

Abstract
Differential regulation of transcript stability is an effective means by which an organism can modulate gene expression. A well-characterized example is glutamine signalled degradation of specific transcripts in Aspergillus nidulans. In the case of areA, which encodes a wide-domain transcription factor mediating nitrogen metabolite repression, the signal is mediated through a highly conserved region of the 3′ UTR. Utilizing this RNA sequence we isolated RrmA, an RNA recognition motif protein. Disruption of the respective gene led to loss of both glutamine signalled transcript degradation as well as nitrate signalled stabilization of niaD mRNA. However, nitrogen starvation was shown to act independently of RrmA in stabilizing certain transcripts. RrmA was also implicated in the regulation of arginine catabolism gene expression and the oxidative stress responses at the level of mRNA stability. ΔrrmA mutants are hypersensitive to oxidative stress. This phenotype correlates with destabilization of eifE and dhsA mRNA. eifE encodes eIF5A, a translation factor within which a conserved lysine is post-translationally modified to hypusine, a process requiring DhsA. Intriguingly, for specific transcripts RrmA mediates both stabilization and destabilization and the specificity of the signals transduced is transcript dependent, suggesting it acts in consort with other factors which differ between transcripts.
Introduction
Transcript stability is fundamental to gene expression, with transcript abundance being a product of both the rate of synthesis and degradation. In particular, the ability to respond rapidly to changes in environmental stimuli is limited by the rate of mRNA decay. Consequently decay rates differ significantly between transcripts with specific functional classes having either short or long half-lives, and the stability of transcripts encoding stoichiometric complexes are often closely matched (Wang et al., 2002; Yang et al., 2003). Importantly, the stability of individual transcripts can vary significantly in response to specific stimuli (Iwai et al., 1993; Stoecklin et al., 1994; Morozov et al., 2001; Shim and Karin, 2002; Bevilacqua et al., 2003; Puig et al., 2005) and groups of transcripts can be co-ordinately regulated by specific RNA binding proteins (Parker and Song, 2004). In eukaryotes distinct pathways mediate transcript degradation, with the process being initiated by either de-capping, endonuclease cleavage or, most frequently, commencing with degradation of the 3′ poly(A) tail (Parker and Song, 2004). The rate of deadenylation differs widely between mRNAs and appears to be the rate-limiting step in many decay pathways (Tucker et al., 2001). When the poly(A) tail is reduced to a critical length, which in fungi and plants is ∼A15, transcripts are rendered susceptible to degradation either through decapping and subsequent 5′ to 3′ exonuclease activity or exosome mediated 3′ to 5′ degradation (Morozov et al., 2010a; 2012). Since the poly(A) tail is also involved in the initiation and particularly re-initiation of translation (Sachs et al., 1997), the control of deadenylation is a critical point for regulating gene expression. In Aspergillus nidulans, two active deadenylases Ccr4 and Caf1, which form a complex with Not proteins, are primarily responsible for cytoplasmic deadenylation of transcripts (Morozov et al., 2010b). Ccr4 mediates basal degradation, while Caf1 appears to be responsible for accelerated degradation of transcripts in response to specific signals. Similar processes occur in other eukaryotes. Drosophila melanogaster CAF1 has been identified as the target for an RNA binding protein, NANOS, which stimulates deadenylation (Kadyrova et al., 2007). In Saccharomyces cerevisiae, where, atypically, Caf1 appears to be catalytically inactive, it is also the target of some RNA binding proteins which stimulate RNA degradation (Goldstrohm et al., 2006; Hook et al., 2007).
