

Abstract
Heart failure induced by myocardial infarction (MI) causes diaphragm muscle weakness, with elevated oxidants implicated. We aimed to determine whether diaphragm muscle weakness is 1) early-onset post-MI (i.e., within the early left ventricular remodeling phase of 72 h); and 2) associated with elevated protein oxidation. Ligation of the left coronary artery to induce MI (n = 10) or sham operation (n = 10) was performed on C57BL6 mice. In vitro contractile function of diaphragm muscle fiber bundles was assessed 72 h later. Diaphragm mRNA and protein expression, enzyme activity, and individual carbonylated proteins (by two-dimensional differential in-gel electrophoresis and mass spectrometry) were subsequently assessed. Infarct size averaged 57 ± 1%. Maximal diaphragm function was reduced (P < 0.01) by 20% post-MI, with the force-frequency relationship depressed (P < 0.01) between 80 and 300 Hz. The mRNA expression of inflammation, atrophy, and regulatory Ca2+ proteins remained unchanged post-MI, as did the protein expression of key contractile proteins. However, enzyme activity of the oxidative sources NADPH oxidase and xanthine oxidase was increased (P < 0.01) by 45 and 33%, respectively. Compared with sham, a 57 and 45% increase (P < 0.05) was observed in the carbonylation of sarcomeric actin and creatine kinase post-MI, respectively. In conclusion, diaphragm muscle weakness was rapidly induced in mice during the early left ventricular remodeling phase of 72 h post-MI, which was associated with increased oxidation of contractile and energetic proteins. Collectively, these findings suggest diaphragm muscle weakness may be early onset in heart failure, which is likely mediated in part by posttranslational oxidative modifications at the myofibrillar level.
Keywords: chronic heart failure, MI: protein carbonylation, skeletal muscle
myocardial infarction (mi) is a leading cause of global morbidity and mortality and represents a primary risk factor for chronic heart failure (CHF) (13). Post-MI, two stages of left ventricular (LV) remodeling have been defined: an “early” stage (within 72 h) that involves the expansion of the infarct area, and a “late” stage (beyond 72 h) where global LV changes associated with CHF development ensue (41). CHF is characterized by symptoms of breathlessness, muscle weakness, and exercise intolerance, the latter being a strong predictor of mortality (21, 31). The pathophysiology underlying these symptoms is complex and remains poorly understood, an issue largely complicated by the fact that central hemodynamics are poorly associated with exercise limitation in CHF (7). In contrast, limb skeletal muscle and ventilatory impairments have been shown to be strong predictors of the former (7). It is not surprising, therefore, that the most important muscle of inspiration, the diaphragm, has reduced function following MI-induced CHF (10, 40, 44–47), which correlates strongly to prognosis (22, 26). While diaphragm muscle weakness is likely a key mediator in the overall pathogenesis of CHF, little is also still known about its temporal progression from MI to CHF (i.e., during the early LV remodeling phase). For example, at present, diaphragm muscle weakness post-MI is considered to develop progressively over weeks to months (10, 40, 44–47), but whether it is in fact early onset and mediated by similar mechanisms that are reported to occur in CHF remains unknown.
Diaphragm muscle weakness in MI-induced CHF is mediated by muscle atrophy (i.e., loss of muscle mass) and/or contractile dysfunction (i.e., a decrease in force normalized to muscle mass) (10, 40, 44–47). These represent two distinct mechanisms of muscle weakness that can act in parallel or alone (36). Diaphragm muscle atrophy is likely caused by an increased proteolysis [e.g., via activation of the ubiquitin proteasome system and caspase-3 (44, 47)], whereas contractile dysfunction is likely related to oxidant-induced modifications of key proteins (9, 40). That diaphragm dysfunction is induced only hours within specific pathologies associated with elevated systemic inflammation and reactive oxygen species (ROS) [e.g., sepsis (4) and mechanical ventilation (35)], where carbonylation of myofibrillar and energetic proteins (e.g., actin, myosin, creatine kinase) is reported, suggests a similar scenario may also occur during the early LV remodeling phase post-MI. Hence, the oxidative modifications of proteins may represent an early and critical step leading to contractile dysfunction in MI-induced CHF.
The present study, therefore, aimed to assess in vitro respiratory muscle function in mice after 72 h to determine whether diaphragm muscle weakness was induced during the early LV remodeling phase post-MI. We hypothesized that diaphragmatic contractile dysfunction would be rapidly induced following MI, likely mediated by oxidant-induced intracellular modifications. To the best of our knowledge, no studies have yet been performed to confirm diaphragm dysfunction is developed in mice post-MI, limiting the potential benefits offered by transgenic strains in elucidating novel mechanisms (c.f., Ref. 18). We, therefore, selected the mouse not only on the basis of it representing a well-established animal model of MI (27, 28, 32) that progresses to CHF (6, 15, 48), but also to serve as an important “proof of concept” based on the advantage of future application of transgenic strains.
METHODS
Animals and Procedures
Eight-week-old female C57 BL6 mice (n = 20; body weight ∼20 g) were included in these experiments, as approved by the local Animal Research Council, University of Leipzig (TVV 28/11). Mice underwent surgery to induce MI (n = 10) or sham operation (n = 10). Briefly, mice were anesthetized with pentobarbital sodium, intubated, and underwent pressure-supported mechanical ventilation that lasted on average 15 min (Animal Respirator Advanced 4601-1, TSE Systems, Bad Homburg, Germany). A thoracotomy was performed exposing the left side of the heart, and a surgical silk suture (7-0) was used to ligate the left anterior descending coronary artery. Surgery was concluded by progressively closing deep-to-superficial anatomical layers with sutures. Sham mice underwent the same procedures but without ligation. Mice were killed 72 h later, with the diaphragm, heart, and lungs removed for further analysis.
Heart Function and Histology
Echocardiography (Vevo 770, Visual Sonics Europe, Amsterdam, The Netherlands) was performed in M-mode using a 30-MHz transducer immediately before death. Mice were anesthetized by breathing a 5% isoflurane-O2 balanced mixture (maintained at 2%), with LV end-diastolic (LVEDD) and systolic diameters (LVESD) assessed, allowing calculation of their product: LV fractional shortening [LVFS = (LVEDD − LVESD/LVEDD) × 100]. The heart was also dissected into three transverse sections at death for histological analysis, with the medial section fixed in 4% PBS-buffered formalin. Serial cross sections (2 μm) were subsequently stained with hematoxylin and eosin and mounted on glass slides. Computer imaging software (Analysis 3.0, Olympus Soft Imaging Solutions, Münster, Germany) was then used to demarcate the infarct boundary, defined by a significant loss in LV myocardial tissue (i.e., a thinning in the LV wall >2 SDs of mean wall thickness). The thinning of the LV wall also corresponded to changes in the contrast of the image, which was used to corroborate infarct boundary determination. Average infarct size (%) was then quantified as the ratio of infarct circumference to overall LV circumference (33). Only mice with infarcts >35% were included, as this criterion has been shown to induce CHF in mice (6, 15, 48) and also diaphragm dysfunction in rats with CHF (44, 47).
