

Abstract
Chemokines and their receptors play a crucial role in normal brain function as well as in pathological conditions such as injury and disease-associated neuroinflammation. Chemokine receptor-2 (CCR2), which mediates the recruitment of infiltrating and resident microglia to sites of central nervous system (CNS) inflammation, is upregulated by ionizing irradiation and traumatic brain injury. Our objective was to determine if a deficiency in CCR2 and subsequent effects on brain microglia affect neurogenesis and cognitive function after radiation combined injury (RCI). CCR2 knock-out (−/−) and wild-type (WT) mice received 4 Gy of whole body 137Cs irradiation. Immediately after irradiation, unilateral traumatic brain injury was induced using a controlled cortical impact system. Forty-four days postirradiation, animals were tested for hippocampus-dependent cognitive performance in the Morris water-maze. After cognitive testing, animals were euthanized and their brains snap frozen for immunohistochemical assessment of neuroinflammation (activated microglia) and neurogenesis in the hippocampal dentate gyrus. All animals were able to locate the visible and hidden platform locations in the water maze; however, treatment effects were seen when spatial memory retention was assessed in the probe trials (no platform). In WT animals that received combined injury, a significant impairment in spatial memory retention was observed in the probe trial after the first day of hidden platform training (first probe trial). This impairment was associated with increased neurogenesis in the ipsilateral hemisphere of the dentate gyrus. In contrast, CCR2−/− mice, independent of insult showed significant memory retention in the first probe trial and there were no differences in the numbers of newly born neurons in the animals receiving irradiation, trauma or combined injury. Although the mechanisms involved are not clear, our data suggests that CCR2 deficiency can exert a protective effect preventing the impairment of cognitive function after combined injury.
INTRODUCTION
Radiation exposure due to radiological terrorism, industrial accidents or military missions is a continuing threat for the civilian population (1). For example, uncontrolled radiation exposure in an urban environment will involve a wide range of doses and subsequent tissue and body effects. In addition, radiation effects will likely be exacerbated by other types of injury (trauma, burns, infection, etc.) that might occur at the time of irradiation or at some time thereafter (2). The devastating bombing of Hiroshima and Nagasaki during the Second World War and radiation accidents at Chernobyl and Goiania (1), show the importance of managing radiation combined injuries (RCI) (2). Clinical data from these incidents suggest that victims with RCI suffer worse post injury complications than patients with a single type of injury (3). Despite advances in the understanding of the pathophysiology of radiation injury (4, 5), very little information is available on approaches to treat RCI (6). Thus, a deeper insight into the mechanisms underlying the interactions between radiation and other forms of injury, particularly in the central nervous system (CNS), will be beneficial developing potential therapeutic interventions.
In the CNS, severe tissue injury generally occurs after exposure to high radiation doses (7). Exposure to low-dose radiation may not directly affect brain structure or function, but it may make the tissue more sensitive to a secondary injury (8). Traumatic brain injury is a likely consequence of a nuclear blast or a dirty bomb, and is a frequent cause of death and disability when individuals are exposed to explosive forces (9, 10). With respect to the brain, irradiation and traumatic injury are both known to induce various cognitive impairments (11, 12). Such cognitive dysfunctions are often manifest as deficits in hippocampal learning and spatial memory (13, 14). The mechanisms underlying hippocampus dependent cognitive impairments are not clear but are likely multifactorial and may involve alterations in the neurogenic cell population in the dentate gyrus (DG) or microenvironmental factors including neuroinflammation (i.e. increased numbers of activated microglia).
Chemokines have a variety of physiological functions, including immune system regulation, development and cell growth, as well as cellular migration and inflammatory regulation (15). After CNS injury, chemokine and chemokine receptor expression are upregulated within minutes to hours depending on the severity of the insult (16, 17). Chemokine receptor (CCR)-mediated signaling is thought to mediate post-injury neuroinflammation due to its ability to regulate immune cell activation (18). Chemokine receptor 2 (CCR2) is expressed in many cells including microglia and macrophages (19, 20), astrocytes (21) neuronal progenitors cells (22), and mature granular and pyramidal neurons in the hippocampus (23, 24). CCR2 is also the receptor for monocyte chemoattractant protein 1 (MCP-1), a protein associated with the accumulation/activation of monocytes at sites of injury in the CNS (25, 26). We have shown that both ionizing irradiation (11, 27) or traumatic brain injury (11, 27) increase the expression of CCR2 and activate microglia within the dentate gyrus. There are emerging data on the role of the chemokine receptor CCR2 on inflammation and on the progression of different neurodegerative conditions associated with cognitive dysfunction (28–30). Because CCR2 is upregulated by irradiation and trauma, we used CCR2 knockout mice to get an insight as to whether genetic deletion of CCR2 can affect cognitive functions after irradiation combined with traumatic brain injury.
MATERIALS AND METHODS
Animals
A total of 40 two-month-old CCR2−/− (knockout) male mice on a C57BL/6J background and 40 two-month-old C57BL/6J (WT) were obtained from Jackson Laboratories (Bar Harbor, MA). Mice were housed and cared for in compliance with the U.S. Department of Health and Human Services Guide for the Care and Use of Laboratory Animals and institutional IACUCs. In both the mutant and WT groups, animals were randomly assigned to treatment groups that received sham treatment (n = 10/genotype), radiation only (n = 10/ genotype), trauma only (n = 10/genotype) or RCI (n = 10/genotype).
Whole body irradiation was performed using a 137Cs irradiator (Gamma cell 3000; MDS Nordion Inc.). Animals were irradiated individually in a specially designed restrainer that fit into the irradiator and allowed animals to minimally move around. Dosimetry was performed using film exposure within the cesium irradiator and employing the same geometry used for the animal treatments. The film readings were calibrated against a range of doses obtained using a linear accelerator.
Traumatic Brain Injury
Immediately after irradiation, mice were anesthetized with 4% isoflurane, maintained with a non-rebreathing apparatus connected to a nose cone on the stereotaxic head frame (Kopf, Tujunga, CA). Ointment was applied to the eyes to protect vision and heads were shaved with an electric clipper. The skin was prepped with betadine solution and a midline incision was made through the scalp. A circular craniotomy, 3.5 mm in diameter was made in the left parietal skull between the bregma and lambda, 0.5 mm lateral to the midline. The skullcap was carefully removed without disruption of the dura. All mice, regardless of injury type, were subjected to this surgical procedure. Mice that were randomly selected for the trauma only (no irradiation) or RCI treatment groups were subjected to a controlled cortical impact (8, 11). The lesion was produced with a pneumatic impact device using a 3-mm-diameter convex tip, mounted 20° from the vertical to account for the curvature of the mouse skull. The contact velocity was set at 4.5 m/s with a deformation 1.0 mm below the dura and a sustained depression of 150 ms, producing a moderate lesion to the cortex without encroaching on the hippocampus. After the procedure, the scalp was sutured and each animal received a subcutaneous injection of warm physiologic saline (1 ml) to prevent dehydration. During surgery and subsequent recovery, body temperature was maintained with a circulating water heating pad.
The controlled cortical impact model of traumatic brain injury is widely preferred because it generates many of the motor and cognitive impairments seen in TBI patients (31). In the open head model, a portion of the skull is removed, and an impacting rod is driven into the dura to produce deformation of the cortex. Increasing the depth and velocity of the impact intensifies cortical cavitation as well as deficits in motor and behavioral function (32, 33). The chosen impact depth (1.0 mm) used here was based on the previous studies performed in young adult mice where a 0.50 mm deformation produced mild injury, a 1.0 mm deformation produced moderate injury and a 2.0 mm deformation produced severe injury (33).