One well-studied example of regulation mediated at the level of transcript stability is nitrogen metabolism in A. nidulans. Modulation of the GATA transcription factor AreA, involves the rapid degradation of the areA transcript in response to intracellular Gln levels. This is mediated through a highly conserved region of 218 nucleotides within the 3′UTR of areA mRNA (Platt et al., 1996; Morozov et al., 2000). This sequence is one of the most highly conserved non-coding sequences within Aspergilli and is sufficient to determine regulated stability in a heterologous transcript (Morozov et al., 2000). High levels of intracellular Gln trigger deadenylation of the areA transcript, which promotes de-capping and rapid 5′ to 3′-mediated decay (Morozov et al., 2000). Additionally, a large subset of genes directly involved in nitrogen metabolism is also regulated at the level of transcript stability in response to nitrogen availability (Caddick et al., 2006b). These comprise structural genes involved in nitrate utilization including a high affinity ammonium permease (MeaA) and a second GATA transcription factor, AreB (Conlon et al., 2001). Like areA, these transcripts become significantly less stable in the presence of ammonium or Gln. The number of genes involved indicates that this represents a major regulatory mechanism. Furthermore, it opens the possibility that in ascomycetes other major biological processes are also regulated at the level of transcript stability.
Surprisingly, with respect to nitrate metabolism there is another level of complexity (Caddick et al., 2006b). The transcripts encoding genes involved in nitrate assimilation, nitrate reductase (niaD) and nitrite reductase (niiA), but not the nitrate transporter structural genes (crnA and nrtB) are stabilized by the substrates for the respective enzymes, nitrate and nitrite. Nitrate stabilization overrides Gln destabilization and for niaD transcripts, both signals are mediated via the 3′UTR. Also, Gln signalled degradation and nitrate dependent stabilization are both mediated via the poly(A) tail since analysis of niaD transcript deadenylation revealed that Gln accelerates the rate of poly(A) shortening, as is the case for areA. However, intracellular nitrate inhibits the degradation of the poly(A) tail even in the presence of Gln. This divergent regulation makes good physiological sense, as it is important that nitrate and nitrite reductase are retained by the cell until the potentially toxic metabolites (nitrate and nitrite) are removed from the cell.
The regulated degradation of niaD and areA transcripts is consistent with deadenylation being the trigger for mRNA decay. In A. nidulans, when the poly(A) tail reaches a critical threshold length of A15, a 3′ pyrimidine tag is added to transcripts which promotes decapping and transcript degradation (Morozov et al., 2010a,b), mediated by the general eukaryotic mRNA degradation pathways (Parker and Song, 2004). Therefore, deadenylation, as the critical rate limiting step, is the likely target of regulation (Tucker et al., 2001). We hypothesized that this regulation will involve trans-acting factors which modulate the activity of the deadenylase, Caf1 (Morozov et al., 2010a). The aim of this study was to identify trans-acting factors involved in regulating mRNA deadenylation.
Utilizing the areA 3′UTR, which is known to mediate Gln signalled regulation of transcript stability, we conducted pull down analysis and identified an RNA binding protein, RrmA. Disruption of rrmA resulted in loss of both Gln signalled degradation of niaD and areA mRNAs and nitrate signalled stabilization of the niaD mRNA. However, a distinct response to nitrogen starvation was not affected. RrmA was already known to have a role in amino acid metabolism and we have shown that it regulates the stability of the arginine catabolism gene transcripts, agaA and otaA. The rrmA deletion mutant is highly pleiotropic. A key aspect of rrmA− strains is hypersensitivity to oxidative stress. We have shown that this correlates with the destabilization of dhsA and eifE transcripts, which encode deoxyhypusine synthase and eIF5A respectively. RrmA appears to target a diverse array of transcripts and can mediate a variety of environmental signals at the level of both stabilization and destabilization in a transcript dependent manner.
Results
(1) Identification of RrmA as an AreA mRNA binding protein
We have previously determined that the accelerated degradation of areA mRNA in response to increased intracellular Gln levels is dependent on the transcript's 3′ UTR (Platt et al., 1996; Morozov et al., 2000). One possibility is that specific proteins mediate this response. We therefore expressed the 3′ UTR in vitro and utilized this RNA element in electrophoretic mobility shift assays (EMSAs) (Fig. 1A). From this analysis it appeared that crude protein extracts led to gel retardation, indicative of protein binding to the RNA.