Diaphragm Preparation and Function
A laparotomy and thoracotomy were performed to allow complete excision of the diaphragm. The right costal diaphragm muscle was immediately snap-frozen in liquid N2 for subsequent molecular analyses, while the left costal diaphragm muscle was prepared in a Krebs-Hanseleit buffer solution (120.5 NaCl, 4.8 KCl, 1.2 MgSO4, 1.2 NaH2PO4, 20.4 NaHCO3, 1.6 CaCl2, 10 dextrose, 1 pyruvate; in mmol/l at a pH of ∼7.40) at room temperature, equilibrated with 95% O2/5% CO2 for contractile measurements. Briefly, a muscle bundle connected from rib to central tendon was dissected, attached to silk sutures (4-0) at either end, and mounted vertically in a buffer-filled organ bath. The suture connected to the rib was secured to a hook at the bottom of the bath while the tendon was tied to a length-controlled lever system (301B, Aurora Scientific, Aurora, Canada). In vitro muscle function was assessed by platinum electrodes stimulating the muscle with a supramaximal current (600 mA; 500-ms train duration; 0.25-ms pulse width) via a high power bipolar stimulator (701B, Aurora Scientific, Aurora, Ontario, Canada). The muscle bundle was set at an optimal length equivalent to the maximal twitch force produced, after which bath temperate was increased to 37°C, and a 15-min thermo-equilibration period followed. A force-frequency protocol was then performed at 1, 15, 30, 50, 80, 120, 150, and 300 Hz, respectively, separated with 1-min rest intervals. Following a 5-min period in which muscle length was measured using digital calipers, the muscle underwent a fatigue protocol over 10 min (40 Hz every 2 s). The muscle was subsequently detached, trimmed free from rib and tendon, blotted dry, and weighed. Muscle force (N) was normalized to muscle cross-sectional area (cm2) by dividing muscle mass (g) by the product of optimal length (cm) and estimated muscle density (1.06) (8), which allowed specific force in N/cm2 to be calculated.
Molecular Analyses
Quantification of mRNA expression, enzyme activity, and protein expression.
The relative expression of mRNA was quantified as previously described (23). Briefly, total RNA was isolated from muscle tissue using RNeasy and reverse transcribed into cDNA using random hexamers and Sensiscript reverse transcriptase (Qiagen, Hilden, Germany), where an aliquot of the cDNA was used for quantitative RT-PCR (LightCycle 2.0, Roche Diagnostics, Mannheim, Germany). The mRNA expression in the diaphragm and heart was assessed for markers of inflammation (i.e., TNF-α, IL-6, IL-1β) and also in the diaphragm for markers of atrophy [i.e., key ubiquitin proteasome E3 ligases muscle RING finger-1 (MuRF-1) and muscle atrophy F-box (MAFbx)] and regulatory Ca2+ proteins [i.e., ryanodine receptor 1 (RyR1), dihydropyridine receptor (DHPR), sarcoplasmic reticulum Ca2+-ATPase 2A (SERCA2A)]. The relative expression of target genes was normalized to hypoxanthine-guanine-phosphoribosyl-transferase via the 2−ΔΔCT [arbitrary units (AU)] quantification method (20), with serial dilution curves confirming similar reaction amplification efficiencies.
In addition, frozen muscle samples were also homogenized in lysis buffer (50 mM Tris, 150 mM sodium chloride, 1 mM EDTA, 1% NP-40, 0.25% sodium-deoxycholate, 0.1% SDS, 1% Triton X-100; pH 7.4) containing a protease inhibitor mix (Inhibitor mix M, Serva, Heidelberg, Germany), sonicated, and centrifuged at 16,000 g for 5 min. The supernatant was isolated, and protein content determined (BCA assay, Pierce, Bonn, Germany). Thereafter, enzymatic activities of glutathione peroxidase (GPX), catalase, superoxide dismutase (SOD), xanthine oxidase (XO), and NADPH oxidase were measured photometrically from diaphragm homogenates by commercially available kits, in accordance with the manufacturer's instructions (BioVision, Milpitas, CA). Western blot of diaphragm homogenates was used to quantify protein expression of myosin light chain, α-actin, and troponins T, I, and C, as previously described (12, 19).
Quantification of Protein Carbonylation
Sample preparation.
For each muscle sample, total protein was extracted by lysis buffer {7 M urea, 2 M thiourea, 30 mM Tris, 4% (wt/vol) 3-([3-cholamidopropyl]dimethylammonio)-2-hydroxy-1-propanesulfonate (CHAPS), 1 mM phenylmethylsulfonyl fluoride, and 1% protease inhibitor cocktail; Sigma-Aldrich, St. Louis, MO}, and protein concentration was determined (2-D Quant Kit, GE Healthcare, Uppsala, Sweden). Protein samples were subsequently purified (2-D Clean-Up Kit, GE Healthcare). Fluorescent labeling of carbonyls was achieved by incubating 150 μg of acetone precipitated protein with 100 μl of Alexa 488 fluorescent hydroxylamine (FHA; Invitrogen; 1 mg/ml) at room temperature for 2 h in darkness. FHA-labeled proteins were then precipitated with ice-cold trichloroacetic acid, and, after centrifugation, the protein pellet was washed three times with ethyl acetate-ethanol (1:1) solution (34). Total protein of the FHA-labeled protein sample was accomplished by additional staining using Cy5 Fluor minimal dye (400 pmol per 50 μg protein sample) in accordance with the manufacturer's recommendations (GE Healthcare). Thereafter, the double-labeled protein sample was adjusted to 450 μl with rehydration buffer [7 M urea, 2 M thiourea, 2% CHAPS, 0.5% immobilized pH gradient (IPG) buffer, pH 3–10, and a trace of bromophenol blue] before the two-dimensional differential in-gel electrophoresis (2D-DIGE) analysis, which included both isoelectric focusing (IEF) and sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE).
