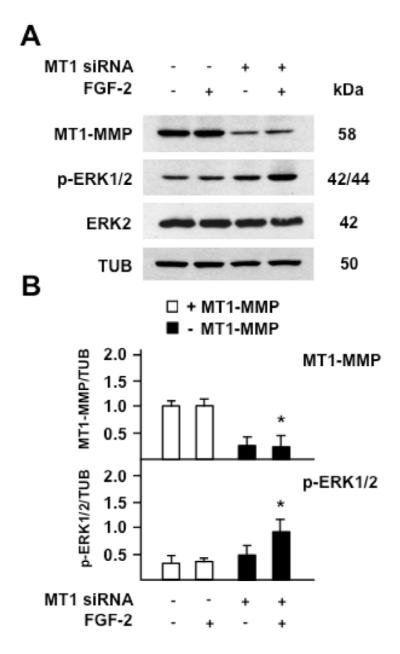
Western blotting analysis of MT1-MMP expression and ERK1/2 activation (p-ERK1/2). A. Res186 cells transiently transfected with the MT1-MMP siRNAs pool or control scrambled siRNAs were grown for 24 h in medium containing 0.5% FBS and incubated with FGF-2 (30 ng/ml) for 15 min. Tubulin (TUB) and ERK2 are shown as loading control. B. Mean ± S.E. of densitometric readings normalized to the corresponding loading control; *, p ≤ 0.05; MT1-MMP siRNA transfectants + FGF-2 vs. control siRNA transfectants + FGF-2. This experiment was repeated twice with similar results.
MT1-MMP does not degrade FGF-2 or FGFR-2
Our finding that the proteolytic activity of MT1-MMP decreases FGF-2 signaling generated the hypotheses that MT1-MMP degrades either FGF-2 or its tyrosine kinase receptor(s), or the ECM- and cell membrane-associated HSPG that serve as low-affinity binding sites for FGF-2.
To investigate the potential cleavage of FGF-2 by MT1-MMP we incubated our Tet-Off MT1-MMP transfectants with FGF-2 and analyzed it in cell extracts and conditioned medium. Because the FGF-2 concentration we used in our previous experiments (0.75 ng/ml) did not allow Western blotting detection of FGF-2 in the cell extracts or conditioned medium, in this experiment we treated cells with 30 ng/ml of FGF-2. This FGF-2 concentration induced ERK1/2 activation more rapidly than 0.75 ng/ml (Fig. 5). However, consistent with our previous results, the level of ERK1/2 activation was 2- to 3-fold higher in cells devoid of MT1-MMP than in MT1-MMP-expressing cells. Western blotting analysis of FGF-2 in the cell extracts showed one band with the expected Mr of human recombinant FGF-2 (18,000). The intensity of this band was 2- to 3-fold lower in MT1-MMP-expressing cells than in cells devoid of MT1-MMP, suggesting that a lower amount of FGF-2 bound to MT1-MMP-expressing cells (Fig. 5). This effect could result from proteolytic degradation of either FGF-2 or its cell membrane or ECM receptors. The analysis of FGF-2 in the conditioned medium showed one band with the expected Mr, whose intensity was comparable in the conditioned medium of cells with or without MT1-MMP. No lower-Mr bands consistent with potential degradation products of FGF-2 were detected, indicating that MT1-MMP does not degrade FGF-2. To confirm this indication, extracts of Tet-Off MT1-MMP transfectants grown in the presence or absence of DOX were incubated overnight with FGF-2 and analyzed by Western blotting with FGF-2 antibody. The results (Fig. 6 A) showed one band consistent with the Mr of human recombinant FGF-2 and no degradation products. It is possible that potential degradation products of FGF-2 are short-lived, or were not recognized by our antibody. However, the size and intensity of the Mr 18,000 FGF-2 band were comparable in samples incubated with extracts of cells expressing, or devoid of MT1-MMP. Therefore, these results showed that MT1-MMP does not degrade FGF-2.
Figure 6. MT1-MMP does not cleave FGF-2, FGFR-2 or ADAM-9 in MCF-7 cells.

A. MT1-MMP Tet-Off MCF-7 cells were grown for 24 h in the presence or absence of DOX (1 μg/ml) in medium containing 0.5% FBS. The cells were lysed and 100 μg of cell extract protein was incubated with FGF-2 (1 ng) in a total volume of 35 μl for 18 h at 37° C with gentle rotation. As a control for proteolysis-independent degradation of FGF-2 1 ng of FGF-2 was loaded onto the gel without (leftmost lane) or with incubation with lysis buffer alone for 18 h at 37° C (lane 4 from left). As a control for potential endogenous FGF-2, cell extracts were incubated without addition of FGF-2 (rightmost two lanes). At the end of the incubation the samples were analyzed by Western blotting with antibodies to FGF-2 and MT1-MMP. Tubulin (TUB) is shown as a loading control. This experiment was repeated twice with comparable results. B. MT1-MMP Tet-Off MCF-7 cells grown for 24 h in the presence or absence of DOX (1 μg/ml) in medium containing 0.5% FBS were incubated with FGF-2 (1 ng/ml for 15 min). Cell extracts were analyzed by Western blotting with antibodies to FGFR-2, ADAM-9, phospho-ERK1/2 and MT1-MMP. Tubulin (TUB) is shown as a loading control. C. Densitometric analysis of FGF-2 after incubation with cell extracts as described for panel A, and of FGFR-2, ADAM-9 and ERK1/2 activation as described for panel B. The top panel shows mean ± S.E. of densitometric readings normalized to the reading of the FGF-2 control in lane 4 of panel A, made equal to 1. The other histograms show mean ± S.E. of densitometric readings normalized to the corresponding loading control; *, p ≤ 0.05; - MT1-MMP + FGF-2 sample vs. + MT1-MMP + FGF-2 sample. These experiments were repeated over five times with similar results.