BrdU Injection
Fourteen days after sham injury or traumatic brain injury, all mice received daily injections of BrdU (100 mg/kg) for 7 consecutive days. Four weeks after the first BrdU injection, mice underwent Morris water maze training and testing and then they were euthanized by cervical dislocation, and tissues were collected for analysis of neurogenesis (Fig. 1).
FIG. 1.
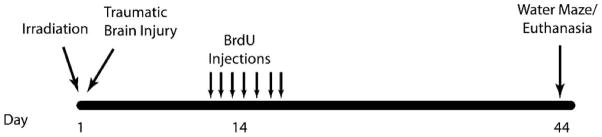
Schematic diagram showing experimental design. Two-month-old CCR2−/− and C57BL/6J male mice received whole body irradiation (4 Gy) and immediately after (~15 min) received either focal traumatic brain injury or sham injury. Two weeks later, animals were injected daily for 7 days with BrdU (100 mg/kg). Four weeks after BrdU injections, animals underwent Morris water maze testing for 5 days.
Morris Water Maze
Assessment of hippocampus-dependent cognitive performance was performed 6 weeks after irradiation using the Morris water maze test as previously described in detail (8). Briefly, a circular pool (diameter 140 cm) was filled with opaque water (24°C) and mice were trained to locate a submerged platform (luminescence: 200 lux). To determine if treatment affected the ability to swim or learn the water maze task, mice were first trained to locate a clearly marked platform (visible platform, days 1 and 2). Mice were subsequently trained to locate the platform when it was hidden beneath the surface of the opaque water (days 3–5). With the escape platform removed from the water maze (i.e., the probe trial), it was possible to determine the ability of animals to remember its previous location. The time spent searching in the target quadrant compared to the time spent in the three nontarget quadrants was a measure of spatial memory retention and probe trials were conducted 1 h after the last hidden trial of each mouse on each day of hidden platform training (i.e., three separate probe trials). Thirty minutes after the last probe trial, mice were killed by cervical dislocation and decapitated. Brains were removed quickly (within 60 s) and frozen in −70°C isopentane, transported in dry ice and stored at −80°C until sectioning.
Histological Procedures
Brain sections were taken from the medial portion of the dorsal hippocampus (anteroposterior ~2.92–4.0 mm from bregma). Tissues from 4 animals were blocked together and cryosectioned (34). Each slide contained a 20 μm thick sample from each of the experimental conditions: 0 Gy/sham treatment; 4 Gy/sham treatment; 0 Gy/trauma; 4 Gy/trauma. All slides were stored at −70°C until processed for immunocytochemical analysis.
Neurogenesis
To determine the effects of single or combined treatment on the survival of newly born cells in the dentate subgranular zone (SGZ) a double labeling protocol was used to identify newly born cells independent of phenotypes (BrdU+ only), and newly born neurons (BrdU+/NeuN+). First, the tissues were stained for BrdU. The sections were fixed for 8 min in 2% paraformaldehyde and then rinsed in Tris buffered saline (TBS) (pH 7.4). Endogenous peroxidase activity was quenched by 30 min incubation in freshly prepared 3% H2O2 solution. After 2 × 5 min washes in TBS buffer, the tissue was treated with 2N hydrochloric acid for 30 min at 37°C to denature DNA. The slides were immersed in 0.1 M Na2B4O7 to neutralize the acid, followed by eight rinses in TBS for 5 min each to return the pH to approximately 7.4. Nonspecific antigen binding was blocked with TBS containing TSA blocking reagent (PerkinElmer life Sciences, Emeryville, CA) at ambient temperature for 30 min. Newly born cells were stained using rat anti-BrdU (1:50 Accurate Chemical & Scientific Corporation) incubated overnight at 4°C. The primary antibody was then detected by 2 h incubation with an anti-rat Red-X (Jackson ImmunoResearch). Neuronal staining was performed using an antibody for the neuron-specific nuclear protein NeuN (1:500; Millipore MAB377; Billerica, MA). The primary antibody was then detected by 2 h incubation with an anti-mouse Alexa Fluor 488 (Molecular Probes, Eugene, OR). BrdU and NeuN positive cells were double labeled in the same section.
Total Activated and Newly Born Activated Microglia
The numbers of total activated microglia was determined using an anti CD68 antibody and the number of newly born activated microglia was determined by counting cells double labeled for CD68 and BrdU. Sections were fixed for 10 min in 4% paraformaldehyde, washed with TBS and endogenous peroxidase activity was quenched in 1% H2O2 solution. Next, the sections were incubated for 30 min in TSA blocking buffer containing 3% normal rabbit serum to block nonspecific antigen binding. The sections were then incubated with rat anti-mouse CD68 antibody (1:1000, Abcam) overnight at 4°C after incubation with rabbit anti-rat IgG (1:200, Vector) for 2 h at room temperature. Staining signals were further amplified with an avidin/biotin amplification system (Vector) after Cy3 tyramide amplification (Perkin Elmer). To label newly born activated microglia (CD68+/BrdU+), sections were further washed with TBS-Tween and treated for DNA denaturation as described above. Rat anti-BrdU primary antibody (1:50, Accurate Chemical & Scientific Corp.) was applied for overnight at 4°C and detected with anti-rat FITC (Jackson ImmunoResearch Laboratories, West Grove, PA) secondary antibody. Only those cells for which the BrdU nucleus was unambiguously associated with the marker for activated microglia (CD68) were scored as positive for newly born activated microglia. The results were expressed as number of cells/mm2.
Data Analysis
Statistical analyses for the immunohistochemistry cell counts were performed using GraphPad Prism software (La Jolla, CA). Two-way ANOVA was used to test the main effects of genotype (WT or CCR2−/−) and treatment (sham, irradiation, trauma or RCI). When the interaction between genotype and treatment was significant (P < 0.05), a step-down procedure for multiple comparisons was followed with two separate one-way ANOVAs. When the overall one-way ANOVA was significant (P < 0.05), individual between-groups comparisons were performed with Newman-Keuls post-hoc test. Data for the ipsilateral and contralateral hemispheres were analyzed separately.
Statistical analyses for the behavioral data were generated using R statistical programming language. Visible and hidden water maze learning curves were analyzed using 2 mixed model repeated measures ANOVAs. Model 1 was used to assess whether each genotype learned over time. Here radiation and treatment were between group factors. Day was treated as a continuous variable. This model was fit separately to CCR2 and WT data. Model 2 was used to compare performance of each group at each day. Radiation and treatment were between group factors and day was treated as categorical to better allow for day specific comparisons among groups, and eliminate the assumption of a linear trend in performance over time. The Holm’s correction was used to control for multiple comparisons. Separate analyses were conducted for the visible and hidden platform learning curves. For analysis of performance in the water maze probe trials, one-way ANOVAs were used along with Newman-Keuls post-hoc test when appropriate. Differences were considered to be statistically significant when P < 0.05.