First- and Second-Dimensional Protein Separation
First-dimensional protein separation required labeled proteins being loaded into 24-cm IPG dry-strips (pH 3–10; GE Healthcare) and rehydrated for 12 h in the dark. The IPG strips were focused at 20°C using an Ettan IPGPhor 3 IEF unit (GE Healthcare) at 500 V for 1 h, a 7.5-h gradient to 1,000 V, a 3-h gradient to 8,000 V, a 5-h plateau at 8,000 V, a 3-h gradient to 10,000 V, and a final plateau at 10,000 V for 4 h, as previously described (30). For the second-dimensional separation, each IPG strip was equilibrated twice for 15 min in equilibration buffer (6 M urea, 30% glycerol, 2% SDS, 50 mM Tris·HCl; pH 8.8), with the exception that the first period contained 1% wt/vol DTT, while the second period contained 2.5% wt/vol iodoacetamide and a trace of bromophenol blue. The equilibrated IPG strips were then carefully positioned on 12% acrylamide gels and covered with 1% wt/vol agarose. Second-dimension separation by SDS-PAGE was performed using a PROTEANPlus Dodeca Cell (Bio-Rad, Hercules, CA), where proteins were separated at 1 W/gel until the dye front reached the bottom of the gel. 2D-DIGE gels were subsequently scanned within the gel cassettes using the Typhoon Trio Scanner (GE Healthcare). The Alexa 488 hydroxylamine dye was excited at 488 nm and emission spectra obtained at 520 nm, whereas the Cy5 dye was excited at 633 nm and emission spectra obtained at 670 nm. The gels were visualized and first evaluated with the Image Quant Software (GE Healthcare, Uppsala, Sweden), where the two images and an overlay image underwent individual inspection. After scanning, gels were removed from the cassette and underwent blue silver-staining overnight. 2D-DIGE gel analysis was performed using Delta2D 4.0 software (Decodon, Greifswald, Germany) with advanced image processing algorithms that permits image fusion, background subtraction, normalization, and relative quantitation of proteins from different images. Following automated spot detection, each spot was manually verified and edited using a three-dimensional view algorithm. For each gel, the spot ratios of the Alexa 488 hydroxylamine and the Cy5 dye staining were determined. For further identification, only spots that passed a between-group statistical comparison (P < 0.05) and showed a spot ratio lower than 0.5 or higher than 2.0 were considered.
Gel Preparation
Carbonylated protein spots of interest were cut from polyacrylamide gels and digested overnight using trypsin to allow peptide extraction for subsequent mass spectrometry (MS) analysis, as previously described (17). The gel spots were then 1) washed two times for 1 h with water, acetic acid, and ethanol (50:5:45 vol/vol); 2) reduced by the addition of acetonitrile and alkylated with DTT and iodoacetamide; 3) incubated with 5 mM ammonium bicarbonate and 100% acetonitrile; and 4) dried by vacuum centrifugation. Dried gel pieces were swollen by adding trypsin in ammonium carbonate buffer (100 mM, pH 8.5) and incubated at 37°C for 12 h. Extractions of peptides were subsequently performed with an extraction buffer containing acetonitrile, water, and formic acid (5:4.4:0.6 vol/vol), followed by the extracted peptide solutions being dried using vacuum centrifugation. Thereafter, the precipitates were reconstituted with 10 μl of 5% acetonitrile in 0.1% trifluoroacetic acid, with 0.5 μl of this peptide extract and 0.5 μl of α-cyano-4-hydroxycinnamic acid mixed and spotted on ground steel target plates (Bruker Daltonics, Bremen, Germany).
Identification of Carbonylated Proteins by MS
Matrix-assisted laser desorption/ionization time-of-flight MS was performed to identity peptides (Ultra Flex III, Bruker Daltonics, Bremen, Germany), using a ground steel MTP 384 target plate (Bruker Daltonics), in accordance with the manufacturer's instructions. In brief, samples were prepared by mixing equal volumes (1 μl) of sample and α-cyano-4-hydroxycinnamic acid matrix (0.1 g/l), which were then spotted onto the ground steel MTP 384 target plate. The solution was allowed to evaporate at room temperature. Spectra were obtained on a matrix-assisted laser desorption/ionization time-of-flight/time-of-flight mass spectrometer (Ultra Flex III, Bruker Daltonics, Bremen, Germany) using FlexControl software version 3.0 (Bruker Daltonics, Bremen, Germany), as described previously (17). In brief, the instrument was operated at pulse rates of 100 Hz, with the pulse ion extraction delay set to 400 ns (17). Measurements were carried out in positive reflector mode using an acceleration voltage of 25.0 kV (ion source 1) and 21.85 kV (ion source 2). Lens voltage was set at 9.5 kV, and reflector voltages at 26.3 and 13.7 kV. Mass spectra were recorded in the mass-to charge ratio range between 200 and 3,500. When possible, up to 10 MS2 spectra were performed. The spectra were processed by the software FlexAnalysis (Bruker Daltonics, Bremen, Germany). A database search was finally conducted using the MS/MS ion search (MASCOT v 2.2, Matrix Science, London, UK) against all metazoan (animals) entries of NCBInr (GenBank) for the subsequent parameters: trypsin digestion, up to one missed cleavage and fixed modifications for carbamidomethyl (C) with the following variable modifications: oxidation (M); peptide tolerance: ±1.2 Da; MS/MS tolerance: ±0.6 Da; peptide charge: +1, +2 and +3.
Statistical Analysis
Data are presented as means ± SE. Between-group differences were assessed by independent t-test, while the force-frequency and fatigue relationships were assessed by two-way repeated-measures ANOVA. Significance was accepted as P < 0.05. Analyses were performed by SPSS version 22 (SPSS, Chicago, IL).
RESULTS
Mice Characteristics
Physical, histological, and echocardiography data collectively confirmed the operation was successful for inducing a successful MI (Table 1 and Fig. 1). The histology revealed a mean infarct size of 57 ± 1% (Table 1 and Fig. 1), with echocardiography demonstrating LVFS was reduced by 65% postinfarct relative to sham (Table 1; P < 0.01). These findings are also supported by the significantly increased heart and lung weights post-MI (both absolute and relative to body weight) (Table 1). Diaphragm muscle wet weight was not different between groups (33 ± 1 vs. 32 ± 1 mg; P > 0.05), however.
Table 1.
Mice characteristics for sham and myocardial infarction mice
| Sham | MI | |
|---|---|---|
| n | 10 | 10 |
| Physical | ||
| Heart weight, mg | 94.1 ± 2.5 | 117.0 ± 5.2* |
| Heart-to-body weight ratio | 5.1 ± 0.1 | 7.2 ± 0.3* |
| Lung weight, mg | 103.3 ± 4.9 | 169.5 ± 13.1* |
| Lung-to-body weight ratio | 5.7 ± 0.3 | 10.5 ± 0.9* |
| Histology | ||
| LV infarct size, % | – | 57.0 ± 1.0 |
| Echocardiography | ||
| LVEDD, mm | 3.39 ± 0.16 | 4.25 ± 0.11* |
| LVESD, mm | 2.25 ± 0.14 | 3.77 ± 0.17* |
| LVFS, % | 33 ± 3 | 12 ± 2* |
Values are mean ± SE; n, no. of mice. MI, myocardial infarction; LV, left ventricle; LVEDD, LV end-diastolic diameter; LVESD, LV end-systolic diameter; LVFS, LV fractional shortening.
P < 0.01 vs. sham.
Fig. 1.

Representative examples of hematoxylin and eosin stained transverse sections from a sham mouse (A) and a myocardial infarction (MI) mouse with an infarct size of 57% (B). Note the left ventricular remodeling (i.e., thinning) that occurred in the 72 h following ligation of the left coronary artery, as denoted by the dashed lines.