Previous studies have shown that in calvarial osteoblasts MT1-MMP upregulates FGF signaling indirectly by cleaving ADAM-9, which in turn sheds FGFR-2 from the cell membrane (Chan, et al., 2012). Therefore, we characterized the effect of MT1-MMP on FGFR-2 and ADAM-9 cleavage in our Tet-Off MT1-MMP transfectants. For this purpose cells grown in the presence or absence of DOX were analyzed by Western blotting with antibodies to ADAM-9 and FGFR-2 (Fig. 6 B).
The analysis of FGFR-2 showed bands with the expected Mr of FGFR-2 (Chan, et al., 2012). The size and intensity of these bands were comparable in cells expressing or non-expressing MT1-MMP, and in the presence or absence of FGF-2. In addition, no lower-Mr bands were detected in cell extracts (Fig. 6 B) or conditioned media (not shown). Similarly, Western blotting with antibody to ADAM-9 showed one band with Mr 84,000, consistent with that of the native proteinase (Chan, et al., 2012). The intensity of this band was similar in extracts of MT1-MMP-expressing cells and of cells devoid of MT1-MMP. Lower-Mr bands with Mrs ranging ~ 40,000 and 50,000 – 55,000 were also detected. The Mrs of these bands are comparable to those of ADAM-9 degradation products generated by MT1-MMP (Chan, et al., 2012); however, their size or intensity was similar in samples with and without MT1-MMP. Moreover, no immunoreactive bands were detected in the conditioned media (not shown) as would be expected for MT1-MMP-generated degradation products of ADAM-9 (Chan, et al., 2012). Therefore, these results showed that - unlike in calvarial osteoblasts - in MCF-7 cells MT1-MMP does not cleave ADAM-9. However, consistent with our previous findings, also in this experiment ERK1/2 activation by FGF-2 was lower in cells expressing MT1-MMP than in cells devoid of this proteinase (Fig. 6 B and C). Thus, these results showed that MT1-MMP downregulation of FGF-2 signaling is not mediated by cleavage of FGFR-2.
MT1-MMP expression downregulates FGFR-1 and FGFR-4 and decreases low-affinity binding of FGF-2 to the cell surface
Because our Tet-Off MT1-MMP transfectants also express FGFR-1 and FGFR-4 we characterized the levels of these receptors in cells grown in the presence or absence of DOX. Western blotting analysis (Fig. 7) showed that cells that expressed MT1-MMP (in the absence of DOX) had levels of both FGFR-1 and FGFR-4 lower than cells devoid of MT1-MMP. For both receptors we could not detect immunoreactive bands compatible with degradation products generated by MT1-MMP. Bands with Mrs ~ 40,000 – 80,000 were detected; however, their size or intensity was similar in samples with and without MT1-MMP, suggesting that they represent non-specific bands. As in the case of FGF-2, it is possible that FGFR-1 and -4 degradation products are short-lived or the antibodies we used did not recognize them. However, our previous finding that the proteolytic activity of MT1-MMP is required for downregulating FGF-2 activation of ERK1/2 indicates that the reduced intensity of the Western blot bands derived from MT1-MMP-mediated degradation of the FGFRs. Therefore, these results indicated that MT1-MMP downregulates FGF-2 binding to the cell surface by degrading FGFR-1 and FGFR-4.
Figure 7. MT1-MMP expression inversely correlates with FGFR-1 and FGFR-4 levels.

Western blotting analysis of FGFR-1 and FGFR-4. A. MT1-MMP Tet-Off MCF-7 cells grown for 24 h in the presence or absence of DOX (1 μg/ml) in medium containing 0.5% FBS were treated with 0.75 ng/ml of FGF-2 for 15 min. Tubulin (TUB) is shown as a loading control. B. Mean ± S.E. of densitometric readings normalized to the corresponding loading control; *, p ≤ 0.05; - MT1-MMP + FGF-2 vs. the corresponding + MT1-MMP + FGF-2 sample. This experiment was repeated twice with comparable results.
In addition to its specific tyrosine kinase receptors (FGFRs) FGF-2 binds with lower affinity to the HS and HSPG on the cell surface and in the ECM. These low-affinity, high-capacity binding sites act as co-receptors and provide a reservoir of extracellular FGF-2 that can be readily mobilized in the form of FGF-2•HS complex by proteolytic cleavage of the HSPG. Formation of this complex is required for FGF-2 interaction with FGFRs and activation of intracellular signaling (Plotnikov, et al., 1999, Schlessinger, et al., 2000). Based on these considerations we hypothesized that MT1-MMP cleavage of HSPG results in decreased FGF-2 binding to the cell surface and/or ECM. Therefore, we added FGF-2 (30 ng/ml) to the culture medium of MT1-MMP-expressing or non-expressing cells, and characterized the amount of cell-bound FGF-2 by biotinylation of cell surface proteins. The results (Fig. 8 A) showed that the amount of FGF-2 bound to the surface of MT1-MMP expressing cells was dramatically lower than in cells devoid of MT1-MMP.
Figure 8. MT1-MMP reduces low-affinity binding of FGF-2 to the cell surface.