RESULTS
Cognitive Studies
Distance Moved
Cognitive testing using the water maze was performed 6 weeks after injury (radiation, trauma or RCI). In this test, a decrease in path length (distance) to the platform indicated an improvement in spatial learning and memory. First, the mice were trained to locate a visible platform. For both genotypes there was a decrease in the average distance moved to target as a function of training day [distance moved, effect of day WT: F(1,34) = 240.34, P < 0.001; CCR2−/−: F(1,31) = 144.59, P < 0.001; ANOVA model 1, Fig. 2]. In WT mice, there was an effect of treatment on distance moved to target [F(3,34) = 3.68; P < 0.05; ANOVA model 1]. However, Holm’s correction revealed that there were no significant differences between treatment groups on either training day. For CCR2−/− mice there was no treatment effect on distance moved to target (P = 0.13; ANOVA model 1).
FIG. 2.
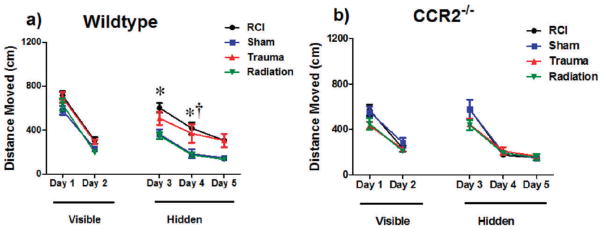
Distance moved to the target platform during visible and hidden training sessions Panel a: WT mice, all groups showed daily improvements in their abilities to locate during the hidden platform training (day 3 and 5). However, both RCI (*P < 0.05; day 3 and 4, Holms) and trauma only († P < 0.05; day 4, Holms) swam longer escape distances compared to sham mice. Panel b: There were no significant group differences in CCR2−/− mice during the visible or hidden platform training. During the visible platform training (day 1 and 2), all experimental groups swam similar distances to the platform. Each datum point represents the mean of 9–10 mice; error bars are standard error of the mean (SEM).
The mixed model repeated measures ANOVA also showed that during hidden platform training, there was a decrease in the average distance moved as a function of training for both genotypes [distance moved, effect of day WT: F(1,72) = 88.19, P < 0.001; CCR2−/−: F(1,66) = 110.10, P < 0.001, ANOVA model 1, Fig. 2]. There was an effect of trauma in WT mice [F(1,65) = 18.77; P < 0.001, ANOVA model 1], and the subsequent post-hoc analysis showed that these mice required longer distances to locate the platform compared to z on day 4 (P < 0.01; Holm’s correction). There was a genotype × trauma interaction [F(1,65) = 13.39, P < 0.001; ANOVA model 1]. Wild-type mice that received trauma moved significantly longer distances to the platform compared to CCR2−/− animals that received trauma (P < 0.01; Holm’s correction). In addition, for WT mice there was a day × radiation × trauma interaction [F(1,138) = 5.20; P < 0.05; ANOVA model 1] and subsequent post-hoc tests showed that after RCI, significantly longer swim paths were required to find the hidden platform compared to sham treated mice on day 3 and day 4 (day 3, sham vs. RCI, P < 0.01; day 4, sham vs. RCI, P < 0.01; Holm’s correction; Fig. 2b) and a trend toward significance on day 5 (day 5, sham vs. RCI, P = 0.061). In the CCR2−/− mice there were no differences in distance moved to locate the hidden platform between any of the treatment groups, suggesting that a deficiency in CCR2 modulates the adverse effects of trauma or RCI on this measure of cognitive function.
Wild-Type Probe Trials
In WT mice, significant group differences were also revealed during the probe trial (memory retention task) after the first day of hidden platform training (Fig. 3b). Sham animals and mice treated with trauma alone or irradiation alone showed spatial memory retention by spending more time in the target quadrant. In contrast, animals that received RCI did not show spatial memory retention (sham: target vs. any other quadrant, P < 0.05; trauma: target vs. any other quadrant, P < 0.05; radiation: target vs. any other quadrant, P < 0.05; RCI: target vs. any other quadrant, P > 0.05; Fig. 3b). During subsequent probe trials 2 and 3, there were no significant differences between sham mice and mice that received trauma, radiation or RCI. All of the treatment groups showed memory retention, spending more time searching in the target quadrant than in any other quadrant (not shown).
FIG. 3.
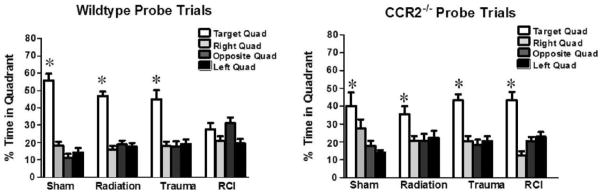
Spatial memory retention in mice during the Morris water maze probe trial after the first day of hidden platform training. Panel a: WT mice showed an impairment of hippocampal-dependent spatial memory during the day 3 probe trial. Panel b: All of the CCR2−/− mice, showed memory retention in the water maze by spending most of their time in the target quadrant which contained the hidden platform. For all 4 treatments, when time spent in the target quadrant was compared to all other quadrants there was a significant (P < 0.05) preference. Each bar represents the mean of 8–10 mice; error bars are standard error of the mean (SEM).
CCR2−/− Probe Trials
In CCR 2−/− mice, spatial memory retention was seen in the first probe trial, regardless of treatment, with mice spending more time in the target quadrant than in any other quadrant (sham: target vs. any other quadrant, P < 0.05; trauma: target vs. any other quadrant, P < 0.05; radiation: target vs. any other quadrant: P < 0.05; RCI: target vs. any other quadrant: P <0.05; Fig. 3b).
Immunohistochemistry
BrdU
Contralateral Hemisphere
The presence of BrdU+ cells 4 weeks after BrdU injection represents the long-term survival of newly generated cells, independent of phenotype. There was an interaction between genotype and injury in the contralateral hemisphere [F(3,50) = 3.79; P < 0.05; two-way ANOVA] for BrdU+ cells, suggesting that genotype influenced the magnitude of how cells responded to injury. In addition, there was an effect of genotype alone [F(3,50) = 7.04; P < 0.001; two-way ANOVA], with WT-sham animals having significantly more BrdU+ cells than CCR2−/− mice. The significant interaction in the two-way ANOVA permits further analysis of each genotype independently. A subsequent analysis of WT mice showed there was a significant injury effect on the number of BrdU+ cells [F(3,28) = 8.38; P < 0.001; one-way ANOVA; Fig. 4a]. There was an average of 179.0 ± 17.3 cells/mm2 in WT-sham animals with a minor reduction after trauma (146.3 ± 12.6 cells/mm2; Fig. 4a) and a significant decrease after irradiation only (126.1 ± 12.2 cells/mm2; P < 0.05). After RCI, the average number of BrdU+ cells (66.9 ± 12.3 cells/mm2) was significantly less than all other treatment groups (RCI vs. sham, P < 0.001; RCI vs. radiation, P < 0.05; sham vs. radiation, P < 0.05; RCI vs. trauma, P < 0.001; Fig. 4a).
FIG. 4.