Diaphragm Muscle Function
In vitro diaphragm muscle function was significantly depressed post-MI (Fig. 2), with specific force reduced at frequencies ≥80 Hz (P < 0.01) and maximal specific force impaired by ∼20% (P < 0.01). No differences in the force-frequency relationship were observed, however, when forces were normalized as percent maximum force (P > 0.05). Twitch contractile dynamics were affected post-MI, with time-to-peak twitch tension slowed (22.3 ± 0.6 vs. 24.1 ± 0.5 ms; P < 0.05) and the twitch-to-tetanus ratio increased (0.18 ± 0.01 vs. 0.22 ± 0.01; P < 0.01), whereas the twitch one-half relaxation time was not different (17.0 ± 1.2 vs. 14.5 ± 1.0 ms; P > 0.05). No differences were discerned at termination of the acute fatigue protocol between sham or MI mice (absolute force: 3.9 ± 0.5 vs. 4.1 ± 0.2 N/cm2; relative to initial contraction: 39 ± 3 vs. 45 ± 2%; P > 0.05), with the rate of force decline not different (0.17 ± 0.01 vs. 0.16 ± 0.01%/s; P > 0.05).
Fig. 2.
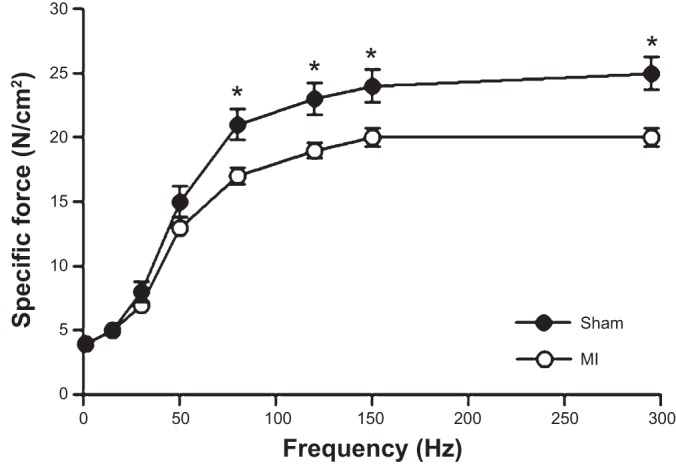
Diaphragm muscle function was reduced 72 h following a MI, as demonstrated by the depressed (P < 0.05) force-frequency relationship. Values are group means ± SE. *P < 0.01 between groups.
mRNA Expression
The mRNA expression of inflammatory cytokines TNF-α, IL-6, and IL-1β was increased in the heart of MI mice compared with sham (Fig. 3, A–C; P < 0.01), but this was not observed in the diaphragm (Fig. 3, D–F; P > 0.05). In addition, the mRNA expression for markers of muscle atrophy (i.e., MuRF-1 and MAFbx) was not different between sham and MI mice in the diaphragm (Fig. 4; P > 0.05), as was the case for key regulatory Ca2+ proteins (RyR1, 100.4 ± 10.6 vs. 106.2 ± 15.2 AU; DHPR, 190.0 ± 15.4 vs. 183.5 ± 17.5 AU; SERCA2A, 3.1 ± 0.2 vs. 2.8 ± 0.2 AU; P > 0.05).
Fig. 3.

Inflammatory cytokines mRNA expression [TNF-α (A), IL-6 (B), and IL-1β (C)] were significantly increased in the heart but not in the diaphragm (D, E, and F, respectively) of mice induced with a MI compared with sham. Values are group means ± SE in arbitrary units (au). *P < 0.01 vs. sham.
Fig. 4.
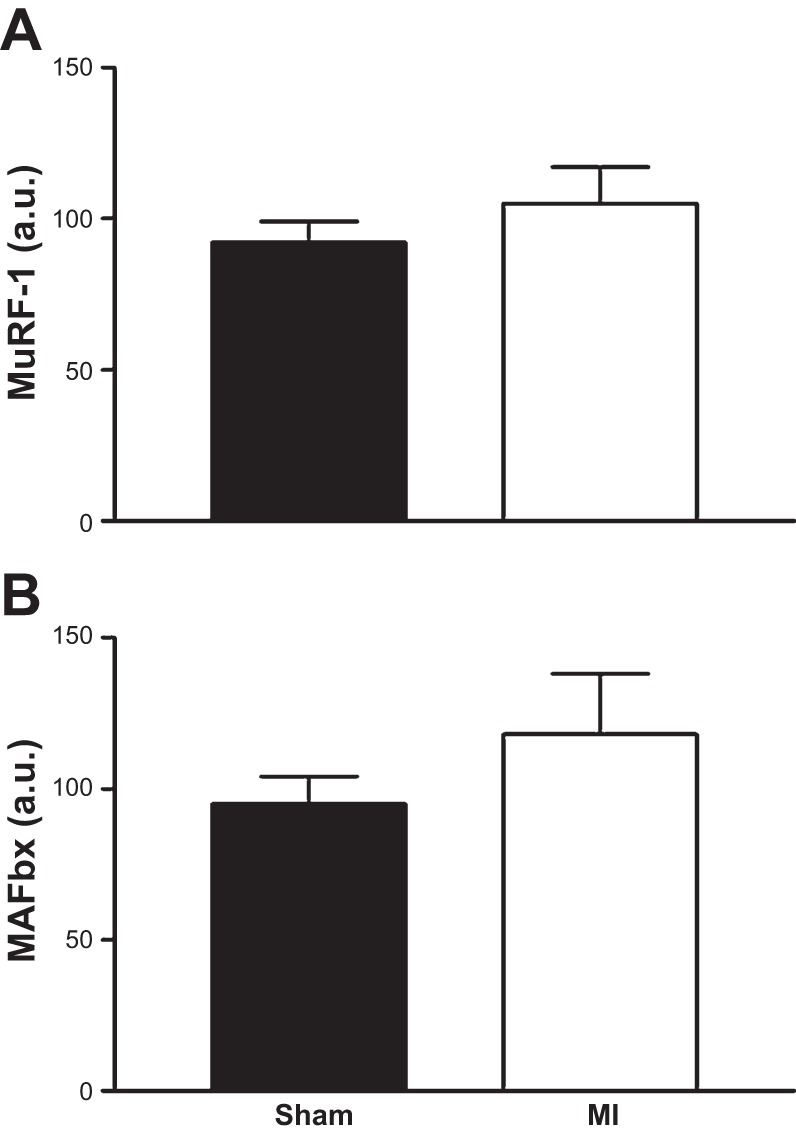
The mRNA expression of muscle RING finger-1 (MuRF-1; A) and muscle atrophy F-box (MAFbx; B), markers of muscle atrophy, were not increased in the diaphragm 72 h following a MI. Values are group means ± SE in au.