A. MT1-MMP Tet-Off MCF-7 cells grown for 24 h in the presence or absence of DOX (1 μg/ml) in medium containing 0.5% FBS were incubated with FGF-2 (30 ng/ml) for 15 min. At the end of the incubation cell surface-associated proteins were biotinylated as described in Materials and Methods. Lysates of biotinylated cells were immunoprecipitated with anti-FGF-2 antibody, and the immunocomplexes detected by Western blotting with HR-streptavidin or anti-FGF-2 antibody. Five nanograms of biotinylated human recombinant FGF-2 (Biot-FGF-2) was also analyzed as a positive control for streptavidin binding to biotin. This experiment was repeated three times with comparable results. B. MT1-MMP Tet-Off MCF-7 cells grown for 24 h in the presence or absence of DOX (1 μg/ml) in medium containing 0.5% FBS were incubated with FGF-2 (30 ng/ml for 15 min). At the end of the incubation the cells were washed 3 times with PBS followed by one washing with 2 M NaCl in 20 mM HEPES, pH 7.5 (Washing buffer: 2M NaCl, pH 7.5) or one washing with PBS as a control (Washing buffer: PBS) as described in Materials and Methods. The washings were pooled, and FGF-2 was characterized in cell extracts, washing buffers and conditioned medium by Western blotting. The levels of FGF-2 in the medium show that the same amount of the growth factor was added to all the samples. Tubulin (TUB) is shown as a loading control. The lower panel of A shows mean ± S.E. of densitometric readings normalized to the reading of the streptavidin MT1-MMP+ FGF-2+ control (top panel), made equal to 1. The lower panel of B shows mean ± S.E. of densitometric readings normalized to the corresponding loading control. Each bar represents the corresponding sample in the upper panel. *, p ≤ 0.05; - MT1-MMP sample vs. the corresponding + MT1-MMP sample. These experiments were repeated three times with comparable results.
To investigate the nature of the FGF-2 binding to the cells we eluted FGF-2 from the cell surface using buffers with different ionic strength. For this purpose FGF-2 was added to the culture medium of cells expressing or not expressing MT1-MMP, and after 15 min incubation at 37° C (a time sufficient for ERK1/2 activation) the cultures were washed with either PBS alone or with PBS followed by 2 M NaCl in 20 mM HEPES, pH 7.5. This NaCl concentration detaches FGF-2 bound to HS or HSPG but does not elute FGF-2 bound to the high-affinity FGFRs (Moscatelli, 1987). FGF-2 was finally characterized in both cell extracts and washing buffers by Western blotting (Fig. 8 B). Consistent with our previous results, when the cells were washed with PBS alone the amount of FGF-2 remaining associated with MT1-MMP expressing cells was slightly lower than in cells without MT1-MMP. However, the amount of FGF-2 eluted with the 2 M NaCl buffer was dramatically higher in cells devoid of MT1-MMP than in MT1-MMP-expressing cells. Washing with this buffer removed most of the FGF-2 and the small amount remaining associated with the cell extracts was 5- to 6-fold higher in cells devoid of MT1-MMP than in MT1-MMP expressing cells (Fig. 8 B). Therefore, these results showed that MT1-MMP reduces low-affinity binding of FGF-2 to the cell surface. Because FGF-2 binding to low-affinity binding sites is required for FGFR activation, downregulation of low-affinity binding results in decreased activation of intracellular signaling. Thus, MT1-MMP downregulates FGF-2 signaling by decreasing its binding to both high- and low-affinity cell surface receptors.
MT1-MMP downregulates cell migration and invasion in response to FGF-2
To investigate the biological effects of MT1-MMP-mediated inhibition of FGF-2 signaling we characterized cell migration in cells with or without expression of MT1-MMP. For this purpose confluent monolayers of MT1-MMP Tet-Off transfectants grown in the presence or absence of DOX were used in a “scratch assay” for cell migration (Liang, et al., 2007) with or without addition of FGF-2 (10 ng/ml) to the culture medium. In the absence of FGF-2 wound closure was slow and progressed at a similar rate in cells expressing or not expressing MT1-MMP. However, in the presence of FGF-2 wound closure occurred more rapidly in cells devoid of MT1-MMP than in cells expressing MT1-MMP (Fig. 9 A and B). MT1-MMP Tet-Off transfectants were also analyzed for their ability to invade matrigel in the presence or absence of DOX. Similarly to the scratch assay (Fig. 9 C), FGF-2 upregulated cell invasion more strongly in cells devoid of than in cells expressing MT1-MMP.
Figure 9. MT1-MMP downregulates cell migration and invasion in response to FGF-2. A. Migration assay.

Confluent monolayers of MT1-MMP Tet-Off MCF-7 cells grown for 24 h in the presence or absence of DOX (1 μg/ml) in medium containing 0.5% FBS were scratched as described in Materials and Methods. After photographing the cells (time 0), incubation was continued in the same media with or without addition of FGF-2 (10 ng/ml). The cultures were observed twice daily, and photographed after 42 h incubation, when wound closure was first observed. Original magnification: 50X. B. Quantitative analysis of the wounds shown in panel A. Wound areas were measured using Adobe Photoshop on photographs of multiple areas taken at 0 and 42 h. The graph shows mean ± S.D. of measurements of 4 to 6 areas per sample. *, p ≤ 0.05; - MT1-MMP + FGF-2 sample vs. + MT1-MMP + FGF-2 sample, both at 42 h. This experiment was repeated twice with similar results. C. Matrigel invasion assay. MT1-MMP Tet-Off MCF-7 cells grown for 24 h in serum-free medium in the presence or absence of DOX (1 μg/ml) were assayed for Matrigel invasion as described in Materials and Methods. *, p ≤ 0.05; - MT1-MMP + FGF-2 sample vs. + MT1-MMP + FGF-2 sample. This experiment was repeated twice with similar results. D. Migration assay. Res186 cells transiently transfected with the MT1-MMP siRNAs pool or control scrambled siRNAs were grown for 24 h in medium containing 0.5% FBS and scratched as described in Materials and Methods. After photographing the cells (time 0), incubation was continued in the same media with or without addition of FGF-2 (10 ng/ml). The cultures were observed twice daily, and photographed after 69 h incubation. Original magnification: 50X. E. Quantitative analysis of the wounds shown in panel, performed as described in Materials and Methods. This experiment was repeated twice with similar results.