Total number of BrdU+ cells per mm2 in the contralateral and ipsilateral dentate subgranular zone of the WT and CCR2−/− mice. Panel a: In the WT mice contralateral hemisphere, there was a significant group difference for BrdU+ cells (P < 0.001). Radiation significantly decreased the numbers of BrdU+ cells in irradiated and RCI mice compared to sham treated and trauma mice (RCI vs. sham, P < 0.001; RCI vs. radiation, P < 0.05; RCI vs. trauma, P < 0.001). Panel b: In CCR2−/− mice there were no significant group differences for BrdU+ cells across treatment groups. Panel c: In the WT mice ipsilateral hemisphere, there was a significant group difference for BrdU+ cells (P < 0.05). RCI significantly increased the numbers of BrdU+ cells compared all treatment groups (WT-RCI vs. WT-sham, P < 0.001; WT-RCI vs. WT-radiation, P < 0.001; WT-RCI vs. WT-trauma, P < 0.05. Panel d: In CCR2−/− mice RCI significantly decreased the number of newly born cells compared to sham (P < 0.05). Each bar represents the mean of 9–10 mice; error bars are standard error of the mean (SEM).
In contrast, in the CCR 2−/− genotype, there were no differences in the numbers of BrdU+ cells between sham animals and mice that received trauma alone or radiation alone with the numbers averaging 118.0 ± 10.9 cells/mm2, 120.8 ± 8.7 cells/mm2, 129.2 ± 15.31 cells/mm2, respectively (Fig. 4b). There was a minor but insignificant decease after RCI (103.6 ± 6.2 cells/mm2; Fig. 4b).
Ipsilateral Hemisphere
There also was an interaction between genotype and injury in the ipsilateral hemisphere for BrdU+ cells [F(3,55) = 6.58; P < 0.001; two-way ANOVA]. In addition, there was an effect of treatment only [F(1,55) = 32.7; P < 0.0001; two-way ANOVA]. WT mice that received trauma only and RCI had significantly increased numbers of newly born cells compared to CCR2−/− mice that received the same treatments (WT-trauma vs. CCR2−/− trauma, P < 0.01; WT-RCI vs. CCR2−/− RCI, P < 0.0001; two-way ANOVA). A subsequent analysis of WT mice showed there was a significant injury effect on the number of BrdU+ cells [F(3,32) = 5.4; P < 0.05; one-way ANOVA]. There was an average of 135.8 ± 12.2 cells/mm2 in sham animals with a minor increase after trauma (167.4 ± 18.7 cells/mm2; Fig. 4c). There also was a minor but insignificant decrease after radiation (122.5 + 7.1 cells/mm2) compared to sham and trauma (Fig. 4c). RCI (226.9 ± 35.11 cells/mm2) significantly increased the number of newly born cells compared to the other treatment groups (WT-RCI vs. WT-sham, P < 0.001; WT-RCI vs. WT-radiation, P < 0.001; WT-RCI vs. WT-trauma, P < 0.05; Fig. 4c).
In CCR2−/− animals, there was a significant injury effect on the number of BrdU+ cells [F(3,23) = 3.06; P < 0.05, one-way ANOVA]. There was an average of 121 ± 11.5 cells/ mm2 in sham animals, and a minor but insignificant decrease after irradiation only (92.8 ± 5.1 cells/mm2) and trauma only (93.9 ± 18.7 cells/mm2; Fig. 4d). After RCI, there was a significant decrease in the number of BrdU+ cells (87.1 ± 12.6 cells/mm2; P < 0.05, Newman-Keuls; Fig. 4d) compared to sham treated animals.
Neurogenesis
Contralateral Hemisphere
With respect to newly born neurons (BrdU+/NeuN+), the results were generally similar to what was seen for BrdU, with an interaction between genotype and injury in the contralateral hemisphere of WT mice [F(3,50) = 3.48; P < 0.05; two-way ANOVA]. However, there was no independent genotype only or treatment only effects. A subsequent analysis of WT mice showed there was a significant effect of injury on the number of BrdU+/NeuN+ cells [F(3,28) = 6.97; P < 0.001; one-way ANOVA; Fig. 5a]. There was an average of 100.0 ± 10.0 cells/mm2 in sham animals. After irradiation alone there was an insignificant decrease (76.2 ± 7.1 cells/mm2; Fig. 5a) relative to sham treatment, but a significant difference with respect to animals that received trauma alone (108.8 ± 11.3 cells/mm2; P < 0.05; Fig. 5a). Compared to sham treated mice and mice that received trauma only, there was a significant decrease in numbers of BrdU+/NeuN+ cells in mice that received RCI (44.3 ± 6.4 cells/mm2) (RCI vs. sham, P < 0.01; RCI vs. trauma, P < 0.05: Newman-Keuls; Fig. 5a).
FIG. 5.

Total number of BrdU+/NeuN+ cells in the contralateral and ipsilateral dentate subgranular zone of the WT and CCR2−/− mice. Panel a: In the WT contralateral hemisphere, there was a significant group difference for BrdU+ cells (P < 0.001). Radiation significantly decreased the numbers BrdU+/NeuN+ neurons in irradiated mice (WT-trauma vs. WT-radiation, P < 0.05) and RCI mice (WT-RCI vs. WT-sham, P < 0.01; WT-RCI vs. WT-trauma, P < 0.01). Panel b: In CCR2−/− mice there were no significant group differences for BrdU+/NeuN+ neurons across treatment groups. Panel c: In the WT ipsilateral hemisphere, there was a significant group difference for BrdU+ cells (P < 0.001). RCI significantly increased the numbers of BrdU+ cells compared all treatment groups (WT-RCI vs. WT-sham, P < 0.001; WT-RCI vs. WT-radiation, P < 0.001; WT-RCI vs. WT-trauma, P < 0.05). Panel d: In the CCR2 animal, there were no significant differences across the various treatment groups. Each bar represents the mean of 9–10 mice; error bars are standard error of the mean (SEM).
In CCR2−/− animals, there were no significant group differences [F(3,22) = 1.7; P = 0.18; one-way ANOVA] in the number newly born neurons (Fig. 5b), suggesting the deficiency in CCR2 negated any effects on single or combined injury on hippocampal neurogenesis in the contralateral hemisphere.
Ipsilateral Hemisphere
As seen in the contralateral hemisphere, there was also an interaction between genotype and injury in the ipsilateral hemisphere for BrdU+/NeuN+ cells [F(3,55) = 8.83; P < 0.001, two-way ANOVA]. In addition, there was an effect of treatment only [F(1,55) = 29.6; P < 0.0001; two-way ANOVA] and genotype only. In WT mice, trauma only and RCI significantly increased the number of newly born neurons compared to CCR2−/− (WT-trauma vs. CCR2−/− trauma, P < 0.05; WT-RCI vs. CCR2−/− RCI, P < 0.0001). There was also an effect of genotype only [F(3,55) = 4.69; P < 0.05; two-way ANOVA], with WT mice generally possessing more newly born cells. Step down analyses showed that in WT mice, there was a significant group difference for cells BrdU+/NeuN+ [F(3,28) = 10.23; P < 0.001; one-way ANOVA, Fig. 5c]. There was an average of 92.4 ± 10.6 cells/mm2 in sham mice with a minor insignificant increase after trauma only (113.7 ± 18.6 cells/mm2; Fig. 5c), and a minor but insignificant decrease after radiation (67.95 ± 4.3 cells/mm2; Fig. 5c). RCI (226.9 ± 35.11 cells/mm2) significantly increased the number of newly born neurons compared to all other treatment groups (WT-RCI vs. Sham, P < 0.001; WT-RCI vs. WT-radiation, P < 0.001; WT-RCI vs. WT-trauma, P < 0.05; Newman-Keuls). In the CCR2−/− animals, there were no significant group differences [F(3,23) = 1.76; P = 0.38] in the number newly born neurons (Fig. 5d).