Figure 1.
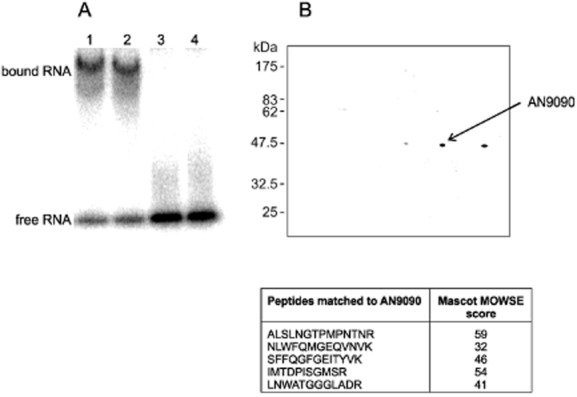
Identification of AN9090 (rrmA) among proteins bound to areA mRNA 3′ UTR.A. Electrophoretic mobility shift assay (EMSA) of the conserved 3′ sequence of areA mRNA incubated in the presence of protein extracts from mycelia grown in the presence of Gln (Lane 1) or −N (Lane 2). As controls a 100× excess of unlabelled RNA was included in the incubation mixture (Lane 3) or the radiolabelled RNA sequence without protein (Lane 4).B. 2D gel electrophoretic separation of proteins bound to areA mRNA 3′ UTR after isolation using oligo-dT immobilized on magnetic beads. The three spots in the 50 kDa range were extracted, subjected to trypsin digestion and analysed using tandem mass spectrometry. MASCOT analysis of peptides from the middle spot identified this as AN9090 from five peptides together giving 16% sequence coverage.
We therefore undertook to isolate these proteins utilizing RNA pull down. The 3′ end of areA mRNA, including a poly(A) tail, was synthesized in vitro and incubated with cell free extract. The RNA and associated proteins were then isolated utilizing oligo-dT immobilized on magnetic beads. After washing, the associated protein was eluted and analysed by gel electrophoresis. One strong band (approximately 50 kD) observed in the RNA pull down was resolved to three spots using 2D gel electrophoresis (Fig. 1B). The same products were not observed using an unrelated RNA target (data not shown).
The three protein spots were extracted from the polyacrylamide gel, subjected to in-gel trypsin digestion and analysed using tandem mass-spectrometry. One of the three samples was identified using MASCOT, with five peptides mapping to AN0909 (Fig. 1). This had previously been identified as RrmA, a putative RNA binding protein, disruption of which influenced the expression of arginine catabolism genes and led to suppression of proA1 with respect to proline auxotrophy (Olszewska et al., 2007).
(2) RrmA is critical for the Gln signalled destabilization of specific transcripts
In order to determine the function of RrmA in Gln signalled mRNA degradation, we disrupted the gene utilizing homologous integration of a deletion cassette in which the rrmA coding region was replaced with Af-pyrG. The resulting transformants were confirmed by PCR and Southern analysis and out-crossed. Utilizing a ΔrrmA strain we undertook RNA degradation studies, monitoring transcript levels over a 30 min time-course after the inhibition of transcription. Quantitative northern analysis revealed that the rapid rate of areA and niaD transcript degradation observed in the wild type in response to the addition of Gln was suppressed in the ΔrrmA strains (Fig. 2, Tables S1 and S2). This implicates RrmA in mediation of the Gln response for these two transcripts. However, with regard to a third transcript, meaA, the significant increase in stability observed under nitrogen starvation was not disrupted in the ΔrrmA strain (Fig. 2, Table S3).
Figure 2.