To validate these results we characterized the migration of cells in which the endogenous expression of MT1-MMP was downregulated by gene silencing (Fig. 9 D and E). For this purpose we used the Res186 glioma cells and MT1-MMP siRNA we used in our previous experiments (Figs. 3 and 4). With these cells FGF-2 did not significantly upregulate cell migration in scratch assays, consistent with our previous finding that Res186 cells have a high level of endogenous FGF-2 and exogenous FGF-2 does not upregulate ERK1/2 activation (Fig. 3). However, downregulation of MT1-MMP expression enhanced cell migration by approximately 80% both in the absence and in the presence of FGF-2. Thus, although MT1-MMP plays an important role in cell migration, it also downregulates the cell’s migratory and invasive response to FGF-2.
DISCUSSION
In addition to degrading the ECM, MT1-MMP controls a variety of cell functions through direct or indirect mechanisms mediated by proteolytic and non-proteolytic interactions with extracellular, intracellular and transmembrane proteins. Here we provide evidence that in tumor cells MT1-MMP downregulates FGF-2 binding to the cell surface and activation of intracellular signaling by a proteolytic mechanism, and decreases cell migration and invasion in response to FGF-2. These conclusions are based on the following observations.
Addition of FGF-2 to the culture medium induces stronger activation of ERK1/2 in cells devoid of MT1-MMP than in cells that express MT1-MMP. This effect is abolished in cells that express proteolytically inactive MT1-MMP but persists in cells expressing proteolytically active MT1-MMP mutants devoid of PEX or cytoplasmic domain. MT1-MMP expression also results in decreased amounts of cell surface-associated FGF-2 which can be eluted with a buffer that detaches FGF-2 from low-affinity binding sites (Moscatelli, 1987). In addition, FGF-2 upregulates cell migration and invasion in vitro more strongly in cells devoid of MT1-MMP than in cells that express MT1-MMP.
In contrast with our findings, previous studies have associated MT1-MMP expression with increased FGF-2 signaling. Onguchi et al. (Onguchi, et al., 2009) reported that MT1-MMP potentiates FGF-2-induced corneal neovascularization by upregulating FGF-2 signaling. Accordingly, FGF-2 stimulation of immortalized corneal fibroblasts from MT1-MMP knockout mice results in lower phosphorylation levels of the MAP kinases p38 and JNK than in the corresponding wt cells; however, MT1-MMP does not affect ERK1/2 activation (Onguchi, et al., 2009). MT1-MMP also upregulates FGF-2 signaling in calvarial osteoblasts. In these cells, MT1-MMP forms a complex with FGFR-2 and ADAM-9, and proteolytically inactivates ADAM-9, thus protecting FGFR-2 from ADAM-9-mediated cleavage. Although our MCF-7 cells express ADAM-9, we found no evidence of its cleavage by MT1-MMP. Similarly, we found no evidence of FGFR-2 degradation in the presence or absence of MT1-MMP. These discrepancies can be explained by two non-mutually exclusive hypotheses. As MT1-MMP, ADAM-9 and FGFR-2 are transmembrane proteins, proteinase-substrate interactions can only occur if they colocalize to specific regions of the cell membrane. It is possible that the colocalization of MT1-MMP, ADAM-9 and FGFR-2 on the plasma membrane varies in different cell types or between normal and tumor cells. In addition, all FGFRs occur in multiple splice variants, which may result in the presence or absence of ADAM-9 cleavage sites. Unlike calvarial osteoblasts, our MCF-7 and glioma cells may express an ADAM-9-resistant variant of FGFR-2.
Although we found that MT1-MMP does not cleave FGFR-2, our data indicate that MT1-MMP degrades FGFR-1 and FGFR-4, the other FGFRs expressed by our MCF-7 cells. In addition, we found that MT1-MMP expression strongly reduces the amount of FGF-2 bound to the cell surface with low-affinity. Therefore, the amount of FGF-2 bound to both high- and low-affinity receptors is dramatically lower in the presence than in the absence of MT1-MMP.
Several considerations show that downregulation of low-affinity binding of FGF-2 to the cell surface and ECM is an effective mechanism through which MT1-MMP controls FGF-2 signaling. Low-affinity binding sites for FGF-2, comprised of HS and HSPG in the ECM and on the cell membrane, are fundamental for FGF-2 activation of intracellular signaling. Cell-associated HSPG are required for the formation of HSPG•FGF•FGFR ternary complexes necessary for FGFR activation (Plotnikov, et al., 1999, Schlessinger, et al., 2000). In fact, there is no detectable interaction between FGF-2 and its high-affinity receptor in the absence of heparin (Ornitz, et al., 1992). In addition, HSPG promote FGF-2 internalization (Hsia, et al., 2003, Rusnati, et al., 1993), and some HSPG such as syndecan-4 activate FGF-2 signaling independently of interaction with FGFRs (Simons and Horowitz, 2001). Therefore, even in the presence of functional FGFRs, downregulation of FGF-2 binding to low-affinity receptors results in decreased activation of the high-affinity FGFRs.
The low-affinity binding sites for FGF-2 act as a high-capacity reservoir for this growth factor, which is highly concentrated in insoluble phase on the cell surface and ECM, and provide prolonged stimulation to the cell. ECM-degrading proteinases including plasminogen activators/plasmin and MMPs have been implicated in the mobilization of FGF-2 from the insoluble phase necessary for FGFR activation. Thus, the current consensus is that ECM-degrading proteinases promote FGF-2 activity. In contrast, our data show that MT1-MMP has an opposite effect as it reduces low-affinity binding of FGF-2 and activation of intracellular signaling.