Activated Total and Newly Born Microglia
Contralateral Hemisphere
For total activated microglia, there was no significant interaction between genotype and treatment in the contralateral hemisphere [F(3,58) = 090; P = 0.44; two-way ANOVA]. However, genotype alone significantly influenced the total number of activated microglia (CD68) [F(1,58) = 9.63; P < 0.05; two-way ANOVA], with CCR2−/− generally possessing more activated microglia. With respect to newly born activated microglia (Fig. 6) there was also no significant interaction between genotype and treatment [F(3,58) = 1.81; P = 0.14; two-way ANOVA]. However, genotype alone [F(1,58) = 26.22; P < 0.0001; two-way ANOVA] had an effect on newly born microglia (BrdU+/CD68+), CCR2−/− animals retained more newly born activated microglia (Table 1).
FIG. 6.
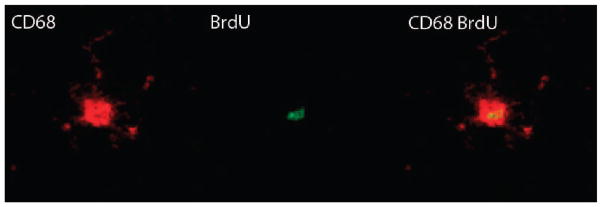
Representative image of a BrdU-positive cells that co-expressed CD68. Neuroinflammation was assessed by counting total activated microglia CD68 (red in image). Proliferating cells were labeled with BrdU (green in image). Four weeks after injections the numbers newly born activated microglia (BrdU+/CD68+) were quantified.
TABLE 1.
Total Number of Activated Microglia (CD68+) and Newly Born Microglia (BrdU+/CD68+) per mm2 in the Dentate Subgranular Zone of the WT and CCR2−/− Mice
| Genotype dose | Sham | Trauma | Radiation | RCI | |
|---|---|---|---|---|---|
|
| |||||
| 0 Gy | 4 Gy | ||||
|
|
|
||||
| CD68 | |||||
| WT | Contra | 212.6 ± 9.8 | 225.6 ± 14.2 | 208.7 ± 11.7 | 205.8 ± 14.5 |
| CCR2−/− | Contra | 234.2 ±16.7 | 243.6 ± 14.0 | 232.4 ± 9.5 | 263.7 ± 17.3 |
| WT | Ipsi | 214.2 ± 12.6 | 278.1 ± 26.7 | 241.3 ± 16.4 | 202.1 ± 13.8 |
| CCR2−/− | Ipsi | 237.6 ±16.4 | 244.6 ± 30.7 | 268.6 ± 14.8 | 270.0 ± 12.2 |
| CD68/BrdU | |||||
| WT | Contra | 18.5 ± 7.0 | 11.1 ± 5.9 | 36.6 ± 10.2 | 34.4 ± 5.6 |
| CCR2−/− | Contra | 65.7 ± 15.5 | 90.7 ± 20.3 | 52.8 ± 11.0 | 87.2 ± 19.2 |
| WT | Ipsi | 30.7 ± 12.7 | 34.5 ± 13.4 | 32.5 ± 9.8 | 61.6 ± 10.4 |
| CCR2−/− | Ipsi | 59.5 ± 9.2 | 73.5 ± 12.4 | 79.3 ± 10.9 | 109.5 ± 15.2 |
Note. Bold numbers represent significance values compared to wild-type mice.
Ipsilateral Hemisphere
As seen in the contralateral hemisphere, there was no interaction between treatment and genotype [F(3,59) = 2.46; P = 0.07; two-way ANOVA] for total activated microglia. There were also no independent genotype only [F(1,58) = 2.69; P = 0.10; two-way ANOVA] or treatment only [F(3,58) = 1.55; P = 0.20; two-way ANOVA] effects. For newly born activated microglia, there was no interaction between genotype and injury [F(3,58) = 0.23; P = 0.87; two-way ANOVA]. Independently, treatment only [F(1,58) = 4.01; P < 0.05; two-way ANOVA] and genotype only [F(3,58) = 21.54; P < 0.001; two-way ANOVA, Fig. 7] affected newly born microglia. CCR2−/− possessed more newly born activated microglia and RCI increased in the numbers of newly born microglia in both genotypes.
FIG. 7.
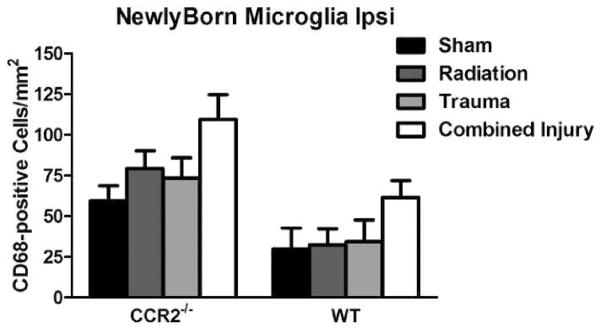
Total number of activated microglia (CD68+) and newly born microglia (BrdU+/CD68+) per mm2 in the dentate subgranular zone of the WT and CCR2−/− mice. Both treatment (P < 0.05) and genotype (P < 0.001) effected newly born microglia in the ipsilateral hemisphere. CCR2−/− had more newly born activated microglia compared to WT. RCI increased in the numbers of newly born microglia in WT and CCR2−/− mice. Each bar represents the mean of 9–10 mice; error bars are standard error of the mean (SEM).
DISCUSSION
The results of this study showed that RCI impaired spatial memory retention in WT mice. In contrast, RCI did not affect spatial memory retention in CCR2−/− mice. Cognitive impairment in WT mice was associated with an increase in the number of BrdU-labeled cells and neurogenesis in the ipsilateral DG, but not in the CCR2−/− mice when compared to their respective sham controls. There were more total activated microglia and newly born activated microglia in the CCR2−/− mice. RCI increased the number of newly born activated microglia in WT and CCR2−/− mice. Although the mechanism behind these finding in not yet clear, our data suggests that a CCR2 deficiency can exert a protective effect preventing the impairment of cognitive function after combined injury.
Chemokines control inflammation by functioning as chemoattractants that recruit inflammatory cells to the appropriate extravascular sites of inflammation (35). Among the most thoroughly characterized chemokines are the monocyte chemoattractant proteins (MCPs). MCP-1 binds primarily to the G-protein-coupled receptor CCR2 (36). In the CNS, CCR2 is expressed by neural progenitors and by mature granular and pyramidal neurons of the hippocampus and could therefore modulate both neurogenesis and neuronal function in the hippocampus (22, 23). The expression patterns of CCR2 are altered in neuropathological conditions including multiple sclerosis, HIV encephalopathy, Alzheimer’s disease and epilepsy and absence of CCR2 has also been shown to protect against insults like cerebral ischemia/reperfusion injury (29, 37, 38). Deficiency or inhibition of CCR2, results in a marked reduction in the macrophage recruitment from the periphery to lesions in the CNS (39). Given the reported role of CCR2 in the pathogenesis of a variety of diseases or insults that can induce cognitive sequelae, we used CCR2−/− mice to get an insight as to whether deficiency in CCR2 can affect neurocognitive functioning following combined radiation injury.