Enzyme Activity
Compared with sham, mice with MI had a higher enzyme activity in the diaphragm of putative oxidant sources, with a 45% increase in NADPH oxidase activity (Fig. 5A; P < 0.01) and 33% higher XO activity (Fig. 5B; P < 0.01), both inversely correlated to the reduction in maximal diaphragmatic force (NADPH oxidase and XO: R2 = 0.50 and R2 = 0.43, respectively; Fig. 5, C and D; both P < 0.05). In addition, antioxidant enzyme activity was increased post-MI, with catalase higher compared with sham (1.60 ± 0.18 vs. 2.75 ± 0.38 U/mg; P < 0.01), which inversely correlated to the reduction in maximal diaphragm force (R2 = 0.74; P < 0.01). SOD and GPX demonstrated a trend to increase post-MI (39 ± 1 vs. 44 ± 3% and 2.2 ± 0.4 vs. 2.9 ± 0.5 milli-optical density·min−1·mg−1; respectively; P > 0.05), with both associated with reductions in maximal force (SOD: R2 = 0.27; P > 0.05; and GPX: R2 = 0.60; P < 0.01). No strong correlations were observed in the sham group, however.
Fig. 5.

The enzyme activities of the putative oxidant sources NADPH oxidase (A) and xanthine oxidase (XO; B) were significantly increased in the diaphragm following a MI. In MI mice, the higher NADPH enzyme activity inversely correlated to impaired muscle function (R2 = 0.50; P < 0.05; dashed line; C), which was also observed for XO (R2 = 0.43; P < 0.05; dashed line; D). In contrast, no correlations were observed in sham mice (solid lines). Values are group means ± SE. mOD, milli-optical density. *P < 0.001 vs. sham.
Protein Expression
Western blot analyses revealed protein expression of key contractile proteins (i.e., α-actin, myosin light chain, troponin I, troponin C, and troponin T) were not different (P > 0.05) between sham and MI in the diaphragm (in arbitrary units; α-actin, 0.53 ± 0.07 vs. 0.67 ± 0.09; myosin light chain, 0.33 ± 0.07 vs. 0.47 ± 0.15; troponin I, 0.37 ± 0.18 vs. 0.47 ± 0.11; troponin C, 0.34 ± 0.07 vs. 0.52 ± 0.07; troponin T, 0.82 ± 0.23 vs. 0.48 ± 0.09, respectively).
Protein Carbonylation
To investigate protein oxidation, we utilized 2D-DIGE and MS (n = 5 per group), which allowed specific carbonylated proteins to be identified. This analysis revealed key contractile and energetic proteins in the diaphragm were significantly more oxidized in the MI group compared to sham, with a significant increase in the carbonylation of α-actin by 57% and creatine kinase by 45% (Fig. 6).
Fig. 6.

A: a representative example of a gel image from a diaphragm muscle sample, where two-dimensional (2D) differential gel electrophoresis was used to detect significant differences (P < 0.05) in protein carbonylation between sham and MI mice. Our results found the contractile protein α-actin (B and D) and the energetic protein creatine kinase (C and E) were more carbonylated in the diaphragm 72 h after a MI (P < 0.05). Protein samples were subjected to isoelectric focusing (24 cm nonlinear immobilized pH gradient strip with pH range 3–10) followed by being positioned on 12% acrylamide gels to undergo SDS-PAGE to allow separation of proteins. Fluorescent labeling of carbonylated proteins with Alexa-488-hydroxylamine (A) and total protein with Cy5 dye allowed carbonylated proteins in muscle samples to be detected (B and C) and quantified as a ratio (D and E). Mass spectrometry was used to confirm the identified proteins. pI, isoelectric point; MW, molecular weight. Values are group means ± SE. *P < 0.05.
DISCUSSION
This study in mice provides novel evidence that diaphragm muscle weakness is rapidly induced post-MI, within 72 h of the early LV remodeling phase. We also demonstrated that this reduction in diaphragm muscle function was associated with posttranslational oxidative modifications of key contractile and energetic proteins (i.e., α-actin and creatine kinase). Collectively, therefore, these data suggest diaphragm muscle weakness may be early onset in MI-induced CHF and mediated in part by intracellular protein oxidation.
Diaphragm Muscle Weakness Post-MI
MI is a leading cause of global morbidity and mortality, with a prevalence of ∼20% for subsequent CHF development (13). Following MI, two phases of LV remodeling have been defined: an early phase (within 72 h) or late phase (after 72 h) (41). It is well established that, during the late ventricular remodeling phase, where CHF is developed, diaphragm muscle weakness is manifested, as reported in animal models from fiber bundles (40, 45, 46) and single fibers (44, 47), and also in patients (22, 26). In general, maximal diaphragmatic force in animal models is impaired by ∼20%, measured months after inducing CHF following MI. Our findings are, therefore, surprising, in that we demonstrate a similar reduction in maximal diaphragm function during the early LV remodeling phase 72 h post-MI. In addition, these data (to the best of our knowledge) are also the first to show diaphragm muscle weakness occurs in a mouse model post-MI. This study, therefore, serves as an important proof of concept for future research, where transgenic strains can be used to test specific mechanisms related to diaphragm dysfunction following MI.
We selected the ligation procedure in the present study as it represents an established mouse model of MI that progresses to CHF (6, 15, 27, 28, 32, 48). Our infarct size of 57% is similar to that previously reported in mice after 3 days (37); however, it should be noted that this infarct is relatively severe and likely predisposes to an increased mortality over the long term compared with more moderate infarcts (6). Nevertheless, evidence suggests that ∼15% of patients who suffer MI will have a LV infarct size greater than 40% (25, 29), suggesting our model is of clinical relevance. Indeed, our findings have important clinical implications for MI patients, whose major symptoms of breathlessness, muscle weakness, and exercise limitation are likely exacerbated by early decrements in diaphragm muscle function. For example, our data suggest patients who suffer MI may be at immediate risk of diaphragm muscle weakness, a question that remains to be answered but warrants further investigation. Furthermore, our findings are also relevant to CHF patients, suggesting diaphragm muscle weakness may be early onset in this disease. That diaphragm function was reduced within hours after the initial cardiac insult suggests diaphragm dysfunction in CHF may not be progressive, as previously assumed (10, 22, 26, 40, 44–47), and potentially represents an early therapeutic target in the treatment of CHF.
It remains likely, however, that two processes may be acting to induce diaphragm muscle weakness in CHF: the first is related to the initial acute cardiac insult of the MI, whereas the second is related to the slower progressive pathophysiology associated with CHF [e.g., reductions in central and peripheral blood flow, ventilatory impairments, impaired endothelial function, and systemic and local inflammation (7)]. This may help explain why we observed no differences between groups in forces at the lower frequencies and during the fatigue protocol, as the diaphragm may be differentially impacted over time. However, of note, we employed a frequency-matched rather than force-matched protocol (11), and whether the diaphragm would have fatigued more rapidly at similar forces requiring an increased motor unit recruitment remains unknown.