We did not identify the FGF-2 low-affinity binding site(s) degraded by MT1-MMP. A variety of molecules bind FGF-2; they are heterogeneous in nature and the identification of the one(s) degraded by MT1-MMP is beyond the scope of the present study. An FGF-2-binding HSPG known to be cleaved by MT1-MMP is syndecan-1 (Endo, et al., 2003, Su, et al., 2008). Therefore, we characterized syndecan-1 levels in our MCF-7 Tet-Off transfectants with or without MT1-MMP. Both flow cytometry and immunofluorescence analyses showed no difference in syndecan-1 levels between MT1-MMP-expressing and non-expressing cells (Supplemental Figs. 1 and 2). These results are at variance with published data obtained with different cells (human T47D breast carcinoma and HT1080 fibrosarcoma cells, and HEK293T embryonic kidney cells). Two hypotheses can explain this discrepancy. All these cell lines express varying levels of MMP-2, whereas our MCF-7 cells do not (D’Alessio, et al., 2008, Rozanov, et al., 2001). It is possible that MT1-MMP cleaves syndecan-1 indirectly through MMP-2, which is not produced by our MCF-7 cells. It should also be noted that syndecan-1 cleavage by MT1-MMP was detected in cells transfected with (i. e. overexpressing) syndecan-1, whereas we analyzed endogenous syndecan-1. It is possible that overexpression facilitates syndecan-1 interaction with MT1-MMP.
Another cell surface binding site for FGF-2 is integrin αvß3 (Rusnati, et al., 1997, Tanghetti, et al., 2002). FGF-2 binds to αvβ3 integrin, and forms a trimolecular complex with FGFR-1 necessary for ERK1/2 activation. However, MT1-MMP cleavage results in activation – not degradation – of αvβ3 integrin (Deryugina, et al., 2002). Therefore, we rule out the hypothesis that the effect of MT1-MMP on FGF-2 binding to the cell surface is mediated by αvβ3 cleavage.
FGF signaling is mediated by three distinct but interactive signaling cascades, the MAP kinase, AKT and PLCγ pathways, which control diverse cell responses (Goetz and Mohammadi, 2013). The RAS - MAP kinase cascade primarily controls cell proliferation but can also modulate cell differentiation, migration and other cellular functions. Activation of the PI3 kinase – AKT pathway mediates pro-survival signaling through inactivation of pro-apoptotic effectors such as the BCL-2 antagonist of cell death (BAD). Phosphorylation of phospholipase C gamma (PLCγ1) by FGFR kinase results in the hydrolysis of the membrane phospholipid phosphatidylinositol-4,5-bisphosphate (PtdIns(4,5)(P2) into diacylglycerol (DAG) and inositol-1,4,5,-trisphosphate (IP3). DAG activates protein kinase C (PKC), which phosphorylates a variety of proteins that control cell motility. IP3 induces release of calcium ions from intracellular stores, which results in the activation of a variety of calcium-dependent proteins that also control several cell functions including cell motility (Goetz and Mohammadi, 2013). We only characterized the effect of MT1-MMP on FGF-2 activation of the MAPK ERK1/2 signaling pathway. However, because MT1-MMP acts at the level of FGF – FGFR interaction - at the top of all the FGF signaling cascades – it is most likely that the mechanism we described downregulates all the signaling pathways activated by FGF-2.
Therefore, MT1-MMP mediated downregulation of FGF-2 signaling can have significant effects on a variety of cell functions. Our data show that, whereas MT1-MMP promotes cell migration (Wang and McNiven, 2012), it also inhibits the promigratory action of FGF-2. This effect of MT1-MMP may represent a physiological mechanism for the control of cell migration and other cell functions modulated by FGF-2. FGF-2 is a potent inducer of angiogenesis, a process in which MT1-MMP plays a fundamental role (Chun, et al., 2004). MT1-MMP mediated downregulation of FGF-2 signaling can provide a mechanism for the control of endothelial functions important for angiogenesis. High levels of FGF-2 and MT1-MMP are also expressed in a variety of malignant tumors, and expression of these proteins has been associated with adverse prognosis (Katayama, et al., 2004, Kwabi-Addo, et al., 2004, Matsuda, et al., 2012, Metzner, et al., 2011, Perentes, et al., 2011). The crosstalk between MT1-MMP and FGF-2 in tumors can modulate endothelial cell functions involved in tumor angiogenesis. Importantly, because all members of the FGF family have affinity for heparin, although to variable extents, the mechanism we described can also control the activity of other FGFs, which mediate a variety of physiological and pathological processes.
Supplementary Material
Supplemental Figure 1. Immunofluorescence analysis of syndecan-1 levels in MCF-7 cells with or without MT1-MMP. MT1-MMP Tet-Off MCF-7 cells grown for 24 h in the presence or absence of DOX (1 μg/ml) were analyzed by immunofluorescence as described in Materials and Methods. This experiment was repeated three times with comparable results.
Supplemental Figure 2. Flow cytofluorometric analysis of syndecan-1 levels in MCF-7 cells with or without MT1-MMP. MT1-MMP Tet-Off MCF-7 cells grown for 24 h in the presence or absence of DOX (1 μg/ml) were analyzed by flow cytometry as described in Materials and Methods. This experiment was repeated three times with comparable results.
ACKNOWLEDGMENTS
We are grateful to Drs. D. Moscatelli, M. Cioce and C. Canino (NYU School of Medicine) for their advice and critical reading of the manuscript, C. Jones (The Institute of Cancer Research, Sutton, UK) for the generous gift of the pediatric glioma cell lines, and J. Cao (Stony Brook School of Medicine) for the plasmid encoding the MT1-MMP mutant devoid of PEX domain.
Contract grant sponsor: NIH; Contract grant number: R01 CA136715 and R21 AG033735
Footnotes
The authors declare no conflict of interest.