Hippocampus-dependent cognitive impairments were assessed using a water maze involving a probe trial at the end of each day of multiple hidden platform training sessions (40). Our data show that trauma and RCI affected hippocampal-dependent learning. In addition, a significant impairment was also observed after the first probe trial in WT mice that received RCI, but the impairment was not present in CCR2−/− mice. Overall these data suggest that CCR2 deficiency is able to prevent the hippocampal-dependent cognitive impairments induced by a combined radiation-traumatic brain injury situation.
In humans and animals, cognitive changes after irradiation alone or trauma alone often involve changes in hippocampus-dependent learning and spatial information processing (11, 13, 27, 41–44). While the mechanisms responsible for such changes remain elusive, they are likely multifactorial and may involve the loss of mature neurons in the DG (41), genetic risk factors (45), alterations in NMDA subunits (46) and alterations in the neurogenic cell populations in the DG (27, 43, 47). Within the hippocampus, the subgranular zone of the DG harbors neural stem/ progenitor cells that continuously divide and give birth to new cells that differentiate into neurons in the adult brain (48). Increased neurogenesis results in improved performance in hippocampal-dependent memory tasks (49), while disruption of hippocampal neurogenesis can result in decreased performance in hippocampal-dependent tasks. Data from our laboratory have shown that cells in the neurogenic zone of the hippocampal DG are extremely sensitive to low doses of radiation, (47, 50–52) and that such changes are associated with hippocampal dependent cognitive impairments (27, 43). In addition, data from rodent models have shown that focal traumatic brain injury also induces significant changes in neurogenesis that can be associated with altered cognitive outcome (53–55).
Our present data show that in WT mice there was a decrease in neurogenesis in the ipsilateral hemisphere after irradiation only. These results are in general agreement with our previous published data and show a reduction in the number of newly generated neurons after irradiation (43). However, in the current study the decrease in neurogenesis was not as large as seen after doses generally associated with cognitive impairment (27, 43). In contrast, neurogenesis was increased in the ipsilateral hemisphere of animals that received trauma only and those animals displayed no cognitive deficits. The increase in neurogenesis might be an adaptive response that contributes to the recovery of learning and memory after trauma (56). However, in those animals showing significant cognitive impairments (RCI) the numbers of newly born neurons were significantly higher than those seen in sham controls and animals that received irradiation only. It is possible that the significant increase in the absolute numbers of newly born neurons is unable to adequately compensate for the factors responsible for cognitive impairment.
A number of studies suggest that inflammation may contribute to alterations in neurogenesis and cognitive function after CNS irradiation or trauma (43, 47, 57, 58). One of the hallmarks of inflammation is the activation of microglia (59). In an un-injured brain, microglia monitor the microenvironment to ensure that homeostasis is maintained (60, 61). After injury, activated microglia play an important role in the phagocytosis of dead cells, and sustained microglial activation contributes to the chronic inflammatory state in the brain (62). In the present study, we quantified the numbers of total activated microglia (CD68+ only) and the numbers of newly born activated microglia (BrdU+/CD68+). The data show a significant genotype effect (P < 0.0001) with more newly born activated microglia in the CCR2−/− mice (Fig. 7). However, none of the CCR2−/− mice displayed any cognitive deficits suggesting that these newly born cells may have a dual role in the current injury paradigm. Microglia, depending on their functional phenotype can be either detrimental or supportive for neurogenesis in the injured brain (61, 63). Classically activated microglia (M1) are cytotoxic due to the secretion of reactive oxygen species (ROS) and proinflammatory cytokines. In contrast, alternatively activated microglia (M2) release high levels of anti-inflammatory cytokines and neurotrophic factors that can promote repair processes such as angiogenesis and extracellular matrix remodeling (61, 64). Due to technical limitations it was not possible to determine the phenotype of the newly born activated microglia in the current study. Further work is warranted to delineate how CCR2 deficiency impacts the developmental phenotypes of these cells and how those changes may influence the effects of radiation and RCI on cognitive performance.
In conclusion, the data from the current show that radiation combined injury impaired spatial memory acquisition and consolidation in WT mice. CCR2 deficiency exerts a protective effect preventing the impairment of cognitive function after combined injury. Additional experiments are necessary to understand how CCR2 deficiency impacts various mediators of neuroinflammation and whether those changes influence the effects of RCI on cognition and cellular/molecular markers of learning and memory.
Acknowledgments
The research described was supported by award number K12GM081266 from the National Institute of General Medical Sciences/National Institutes of Health and National Institutes of Health (NIH) grant R33 AI080531.
References
- 1.Chin FK. Scenario of a dirty bomb in an urban environment and acute management of radiation poisoning and injuries. Singapore Med J. 2007;48(10):950–7. [PubMed] [Google Scholar]
- 2.DiCarlo AL, Hatchett RJ, Kaminski JM, Ledney GD, Pellmar TC, Okunieff P, et al. Medical countermeasures for radiation combined injury: radiation with burn, blast, trauma and/or sepsis. Radiat Res; Report of an NIAID Workshop; March 26–27, 2007; 2008. pp. 712–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Manthous CA, Jackson WL., Jr The 9–11 Commission’s invitation to imagine: a pathophysiology-based approach to critical care of nuclear explosion victims. Critical Care Med. 2007;35(3):716–23. doi: 10.1097/01.CCM.0000257328.31668.22. [DOI] [PubMed] [Google Scholar]
- 4.Chao NJ. Accidental or intentional exposure to ionizing radiation: biodosimetry and treatment options. Experimental Hematol. 2007;35(4 Suppl 1):24–7. doi: 10.1016/j.exphem.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 5.Marazziti D, Baroni S, Catena-Dell’Osso M, Schiavi E, Ceresoli D, Conversano C, et al. Cognitive, psychological and psychiatric effects of ionizing radiation exposure. Current Med Chem. 2012;19(12):1864–9. doi: 10.2174/092986712800099776. [DOI] [PubMed] [Google Scholar]