Mechanisms of Diaphragm Muscle Weakness
Diaphragm muscle weakness in CHF post-MI is generally considered to be consequent to atrophy and/or contractile dysfunction. Muscle atrophy has been reported in CHF diaphragm (16, 38), which is consistent with reports of increased protein degradation (44, 47). For example, the mRNA expression of key ubiquitin proteasome E3 ligases MuRF-1 and MAFbx, as well as increased proteasome and caspase activation, has been shown to be upregulated in CHF diaphragm. However, that we found diaphragm mRNA expression of MuRF-1 and MAFbx to be unchanged post-MI, and also the protein expression of contractile proteins and diaphragm muscle mass unaltered, collectively indicate atrophy and increased protein degradation were unlikely to have contributed to the loss of diaphragm function. This is possibly explained by the short time frame used in the present study (i.e., 72 h), which may have limited the “downstream” effects MI has on the diaphragm, thus being too acute to activate proteases.
In contrast, our findings implicate contractile dysfunction as the mechanism leading to diaphragm muscle weakness 72 h post-MI. Diaphragm force was reduced across a range of frequencies, independent of muscle mass, and this was associated with increased oxidative modifications of cellular proteins. Specifically, we found a significant increase post-MI in the oxidation of individual proteins important for contractile energetics (i.e., α-actin and creatine kinase) (Fig. 6). Interestingly, these findings parallel other studies that used similar analytical techniques, which also reported the same proteins to be oxidized in the diaphragm following induction of sepsis (4), chronic obstructive pulmonary disease (24), and after TNF-α treatment (23), conditions also accompanied by reductions in muscle function. Overall, therefore, our data and that of others (4, 23, 24) suggest α-actin and creatine kinase are appreciably susceptible to oxidation. Carbonylation can thus be considered an important determinant of protein structure and function (5), which in turn influences diaphragm muscle force.
The mechanism for how oxidants are increased in the diaphragm, despite the initial insult being located at the heart, remains unclear. We propose that an endocrine-mediated increase in local oxidants may be responsible (18), whereby increased production of inflammatory cytokines in the heart “spill over” into the circulation (42), which leads to an increased oxidant production in the diaphragm. This process may then initiate carbonylation of key contractile proteins, which in turn induces muscle weakness. This hypothesis is supported by evidence where cardiac-specific overexpression of TNF-α in transgenic mice led to increased levels of the cytokine in the serum, but not in the diaphragm (18). Interestingly, however, the diaphragm of transgenic mice had significantly higher levels of oxidants than that observed in wild-type mice, which was associated with large reductions (∼50%) in diaphragm fiber bundle force, an effect partially abolished if muscle fibers were pretreated with the reduced thiol donor N-acetyl-cysteine (a general antioxidant). Our data are in line with these findings, whereby we found no change in diaphragm mRNA expression of TNF-α, IL-6, and IL-1β post-MI, but did so in the myocardium, therefore increasing the likelihood for an increased systemic inflammation. In addition, the increased enzyme activity of NADPH oxidase and XO in the diaphragm, as well as the greater carbonylation of proteins, further supports the hypothesis that an increase in local oxidants occurred post-MI. Interestingly, in the described study (18), the levels of TNF-α were too low to induce muscle atrophy, reinforcing our conclusions that contractile dysfunction was likely the main mechanism causing muscle weakness in the present study. Circulating TNF-α may activate an increased local oxidant production via the TNF receptor subtype 1, as diaphragm fiber bundles from transgenic animals not expressing this receptor have been shown to maintain muscle function (unlike wild type) when incubated with exogenous TNF-α (14).
A further question that remains is what source may have contributed to an increased oxidant production in the diaphragm post-MI. ROS are increased in the diaphragm during CHF (9, 40), and our findings of an increased enzyme activity of NADPH and XO in the diaphragm following an infarct suggest these are at least two likely contributors. The close association between an increased activity of these enzymes and reduced diaphragm muscle function further supports that oxidants are linked to diaphragm muscle weakness post-MI (Fig. 5, C and D). Nevertheless, we cannot rule out other putative sites of ROS production, such as the mitochondria, which are known to have increased ROS production in the diaphragm of CHF rats postinfarct (40). Unfortunately, due to tissue limitation, we were unable to address this issue. Finally, we are unable to directly rule out other mechanisms that may have acted to reduce diaphragm muscle function post-MI, such as impairments to Ca2+ regulation (43, 47), redox-sensitive kinases or phosphatases (2), blood flow (39), and/or the work of breathing (3). Although CHF is known to impair Ca2+ homeostasis in the diaphragm (43, 47), this is suggested to be consequent to a reduced sensitivity at the myofilament level rather than impaired sarcoplasmic reticulum function (1). Indeed, we also found no changes in the mRNA expression of key regulatory Ca2+ proteins (e.g., RyR1, DHPR, SERCA2A). Collectively, therefore, our data and that of others support that impairments at the myofibrillar level were likely the main mechanism responsible for diaphragm muscle weakness observed 72 h post-MI.
Conclusions
These data in mice provide evidence that a MI induces diaphragm muscle weakness within 72 h of the early LV remodeling phase, which is associated with posttranslation oxidative modifications of key contractile and energetic proteins. Our findings suggest, therefore, that humans who suffer a MI may be at immediate risk of respiratory muscle dysfunction, while also providing initial evidence that diaphragm muscle weakness may be early onset in CHF.
GRANTS
T. S. Bowen is a recipient of a Postdoctoral Research Fellowship from the Alexander von Humboldt Foundation.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: T.S.B., N.M., and V.A. conception and design of research; T.S.B., S.W., S.G., Y.K., L.S., and V.A. performed experiments; T.S.B., S.W., S.G., Y.K., A.O., L.S., and V.A. analyzed data; T.S.B., N.M., A.S., T.D., A.O., A.L., L.S., G.S., and V.A. interpreted results of experiments; T.S.B., Y.K., and A.O. prepared figures; T.S.B. drafted manuscript; T.S.B., N.M., and V.A. edited and revised manuscript; T.S.B., N.M., S.W., S.G., Y.K., A.S., T.D., A.O., A.L., L.S., G.S., and V.A. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors are grateful for the excellent advice provided by Leonardo F. Ferreira, University of Florida, regarding the preparation of the diaphragm for functional measurements.