REFERENCES
- Atkinson JJ, Holmbeck K, Yamada S, Birkedal-Hansen H, Parks WC, Senior RM. Membrane-type 1 matrix metalloproteinase is required for normal alveolar development. Dev Dyn. 2005;232:1079–1090. doi: 10.1002/dvdy.20267. [DOI] [PubMed] [Google Scholar]
- Bax DA, Little SE, Gaspar N, Perryman L, Marshall L, Viana-Pereira M, Jones TA, Williams RD, Grigoriadis A, Vassal G, Workman P, Sheer D, Reis RM, Pearson AD, Hargrave D, Jones C. Molecular and phenotypic characterisation of paediatric glioma cell lines as models for preclinical drug development. PLoS One. 2009;4:e5209. doi: 10.1371/journal.pone.0005209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beenken A, Mohammadi M. The FGF family: biology, pathophysiology and therapy. Nat Rev Drug Discov. 2009;8:235–253. doi: 10.1038/nrd2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks AN, Kilgour E, Smith PD. Molecular pathways: fibroblast growth factor signaling: a new therapeutic opportunity in cancer. Clin Cancer Res. 2012;18:1855–1862. doi: 10.1158/1078-0432.CCR-11-0699. [DOI] [PubMed] [Google Scholar]
- Chan KM, Wong HL, Jin G, Liu B, Cao R, Cao Y, Lehti K, Tryggvason K, Zhou Z. MT1-MMP inactivates ADAM9 to regulate FGFR2 signaling and calvarial osteogenesis. Dev Cell. 2012;22:1176–90. doi: 10.1016/j.devcel.2012.04.014. [DOI] [PubMed] [Google Scholar]
- Chun TH, Hotary KB, Sabeh F, Saltiel AR, Allen ED, Weiss SJ. A pericellular collagenase directs the 3-dimensional development of white adipose tissue. Cell. 2006;125:577–591. doi: 10.1016/j.cell.2006.02.050. [DOI] [PubMed] [Google Scholar]
- Chun TH, Sabeh F, Ota I, Murphy H, McDonagh KT, Holmbeck K, Birkedal-Hansen H, Allen ED, Weiss SJ. MT1-MMP-dependent neovessel formation within the confines of the three-dimensional extracellular matrix. J Cell Biol. 2004;167:757–767. doi: 10.1083/jcb.200405001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Alessio S, Ferrari G, Cinnante K, Scheerer W, Galloway AC, Roses DF, Rozanov DV, Remacle AG, Oh ES, Shiryaev SA, Strongin AY, Pintucci G, Mignatti P. Tissue inhibitor of metalloproteinases-2 binding to membrane-type 1 matrix metalloproteinase induces MAPK activation and cell growth by a non-proteolytic mechanism. J Biol Chem. 2008;283:87–99. doi: 10.1074/jbc.M705492200. [DOI] [PubMed] [Google Scholar]
- Deryugina EI, Quigley JP. Matrix metalloproteinases and tumor metastasis. Cancer metastasis reviews. 2006;25:9–34. doi: 10.1007/s10555-006-7886-9. [DOI] [PubMed] [Google Scholar]
- Deryugina EI, Ratnikov BI, Postnova TI, Rozanov DV, Strongin AY. Processing of integrin alpha(v) subunit by membrane type 1 matrix metalloproteinase stimulates migration of breast carcinoma cells on vitronectin and enhances tyrosine phosphorylation of focal adhesion kinase. J Biol Chem. 2002;277:9749–9756. doi: 10.1074/jbc.M110269200. [DOI] [PubMed] [Google Scholar]
- Endo K, Takino T, Miyamori H, Kinsen H, Yoshizaki T, Furukawa M, Sato H. Cleavage of syndecan-1 by membrane type matrix metalloproteinase-1 stimulates cell migration. J Biol Chem. 2003;278:40764–40770. doi: 10.1074/jbc.M306736200. [DOI] [PubMed] [Google Scholar]
- Flaumenhaft R, Moscatelli D, Saksela O, Rifkin DB. Role of extracellular matrix in the action of basic fibroblast growth factor: matrix as a source of growth factor for long-term stimulation of plasminogen activator production and DNA synthesis. J Cell Physiol. 1989;140:75–81. doi: 10.1002/jcp.1041400110. [DOI] [PubMed] [Google Scholar]
- Flaumenhaft R, Abe M, Mignatti P, Rifkin DB. Basic fibroblast growth factor-induced activation of latent transforming growth factor beta in endothelial cells: regulation of plasminogen activator activity. J Cell Biol. 1992;118:901–909. doi: 10.1083/jcb.118.4.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florkiewicz RZ, Sommer A. Human basic fibroblast growth factor gene encodes four polypeptides: three initiate translation from non-AUG codons. Proc Natl Acad Sci U S A. 1989;86:3978–3981. doi: 10.1073/pnas.86.11.3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florkiewicz RZ, Baird A, Gonzalez AM. Multiple forms of bFGF: differential nuclear and cell surface localization. Growth Factors. 1991;4:265–275. doi: 10.3109/08977199109043912. [DOI] [PubMed] [Google Scholar]
- Galvez BG, Genis L, Matias-Roman S, Oblander SA, Tryggvason K, Apte SS, Arroyo AG. Membrane type 1-matrix metalloproteinase is regulated by chemokines monocytechemoattractant protein-1/ccl2 and interleukin-8/CXCL8 in endothelial cells during angiogenesis. J Biol Chem. 2005;280:1292–1298. doi: 10.1074/jbc.M408673200. [DOI] [PubMed] [Google Scholar]
- Goetz R, Mohammadi M. Exploring mechanisms of FGF signalling through the lens of structural biology. Nat Rev Mol Cell Biol. 2013;14:166–180. doi: 10.1038/nrm3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmbeck K, Bianco P, Caterina J, Yamada S, Kromer M, Kuznetsov SA, Mankani M, Gehron Robey P, Robin Poole A, Pidoux I, Ward JM, Birkedal-Hansen H. MT1-MMP-Deficient Mice Develop Dwarfism, Osteopenia, Arthritis, and Connective Tissue Disease due to Inadequate Collagen Turnover. Cell. 1999;99:81–92. doi: 10.1016/s0092-8674(00)80064-1. [DOI] [PubMed] [Google Scholar]