- 6.Jacob A, Shah KG, Wu R, Wang P. Ghrelin as a novel therapy for radiation combined injury. Molec Med. 2010;16(3–4):137–43. doi: 10.2119/molmed.2009.00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tofilon PJ, Fike JR. The radioresponse of the central nervous system: a dynamic process. Radiat Res. 2000;153(4):357–70. doi: 10.1667/0033-7587(2000)153[0357:trotcn]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 8.Rosi S, Belarbi K, Ferguson RA, Fishman K, Obenaus A, Raber J, et al. Trauma-induced alterations in cognition and Arc expression are reduced by previous exposure to 56Fe irradiation. Hippocampus. 2012;22(3):544–54. doi: 10.1002/hipo.20920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taber KH, Warden DL, Hurley RA. Blast-related traumatic brain injury: what is known. J Neuropsychiatry Clin Neurosci. 2006;18(2):141–5. doi: 10.1176/jnp.2006.18.2.141. [DOI] [PubMed] [Google Scholar]
- 10.Warden D. Military TBI during the Iraq and Afghanistan wars. J Head Trauma Rehabilitation. 2006;21(5):398–402. doi: 10.1097/00001199-200609000-00004. [DOI] [PubMed] [Google Scholar]
- 11.Rola R, Mizumatsu S, Otsuka S, Morhardt DR, Noble-Haeusslein LJ, Fishman K, et al. Alterations in hippocampal neurogenesis following traumatic brain injury in mice. Experimental Neurology. 2006;202(1):189–99. doi: 10.1016/j.expneurol.2006.05.034. [DOI] [PubMed] [Google Scholar]
- 12.Raber J. Unintended effects of cranial irradiation on cognitive function. Toxicol Pathol. 2010;38(1):198–202. doi: 10.1177/0192623309352003. [DOI] [PubMed] [Google Scholar]
- 13.Abayomi OK. Pathogenesis of irradiation-induced cognitive dysfunction. Acta Oncologica. 1996;35(6):659–63. doi: 10.3109/02841869609083995. [DOI] [PubMed] [Google Scholar]
- 14.Crossen JR, Garwood D, Glatstein E, Neuwelt EA. Neurobehavioral sequelae of cranial irradiation in adults: a review of radiation-induced encephalopathy. J Clin Oncol. 1994;12(3):627–42. doi: 10.1200/JCO.1994.12.3.627. [DOI] [PubMed] [Google Scholar]
- 15.Hesselgesser J, Horuk R. Chemokine and chemokine receptor expression in the central nervous system. J Neurovirology. 1999;5(1):13–26. doi: 10.3109/13550289909029741. [DOI] [PubMed] [Google Scholar]
- 16.Kadhim HJ, Duchateau J, Sebire G. Cytokines and brain injury: invited review. J Intensive Care Med. 2008;23(4):236–49. doi: 10.1177/0885066608318458. [DOI] [PubMed] [Google Scholar]
- 17.Jaerve A, Muller HW. Chemokines in CNS injury and repair. Cell Tissue Res. 2012;349(1):229–48. doi: 10.1007/s00441-012-1427-3. [DOI] [PubMed] [Google Scholar]
- 18.Moser B, Wolf M, Walz A, Loetscher P. Chemokines: multiple levels of leukocyte migration control. Trends Immunol. 2004;25(2):75–84. doi: 10.1016/j.it.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 19.Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res. 2009;29(6):313–26. doi: 10.1089/jir.2008.0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, et al. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nature Med. 2007;13(4):432–8. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- 21.Smits HA, Rijsmus A, van Loon JH, Wat JW, Verhoef J, Boven LA, et al. Amyloid-beta-induced chemokine production in primary human macrophages and astrocytes. J Neuroimmunol. 2002;127:160–8. doi: 10.1016/s0165-5728(02)00112-1. [DOI] [PubMed] [Google Scholar]
- 22.Tran PB, Banisadr G, Ren D, Chenn A, Miller RJ. Chemokine receptor expression by neural progenitor cells in neurogenic regions of mouse brain. J Comparative Neurol. 2007;500(6):1007–33. doi: 10.1002/cne.21229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Banisadr G, Queraud-Lesaux F, Boutterin MC, Pelaprat D, Zalc B, Rostene W, et al. Distribution, cellular localization and functional role of CCR2 chemokine receptors in adult rat brain. J Neurochem. 2002;81(2):257–69. doi: 10.1046/j.1471-4159.2002.00809.x. [DOI] [PubMed] [Google Scholar]
- 24.van der Meer P, Ulrich AM, Gonzalez-Scarano F, Lavi E. Immunohistochemical analysis of CCR2, CCR3, CCR5, and CXCR4 in the human brain: potential mechanisms for HIV dementia. Experimental Molecular Pathology. 2000;69(3):192–201. doi: 10.1006/exmp.2000.2336. [DOI] [PubMed] [Google Scholar]
- 25.Ransohoff RM, Liu L, Cardona AE. Chemokines and chemokine receptors: multipurpose players in neuroinflammation. Int Rev Neurobiol. 2007;82:187–204. doi: 10.1016/S0074-7742(07)82010-1. [DOI] [PubMed] [Google Scholar]
- 26.Ma M, Wei T, Boring L, Charo IF, Ransohoff RM, Jakeman LB. Monocyte recruitment and myelin removal are delayed following spinal cord injury in mice with CCR2 chemokine receptor deletion. J Neurosci Res. 2002;68(6):691–702. doi: 10.1002/jnr.10269. [DOI] [PubMed] [Google Scholar]
- 27.Raber J, Rola R, LeFevour A, Morhardt D, Curley J, Mizumatsu S, et al. Radiation-induced cognitive impairments are associated with changes in indicators of hippocampal neurogenesis. Radiat Res. 2004;162(1):39–47. doi: 10.1667/rr3206. [DOI] [PubMed] [Google Scholar]
- 28.Naert G, Rivest S. CC chemokine receptor 2 deficiency aggravates cognitive impairments and amyloid pathology in a transgenic mouse model of Alzheimer’s disease. J Neurosci. 31(16):6208–20. doi: 10.1523/JNEUROSCI.0299-11.2011. 011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Semple BD, Kossmann T, Morganti-Kossmann MC. Role of chemokines in CNS health and pathology: a focus on the CCL2/ CCR2 and CXCL8/CXCR2 networks. J Cerebral Blood Flow Metabolism. 2010;30(3):459–73. doi: 10.1038/jcbfm.2009.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Belarbi K, Jopson T, Arellano C, Fike JR, Rosi S. CCR2 Deficiency Prevents Neuronal Dysfunction and Cognitive Impairments Induced by Cranial Irradiation. Cancer Res. 2013;73:1201–10. doi: 10.1158/0008-5472.CAN-12-2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brody DL, Mac Donald C, Kessens CC, Yuede C, Parsadanian M, Spinner M, et al. Electromagnetic controlled cortical impact device for precise, graded experimental traumatic brain injury. J Neurotrauma. 2007;24(4):657–73. doi: 10.1089/neu.2006.0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saatman KE, Feeko KJ, Pape RL, Raghupathi R. Differential behavioral and histopathological responses to graded cortical impact injury in mice. J Neurotrauma. 2006;23(8):1241–53. doi: 10.1089/neu.2006.23.1241. [DOI] [PubMed] [Google Scholar]
- 33.Yu S, Kaneko Y, Bae E, Stahl CE, Wang Y, van Loveren H, et al. Severity of controlled cortical impact traumatic brain injury in rats and mice dictates degree of behavioral deficits. Brain Res. 2009;1287:157–63. doi: 10.1016/j.brainres.2009.06.067. [DOI] [PubMed] [Google Scholar]
- 34.Rosi S, Ramirez-Amaya V, Vazdarjanova A, Worley PF, Barnes CA, Wenk GL. Neuroinflammation alters the hippocampal pattern of behaviorally induced Arc expression. J Neurosci. 2005;25(3):723–31. doi: 10.1523/JNEUROSCI.4469-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bennett LD, Fox JM, Signoret N. Mechanisms regulating chemokine receptor activity. Immunology. 2011;134(3):246–56. doi: 10.1111/j.1365-2567.2011.03485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dzenko KA, Song L, Ge S, Kuziel WA, Pachter JS. CCR2 expression by brain microvascular endothelial cells is critical for macrophage transendothelial migration in response to CCL2. Microvascular Res. 2005;70(1–2):53–64. doi: 10.1016/j.mvr.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 37.Dragic T, Litwin V, Allaway GP, Martin SR, Huang Y, Nagashima KA, et al. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature. 1996;381(6584):667–73. doi: 10.1038/381667a0. [DOI] [PubMed] [Google Scholar]