REFERENCES
- 1.Andrade FH, Reid MB, Allen DG, Westerblad H. Effect of hydrogen peroxide and dithiothreitol on contractile function of single skeletal muscle fibres from the mouse. J Physiol 509: 565–575, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andrade FH, Reid MB, Westerblad H. Contractile response of skeletal muscle to low peroxide concentrations: myofibrillar calcium sensitivity as a likely target for redox-modulation. FASEB J 15: 309–311, 2001. [DOI] [PubMed] [Google Scholar]
- 3.Barreiro E, Galdiz JB, Marinan M, Alvarez FJ, Hussain SN, Gea J. Respiratory loading intensity and diaphragm oxidative stress: N-acetyl-cysteine effects. J Appl Physiol 100: 555–563, 2006. [DOI] [PubMed] [Google Scholar]
- 4.Barreiro E, Gea J, Di Falco M, Kriazhev L, James S, Hussain SN. Protein carbonyl formation in the diaphragm. Am J Respir Cell Mol Biol 32: 9–17, 2005. [DOI] [PubMed] [Google Scholar]
- 5.Barreiro E, Hussain SN. Protein carbonylation in skeletal muscles: impact on function. Antioxid Redox Signal 12: 417–429, 2010. [DOI] [PubMed] [Google Scholar]
- 6.Bayat H, Swaney JS, Ander AN, Dalton N, Kennedy BP, Hammond HK, Roth DM. Progressive heart failure after myocardial infarction in mice. Basic Res Cardiol 97: 206–213, 2002. [DOI] [PubMed] [Google Scholar]
- 7.Clark AL, Poole-Wilson PA, Coats AJ. Exercise limitation in chronic heart failure: central role of the periphery. J Am Coll Cardiol 28: 1092–1102, 1996. [DOI] [PubMed] [Google Scholar]
- 8.Close RI. Dynamic properties of mammalian skeletal muscles. Physiol Rev 52: 129–197, 1972. [DOI] [PubMed] [Google Scholar]
- 9.Coirault C, Guellich A, Barbry T, Samuel JL, Riou B, Lecarpentier Y. Oxidative stress of myosin contributes to skeletal muscle dysfunction in rats with chronic heart failure. Am J Physiol Heart Circ Physiol 292: H1009–H1017, 2007. [DOI] [PubMed] [Google Scholar]
- 10.Empinado HM, Deevska GM, Nikolova-Karakashian M, Yoo JK, Christou DD, Ferreira LF. Diaphragm Dysfunction in heart failure is accompanied by increases in neutral sphingomyelinase activity and ceramide content. Eur J Heart Fail 16: 519–525, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferreira LF, Moylan JS, Gilliam LA, Smith JD, Nikolova-Karakashian M, Reid MB. Sphingomyelinase stimulates oxidant signaling to weaken skeletal muscle and promote fatigue. Am J Physiol Cell Physiol 299: C552–C560, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gielen S, Adams V, Mobius-Winkler S, Linke A, Erbs S, Yu J, Kempf W, Schubert A, Schuler G, Hambrecht R. Anti-inflammatory effects of exercise training in the skeletal muscle of patients with chronic heart failure. J Am Coll Cardiol 42: 861–868, 2003. [DOI] [PubMed] [Google Scholar]
- 13.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Judd SE, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Mackey RH, Magid DJ, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER 3rd, Moy CS, Mussolino ME, Neumar RW, Nichol G, Pandey DK, Paynter NP, Reeves MJ, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Wong ND, Woo D, Turner MB; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics–2014 update: a report from the American Heart Association. Circulation 129: e28–e292, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hardin BJ, Campbell KS, Smith JD, Arbogast S, Smith J, Moylan JS, Reid MB. TNF-alpha acts via TNFR1 and muscle-derived oxidants to depress myofibrillar force in murine skeletal muscle. J Appl Physiol 104: 694–699, 2008. [DOI] [PubMed] [Google Scholar]
- 15.Hoefer J, Azam MA, Kroetsch JT, Leong-Poi H, Momen MA, Voigtlaender-Bolz J, Scherer EQ, Meissner A, Bolz SS, Husain M. Sphingosine-1-phosphate-dependent activation of p38 MAPK maintains elevated peripheral resistance in heart failure through increased myogenic vasoconstriction. Circ Res 107: 923–933, 2010. [DOI] [PubMed] [Google Scholar]
- 16.Howell S, Maarek JM, Fournier M, Sullivan K, Zhan WZ, Sieck GC. Congestive heart failure: differential adaptation of the diaphragm and latissimus dorsi. J Appl Physiol 79: 389–397, 1995. [DOI] [PubMed] [Google Scholar]
- 17.Jehmlich N, Schmidt F, Taubert M, Seifert J, von Bergen M, Richnow HH, Vogt C. Comparison of methods for simultaneous identification of bacterial species and determination of metabolic activity by protein-based stable isotope probing (protein-SIP) experiments. Rapid Commun Mass Spectrom 23: 1871–1878, 2009. [DOI] [PubMed] [Google Scholar]
- 18.Li X, Moody MR, Engel D, Walker S, Clubb FJ Jr, Sivasubramanian N, Mann DL, Reid MB. Cardiac-specific overexpression of tumor necrosis factor-alpha causes oxidative stress and contractile dysfunction in mouse diaphragm. Circulation 102: 1690–1696, 2000. [DOI] [PubMed] [Google Scholar]
- 19.Linke A, Adams V, Schulze PC, Erbs S, Gielen S, Fiehn E, Mobius-Winkler S, Schubert A, Schuler G, Hambrecht R. Antioxidative effects of exercise training in patients with chronic heart failure: increase in radical scavenger enzyme activity in skeletal muscle. Circulation 111: 1763–1770, 2005. [DOI] [PubMed] [Google Scholar]
- 20.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 25: 402–408, 2001. [DOI] [PubMed] [Google Scholar]
- 21.Mancini DM, Eisen H, Kussmaul W, Mull R, Edmunds LH, Wilson JR. Value of peak exercise oxygen consumption for optimal timing of cardiac transplantation in ambulatory patients with heart failure. Circulation 83: 778–786, 1991. [DOI] [PubMed] [Google Scholar]
- 22.Mancini DM, Henson D, LaManca J, Levine S. Respiratory muscle function and dyspnea in patients with chronic congestive heart failure. Circulation 86: 909–918, 1992. [DOI] [PubMed] [Google Scholar]
- 23.Mangner N, Sandri M, Hoellriegel R, Gielen S, Linke A, Matsumoto Y, Schuler G, Adams V. Exercise training prevents TNF-α induced loss of force in the diaphragm of mice. PLos One 8: e52274, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marin-Corral J, Minguella J, Ramirez-Sarmiento AL, Hussain SN, Gea J, Barreiro E. Oxidised proteins and superoxide anion production in the diaphragm of severe COPD patients. Eur Respir J 33: 1309–1319, 2009. [DOI] [PubMed] [Google Scholar]