- Hotary K, Li XY, Allen E, Stevens SL, Weiss SJ. A cancer cell metalloprotease triad regulates the basement membrane transmigration program. Genes Dev. 2006;20:2673–2686. doi: 10.1101/gad.1451806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsia E, Richardson TP, Nugent MA. Nuclear localization of basic fibroblast growth factor is mediated by heparan sulfate proteoglycans through protein kinase C signaling. J Cell Biochem. 2003;88:1214–1225. doi: 10.1002/jcb.10470. [DOI] [PubMed] [Google Scholar]
- Katayama A, Bandoh N, Kishibe K, Takahara M, Ogino T, Nonaka S, Harabuchi Y. Expressions of matrix metalloproteinases in early-stage oral squamous cell carcinoma as predictive indicators for tumor metastases and prognosis. Clin Cancer Res. 2004;10:634–640. doi: 10.1158/1078-0432.ccr-0864-02. [DOI] [PubMed] [Google Scholar]
- Klint P, Kanda S, Claesson-Welsh L. Shc and a novel 89-kDa component couple to the Grb2-Sos complex in fibroblast growth factor-2-stimulated cells. J Biol Chem. 1995;270:23337–23344. doi: 10.1074/jbc.270.40.23337. [DOI] [PubMed] [Google Scholar]
- Kwabi-Addo B, Ozen M, Ittmann M. The role of fibroblast growth factors and their receptors in prostate cancer. Endocr Relat Cancer. 2004;11:709–724. doi: 10.1677/erc.1.00535. [DOI] [PubMed] [Google Scholar]
- Lehti K, Rose NF, Valavaara S, Weiss SJ, Keski-Oja J. MT1-MMP promotes vascular smooth muscle dedifferentiation through LRP1 processing. J Cell Sci. 2009;122:126–135. doi: 10.1242/jcs.035279. [DOI] [PubMed] [Google Scholar]
- Lehti K, Allen E, Birkedal-Hansen H, Holmbeck K, Miyake Y, Chun TH, Weiss SJ. An MT1-MMP-PDGF receptor-beta axis regulates mural cell investment of the microvasculature. Genes Dev. 2005;19:979–991. doi: 10.1101/gad.1294605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007;2:329–333. doi: 10.1038/nprot.2007.30. [DOI] [PubMed] [Google Scholar]
- Matsuda Y, Hagio M, Seya T, Ishiwata T. Fibroblast growth factor receptor 2 IIIc as a therapeutic target for colorectal cancer cells. Mol Cancer Ther. 2012;11:2010–2020. doi: 10.1158/1535-7163.MCT-12-0243. [DOI] [PubMed] [Google Scholar]
- Mazzieri R, Masiero L, Zanetta L, Monea S, Onisto M, Garbisa S, Mignatti P. Control of type IV collagenase activity by components of the urokinase-plasmin system: a regulatory mechanism with cell-bound reactants. EMBO J. 1997;16:2319–2332. doi: 10.1093/emboj/16.9.2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzner T, Bedeir A, Held G, Peter-Vorosmarty B, Ghassemi S, Heinzle C, Spiegl-Kreinecker S, Marian B, Holzmann K, Grasl-Kraupp B, Pirker C, Micksche M, Berger W, Heffeter P, Grusch M. Fibroblast growth factor receptors as therapeutic targets in human melanoma: synergism with BRAF inhibition. J Invest Dermatol. 2011;131:2087–2095. doi: 10.1038/jid.2011.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscatelli D. High and low affinity binding sites for basic fibroblast growth factor on cultured cells: absence of a role for low affinity binding in the stimulation of plasminogen activator production by bovine capillary endothelial cells. J Cell Physiol. 1987;131:123–130. doi: 10.1002/jcp.1041310118. [DOI] [PubMed] [Google Scholar]
- Moscatelli D. Basic fibroblast growth factor (bFGF) dissociates rapidly from heparan sulfates but slowly from receptors. Implications for mechanisms of bFGF release from pericellular matrix. J Biol Chem. 1992;267:25803–25809. [PubMed] [Google Scholar]
- Ohtake Y, Tojo H, Seiki M. Multifunctional roles of MT1-MMP in myofiber formation and morphostatic maintenance of skeletal muscle. J Cell Science. 2006;119:3822–3832. doi: 10.1242/jcs.03158. [DOI] [PubMed] [Google Scholar]
- Onguchi T, Han KY, Chang JH, Azar DT. Membrane type-1 matrix metalloproteinase potentiates basic fibroblast growth factor-induced corneal neovascularization. Am J Pathol. 2009;174:1564–1571. doi: 10.2353/ajpath.2009.080452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornitz DM, Yayon A, Flanagan JG, Svahn CM, Levi E, Leder P. Heparin is required for cell-free binding of basic fibroblast growth factor to a soluble receptor and for mitogenesis in whole cells. Mol Cell Biol. 1992;12:240–247. doi: 10.1128/mcb.12.1.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perentes JY, Kirkpatrick ND, Nagano S, Smith EY, Shaver CM, Sgroi D, Garkavtsev I, Munn LL, Jain RK, Boucher Y. Cancer cell-associated MT1-MMP promotes blood vessel invasion and distant metastasis in triple-negative mammary tumors. Cancer Res. 2011;71:4527–4538. doi: 10.1158/0008-5472.CAN-10-4376. [DOI] [PubMed] [Google Scholar]
- Plotnikov AN, Schlessinger J, Hubbard SR, Mohammadi M. Structural basis for FGF receptor dimerization and activation. Cell. 1999;98:641–650. doi: 10.1016/s0092-8674(00)80051-3. [DOI] [PubMed] [Google Scholar]