- 38.Semple BD, Bye N, Rancan M, Ziebell JM, Morganti-Kossmann MC. Role of CCL2 (MCP-1) in traumatic brain injury (TBI): evidence from severe TBI patients and CCL2−/− mice. J Cerebral Blood Flow Metabolism. 2010;30(4):769–82. doi: 10.1038/jcbfm.2009.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goser S, Ottl R, Brodner A, Dengler TJ, Torzewski J, Egashira K, et al. Critical role for monocyte chemoattractant protein-1 and macrophage inflammatory protein-1alpha in induction of experimental autoimmune myocarditis and effective anti-monocyte chemoattractant protein-1 gene therapy. Circulation. 2005;112(22):3400–7. doi: 10.1161/CIRCULATIONAHA.105.572396. [DOI] [PubMed] [Google Scholar]
- 40.Raber J, Villasana L, Rosenberg J, Zou Y, Huang TT, Fike JR. Irradiation enhances hippocampus-dependent cognition in mice deficient in extracellular superoxide dismutase. Hippocampus. 2011;21(1):72–80. doi: 10.1002/hipo.20724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fan Y, Liu Z, Weinstein PR, Fike JR, Liu J. Environmental enrichment enhances neurogenesis and improves functional outcome after cranial irradiation. European J Neurosci. 2007;25(1):38–46. doi: 10.1111/j.1460-9568.2006.05269.x. [DOI] [PubMed] [Google Scholar]
- 42.Raber J, Fan Y, Matsumori Y, Liu Z, Weinstein PR, Fike JR, et al. Irradiation attenuates neurogenesis and exacerbates ischemia-induced deficits. Annals Neurology. 2004;55(3):381–9. doi: 10.1002/ana.10853. [DOI] [PubMed] [Google Scholar]
- 43.Rola R, Raber J, Rizk A, Otsuka S, VandenBerg SR, Morhardt DR, et al. Radiation-induced impairment of hippocampal neuro-genesis is associated with cognitive deficits in young mice. Exp Neurol. 2004;188(2):316–30. doi: 10.1016/j.expneurol.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 44.Madsen TM, Kristjansen PE, Bolwig TG, Wortwein G. Arrested neuronal proliferation and impaired hippocampal function following fractionated brain irradiation in the adult rat. Neuroscience. 2003;119(3):635–42. doi: 10.1016/s0306-4522(03)00199-4. [DOI] [PubMed] [Google Scholar]
- 45.Villasana L, Acevedo S, Poage C, Raber J. Sex- and APOE isoform-dependent effects of radiation on cognitive function. Radiat Res. 2006;166(6):883–91. doi: 10.1667/RR0642.1. [DOI] [PubMed] [Google Scholar]
- 46.Shi L, Adams MM, Long A, Carter CC, Bennett C, Sonntag WE, et al. Spatial learning and memory deficits after whole-brain irradiation are associated with changes in NMDA receptor subunits in the hippocampus. Radiat Res. 2006;166(6):892–9. doi: 10.1667/RR0588.1. [DOI] [PubMed] [Google Scholar]
- 47.Mizumatsu S, Monje ML, Morhardt DR, Rola R, Palmer TD, Fike JR. Extreme sensitivity of adult neurogenesis to low doses of X-irradiation. Cancer Res. 2003;63(14):4021–7. [PubMed] [Google Scholar]
- 48.Gage FH. Mammalian neural stem cells. Science. 2000;287(5457):1433–8. doi: 10.1126/science.287.5457.1433. [DOI] [PubMed] [Google Scholar]
- 49.van Praag H, Kempermann G, Gage FH. Neural consequences of environmental enrichment. Nat Rev Neurosci. 2000;1(3):191–8. doi: 10.1038/35044558. [DOI] [PubMed] [Google Scholar]
- 50.Monje ML, Mizumatsu S, Fike JR, Palmer TD. Irradiation induces neural precursor-cell dysfunction. Nature Med. 2002;8(9):955–62. doi: 10.1038/nm749. [DOI] [PubMed] [Google Scholar]
- 51.Rola R, Otsuka S, Obenaus A, Nelson GA, Limoli CL, VandenBerg SR, et al. Indicators of hippocampal neurogenesis are altered by 56Fe-particle irradiation in a dose-dependent manner. Radiat Res. 2004;162(4):442–6. doi: 10.1667/rr3234. [DOI] [PubMed] [Google Scholar]
- 52.Rola R, Zou Y, Huang TT, Fishman K, Baure J, Rosi S, et al. Lack of extracellular superoxide dismutase (EC-SOD) in the microenvironment impacts radiation-induced changes in neurogenesis. Free Radical Biol Med. 2007;42(8):1133–45. doi: 10.1016/j.freeradbiomed.2007.01.020. (discussion 1–2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kleindienst A, McGinn MJ, Harvey HB, Colello RJ, Hamm RJ, Bullock MR. Enhanced hippocampal neurogenesis by intraventricular S100B infusion is associated with improved cognitive recovery after traumatic brain injury. J Neurotrauma. 2005;22(6):645–55. doi: 10.1089/neu.2005.22.645. [DOI] [PubMed] [Google Scholar]
- 54.Lu D, Qu C, Goussev A, Jiang H, Lu C, Schallert T, et al. Statins increase neurogenesis in the dentate gyrus, reduce delayed neuronal death in the hippocampal CA3 region, and improve spatial learning in rat after traumatic brain injury. J Neurotrauma. 2007;24(7):1132–46. doi: 10.1089/neu.2007.0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sun D, McGinn MJ, Zhou Z, Harvey HB, Bullock MR, Colello RJ. Anatomical integration of newly generated dentate granule neurons following traumatic brain injury in adult rats and its association to cognitive recovery. Exp Neurol. 2007;204(1):264–72. doi: 10.1016/j.expneurol.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 56.Kernie SG, Parent JM. Forebrain neurogenesis after focal Ischemic and traumatic brain injury. Neurobiol Disease. 2010;37(2):267–74. doi: 10.1016/j.nbd.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.D’Avila JC, Lam TI, Bingham D, Shi J, Won SJ, Kauppinen TM, et al. Microglial activation induced by brain trauma is suppressed by post-injury treatment with a PARP inhibitor. J Neuroinflammation. 2012;9:31. doi: 10.1186/1742-2094-9-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Monje ML, Palmer T. Radiation injury and neurogenesis. Current opinion in neurology. 2003;16(2):129–34. doi: 10.1097/01.wco.0000063772.81810.b7. [DOI] [PubMed] [Google Scholar]
- 59.Mannix RC, Whalen MJ. Traumatic brain injury, microglia, and Beta amyloid. Int J Alzheimers Dis. 2012;2012:608732. doi: 10.1155/2012/608732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stoll G, Jander S. The role of microglia and macrophages in the pathophysiology of the CNS. Prog Neurobiol. 1999;58(3):233–47. doi: 10.1016/s0301-0082(98)00083-5. [DOI] [PubMed] [Google Scholar]
- 61.Loane DJ, Byrnes KR. Role of microglia in neurotrauma. Neurotherapeutics. 2010;7(4):366–77. doi: 10.1016/j.nurt.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Joo KM, Jin J, Kang BG, Lee SJ, Kim KH, Yang H, et al. Trans-differentiation of neural stem cells: a therapeutic mechanism against the radiation induced brain damage. PloS one. 2012;7(2):e25936. doi: 10.1371/journal.pone.0025936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ekdahl CT, Kokaia Z, Lindvall O. Brain inflammation and adult neurogenesis: the dual role of microglia. Neuroscience. 2009;158(3):1021–9. doi: 10.1016/j.neuroscience.2008.06.052. [DOI] [PubMed] [Google Scholar]
- 64.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8(12):958–69. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]