- 25.McCallister BD Jr, Christian TF, Gersh BJ, Gibbons RJ. Prognosis of myocardial infarctions involving more than 40% of the left ventricle after acute reperfusion therapy. Circulation 88: 1470–1475, 1993. [DOI] [PubMed] [Google Scholar]
- 26.Meyer FJ, Borst MM, Zugck C, Kirschke A, Schellberg D, Kubler W, Haass M. Respiratory muscle dysfunction in congestive heart failure: clinical correlation and prognostic significance. Circulation 103: 2153–2158, 2001. [DOI] [PubMed] [Google Scholar]
- 27.Michael LH, Ballantyne CM, Zachariah JP, Gould KE, Pocius JS, Taffet GE, Hartley CJ, Pham TT, Daniel SL, Funk E, Entman ML. Myocardial infarction and remodeling in mice: effect of reperfusion. Am J Physiol Heart Circ Physiol 277: H660–H668, 1999. [DOI] [PubMed] [Google Scholar]
- 28.Michael LH, Entman ML, Hartley CJ, Youker KA, Zhu J, Hall SR, Hawkins HK, Berens K, Ballantyne CM. Myocardial ischemia and reperfusion: a murine model. Am J Physiol Heart Circ Physiol 269: H2147–H2154, 1995. [DOI] [PubMed] [Google Scholar]
- 29.Miller TD, Christian TF, Hopfenspirger MR, Hodge DO, Gersh BJ, Gibbons RJ. Infarct size after acute myocardial infarction measured by quantitative tomographic 99mTc sestamibi imaging predicts subsequent mortality. Circulation 92: 334–341, 1995. [DOI] [PubMed] [Google Scholar]
- 30.Morbt N, Tomm J, Feltens R, Mogel I, Kalkhof S, Murugesan K, Wirth H, Vogt C, Binder H, Lehmann I, von Bergen M. Chlorinated benzenes cause concomitantly oxidative stress and induction of apoptotic markers in lung epithelial cells (A549) at nonacute toxic concentrations. J Proteome Res 10: 363–378, 2011. [DOI] [PubMed] [Google Scholar]
- 31.Myers J, Prakash M, Froelicher V, Do D, Partington S, Atwood JE. Exercise capacity and mortality among men referred for exercise testing. N Engl J Med 346: 793–801, 2002. [DOI] [PubMed] [Google Scholar]
- 32.Patten RD, Aronovitz MJ, Deras-Mejia L, Pandian NG, Hanak GG, Smith JJ, Mendelsohn ME, Konstam MA. Ventricular remodeling in a mouse model of myocardial infarction. Am J Physiol Heart Circ Physiol 274: H1812–H1820, 1998. [DOI] [PubMed] [Google Scholar]
- 33.Pfeffer MA, Pfeffer JM, Fishbein MC, Fletcher PJ, Spadaro J, Kloner RA, Braunwald E. Myocardial infarct size and ventricular function in rats. Circ Res 44: 503–512, 1979. [DOI] [PubMed] [Google Scholar]
- 34.Poon HF, Castegna A, Farr SA, Thongboonkerd V, Lynn BC, Banks WA, Morley JE, Klein JB, Butterfield DA. Quantitative proteomics analysis of specific protein expression and oxidative modification in aged senescence-accelerated-prone 8 mice brain. Neuroscience 126: 915–926, 2004. [DOI] [PubMed] [Google Scholar]
- 35.Powers SK, Wiggs MP, Sollanek KJ, Smuder AJ. Ventilator-induced diaphragm dysfunction: cause and effect. Am J Physiol Regul Integr Comp Physiol 305: R464–R477, 2013. [DOI] [PubMed] [Google Scholar]
- 36.Reid MB, Moylan JS. Beyond atrophy: redox mechanisms of muscle dysfunction in chronic inflammatory disease. J Physiol 589: 2171–2179, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shiomi T, Tsutsui H, Matsusaka H, Murakami K, Hayashidani S, Ikeuchi M, Wen J, Kubota T, Utsumi H, Takeshita A. Overexpression of glutathione peroxidase prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation 109: 544–549, 2004. [DOI] [PubMed] [Google Scholar]
- 38.Stassijns G, Gayan-Ramirez G, De Leyn P, de Bock V, Dom R, Lysens R, Decramer M. Effects of dilated cardiomyopathy on the diaphragm in the Syrian hamster. Eur Respir J 13: 391–397, 1999. [DOI] [PubMed] [Google Scholar]
- 39.Supinski G, DiMarco A, Dibner-Dunlap M. Alterations in diaphragm strength and fatiguability in congestive heart failure. J Appl Physiol 76: 2707–2713, 1994. [DOI] [PubMed] [Google Scholar]
- 40.Supinski GS, Callahan LA. Diaphragmatic free radical generation increases in an animal model of heart failure. J Appl Physiol 99: 1078–1084, 2005. [DOI] [PubMed] [Google Scholar]
- 41.Sutton MG, Sharpe N. Left ventricular remodeling after myocardial infarction: pathophysiology and therapy. Circulation 101: 2981–2988, 2000. [DOI] [PubMed] [Google Scholar]
- 42.Toth MJ, Ades PA, Tischler MD, Tracy RP, LeWinter MM. Immune activation is associated with reduced skeletal muscle mass and physical function in chronic heart failure. Int J Cardiol 109: 179–187, 2006. [DOI] [PubMed] [Google Scholar]
- 43.van Hees HW, Andrade Acuna G, Linkels M, Dekhuijzen PN, Heunks LM. Levosimendan improves calcium sensitivity of diaphragm muscle fibres from a rat model of heart failure. Br J Pharmacol 162: 566–573, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Hees HW, Li YP, Ottenheijm CA, Jin B, Pigmans CJ, Linkels M, Dekhuijzen PN, Heunks LM. Proteasome inhibition improves diaphragm function in congestive heart failure rats. Am J Physiol Lung Cell Mol Physiol 294: L1260–L1268, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Hees HW, Ottenheijm CA, Granzier HL, Dekhuijzen PN, Heunks LM. Heart failure decreases passive tension generation of rat diaphragm fibers. Int J Cardiol 141: 275–283, 2010. [DOI] [PubMed] [Google Scholar]
- 46.van Hees HW, van der Heijden HF, Hafmans T, Ennen L, Heunks LM, Verheugt FW, Dekhuijzen PN. Impaired isotonic contractility and structural abnormalities in the diaphragm of congestive heart failure rats. Int J Cardiol 128: 326–335, 2008. [DOI] [PubMed] [Google Scholar]
- 47.van Hees HW, van der Heijden HF, Ottenheijm CA, Heunks LM, Pigmans CJ, Verheugt FW, Brouwer RM, Dekhuijzen PN. Diaphragm single-fiber weakness and loss of myosin in congestive heart failure rats. Am J Physiol Heart Circ Physiol 293: H819–H828, 2007. [DOI] [PubMed] [Google Scholar]
- 48.Yang J, Noyan-Ashraf MH, Meissner A, Voigtlaender-Bolz J, Kroetsch JT, Foltz W, Jaffray D, Kapoor A, Momen A, Heximer SP, Zhang H, van Eede M, Henkelman RM, Matthews SG, Lidington D, Husain M, Bolz SS. Proximal cerebral arteries develop myogenic responsiveness in heart failure via tumor necrosis factor-alpha-dependent activation of sphingosine-1-phosphate signaling. Circulation 126: 196–206, 2012. [DOI] [PubMed] [Google Scholar]