- Prats H, Kaghad M, Prats AC, Klagsbrun M, Lelias JM, Liauzun P, Chalon P, Tauber JP, Amalric F, Smith JA, et al. High molecular mass forms of basic fibroblast growth factor are initiated by alternative CUG codons. Proc Natl Acad Sci U S A. 1989;86:1836–1840. doi: 10.1073/pnas.86.6.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presta M, Dell’Era P, Mitola S, Moroni E, Ronca R, Rusnati M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005;16:159–178. doi: 10.1016/j.cytogfr.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Rifkin DB, Moscatelli D, Bizik J, Quarto N, Blei F, Dennis P, Flaumenhaft R, Mignatti P. Growth factor control of extracellular proteolysis. Cell Differ Dev. 1990;32:313–318. doi: 10.1016/0922-3371(90)90045-x. [DOI] [PubMed] [Google Scholar]
- Rozanov DV, Deryugina EI, Ratnikov BI, Monosov EZ, Marchenko GN, Quigley JP, Strongin AY. Mutation analysis of membrane type-1 matrix metalloproteinase (MT1-MMP). The role of the cytoplasmic tail Cys(574), the active site Glu(240), and furin cleavage motifs in oligomerization, processing, and self-proteolysis of MT1-MMP expressed in breast carcinoma cells. J Biol Chem. 2001;276:25705–25714. doi: 10.1074/jbc.M007921200. [DOI] [PubMed] [Google Scholar]
- Rusnati M, Urbinati C, Presta M. Internalization of basic fibroblast growth factor (bFGF) in cultured endothelial cells: role of the low affinity heparin-like bFGF receptors. J Cell Physiol. 1993;154:152–161. doi: 10.1002/jcp.1041540119. [DOI] [PubMed] [Google Scholar]
- Rusnati M, Tanghetti E, Dell’Era P, Gualandris A, Presta M. alphavbeta3 integrin mediates the cell-adhesive capacity and biological activity of basic fibroblast growth factor (FGF-2) in cultured endothelial cells. Mol Biol Cell. 1997;8:2449–2461. doi: 10.1091/mbc.8.12.2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabeh F, Ota I, Holmbeck K, Birkedal-Hansen H, Soloway P, Balbin M, Lopez-Otin C, Shapiro S, Inada M, Krane S, Allen E, Chung D, Weiss SJ. Tumor cell traffic through the extracellular matrix is controlled by the membrane-anchored collagenase MT1-MMP. J Cell Biol. 2004;167:769–781. doi: 10.1083/jcb.200408028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlessinger J, Plotnikov AN, Ibrahimi OA, Eliseenkova AV, Yeh BK, Yayon A, Linhardt RJ, Mohammadi M. Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol Cell. 2000;6:743–750. doi: 10.1016/s1097-2765(00)00073-3. [DOI] [PubMed] [Google Scholar]
- Shamamian P, Schwartz JD, Pocock BJ, Monea S, Whiting D, Marcus SG, Mignatti P. Activation of progelatinase A (MMP-2) by neutrophil elastase, cathepsin G, and proteinase-3: a role for inflammatory cells in tumor invasion and angiogenesis. J Cell Physiol. 2001;189:197–206. doi: 10.1002/jcp.10014. [DOI] [PubMed] [Google Scholar]
- Simons M, Horowitz A. Syndecan-4-mediated signalling. Cell Signal. 2001;13:855–862. doi: 10.1016/s0898-6568(01)00190-5. [DOI] [PubMed] [Google Scholar]
- Su G, Blaine SA, Qiao D, Friedl A. Membrane type 1 matrix metalloproteinase-mediated stromal syndecan-1 shedding stimulates breast carcinoma cell proliferation. Cancer Res. 2008;68:9558–9565. doi: 10.1158/0008-5472.CAN-08-1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama N, Varjosalo M, Meller P, Lohi J, Chan KM, Zhou Z, Alitalo K, Taipale J, Keski-Oja J, Lehti K. FGF receptor-4 (FGFR4) polymorphism acts as an activity switch of a membrane type 1 matrix metalloproteinase-FGFR4 complex. Proc Natl Acad Sci U S A. 2010;107:15786–15791. doi: 10.1073/pnas.0914459107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanghetti E, Ria R, Dell’Era P, Urbinati C, Rusnati M, Ennas MG, Presta M. Biological activity of substrate-bound basic fibroblast growth factor (FGF2): recruitment of FGF receptor-1 in endothelial cell adhesion contacts. Oncogene. 2002;21:3889–3897. doi: 10.1038/sj.onc.1205407. [DOI] [PubMed] [Google Scholar]
- Wang Y, McNiven MA. Invasive matrix degradation at focal adhesions occurs via protease recruitment by a FAK-p130Cas complex. J Cell Biol. 2012;196:375–385. doi: 10.1083/jcb.201105153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarrabi K, Dufour A, Li J, Kuscu C, Pulkoski-Gross A, Zhi J, Hu Y, Sampson NS, Zucker S, Cao J. Inhibition of matrix metalloproteinase 14 (MMP-14)-mediated cancer cell migration. J Biol Chem. 2011;286:33167–33177. doi: 10.1074/jbc.M111.256644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Apte SS, Soininen R, Cao R, Baaklini GY, Rauser RW, Wang J, Cao Y, Tryggvason K. Impaired endochondral ossification and angiogenesis in mice deficient in membrane-type matrix metalloproteinase I. Proc Natl Acad Sci USA. 2000;97:4052–4057. doi: 10.1073/pnas.060037197. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Immunofluorescence analysis of syndecan-1 levels in MCF-7 cells with or without MT1-MMP. MT1-MMP Tet-Off MCF-7 cells grown for 24 h in the presence or absence of DOX (1 μg/ml) were analyzed by immunofluorescence as described in Materials and Methods. This experiment was repeated three times with comparable results.
Supplemental Figure 2. Flow cytofluorometric analysis of syndecan-1 levels in MCF-7 cells with or without MT1-MMP. MT1-MMP Tet-Off MCF-7 cells grown for 24 h in the presence or absence of DOX (1 μg/ml) were analyzed by flow cytometry as described in Materials and Methods. This experiment was repeated three times with comparable results.