RrmA is required to destabilize specific transcripts in response to Gln and nitrate stabilization of niaD mRNA.A. Northern analysis of areA (encoding a global transcription factor mediating nitrogen regulation) and meaA (encoding a high-affinity ammonium transporter) in both wild type and ΔrrmA strains is shown. In order to determine RNA degradation rates under conditions of nitrogen sufficiency (Gln) or nitrogen starvation (−N), transcript levels were monitored over a 30 min time-course after the inhibition of transcription. These two nitrogen regimes were chosen since both areA and meaA transcript stability is differentially regulated under these conditions.B. Northern analysis of niaD (encoding nitrate reductase) transcript levels in rrmA+ and ΔrrmA strains. Both strains include the gpd::crnA construct which constitutively expresses CrnA, a nitrate permease, thus facilitating nitrate uptake in the presence of repressing nitrogen sources such as Gln (Caddick et al., 2006b). Transcript levels were monitored under four nitrogen regimes; glutamine sufficiency (Gln), nitrogen starvation (−N), nitrate (NO3−) or both glutamine and nitrate (Gln + NO3−).Quantitative data from multiple (≥ 3) experiments was utilized to estimate the transcript median half-life (min) under each growth condition for areA (C), meaA (D) and niaD (E). Degradation rates were monitored over a 30 min time-course after transcription was inhibited. 18S rRNA was used as a loading control. Full statistical analysis of these data is presented in Tables S1, S2 and S3. *P < 0.05; **P < 0.01; ***P < 0.001; NS non-significant (P > 0.05).
Table 1.
otaA and agaA transcript stability in ΔrrmA and rrmA+ strains
| Conditions | Strain | otaA | agaA | ||
|---|---|---|---|---|---|
| Half-life (min) | Significance | Half-life (min) | Significance | ||
| A. | |||||
| NO3→NO3 | arcAd47 | 17 | ** | 25 | * |
| ΔrrmA, arcAd47 | 26 | 16 | |||
| NO3→Gln | arcAd47 | 33 | ** | 30 | NS |
| ΔrrmA, arcAd47 | 88 | 33 | |||
| NO 3→NO3+H2O2 | arcAd47 | 23 | ** | 14 | *** |
| ΔrrmA, arcAd47 | 75 | 102 | |||
| B. | |||||
| Arg→−C | wt | 44 | NS | 92 | *** |
| ΔrrmA | 44 | 29 | |||
| Arg→−N | wt | 22 | * | 21 | NS |
| ΔrrmA | 37 | 19 | |||
Both otaA and agaA transcript stability were determined in arcAd47 (A) or wild type (B) backgrounds under conditions of nitrogen repression (Gln), oxidative stress (H2O2), nitrogen starvation (−N) or carbon starvation (−C). Either arginine (Arg) or nitrate (NO3−) were used as nitrogen sources, as indicated. The median half-life was estimated based on quantitative northern analysis of a minimum of three replicate experiments (data not shown). Full statistical analysis of these data is presented in Tables S4B, C and S5B, C. *P < 0.05; **P < 0.01; ***P < 0.001; NS non-significant (P > 0.05).
We have previously demonstrated that for niaD mRNA, in addition to being destabilized by Gln, nitrate leads to stabilization of the transcript (Caddick et al., 2006b). These regulatory effects are lost in the ΔrrmA strain, although stabilization of the niaD transcript was observed in response to nitrogen starvation (Fig. 2, S2). These data indicate that RrmA mediates both the Gln signalled destabilization and nitrate signalled stabilization of niaD mRNA, while nitrogen starvation impacts on stability via a separate mechanism. The distinct nature of nitrogen starvation, as a signal which stabilizes transcripts in an RrmA independent manner, is consistent with data for meaA transcript degradation.
(3) RrmA regulates the rate of areA mRNA deadenylation
We have shown previously that the Gln signalled destabilization of areA mRNA correlates with accelerated deadenylation of the poly(A) tail (Morozov et al., 2000). To determine whether RrmA functions upstream of this event we monitored poly(A) tail length utilizing RNase H analysis. As can be seen in Fig. , the accelerated poly(A) shortening in the presence of Gln observed in the wild type is lost in the ΔrrmA strain. This would be consistent with RrmA being involved in accelerating deadenylation via the Ccr4-Caf1-Not complex. Caf1 has previously been shown to be the key deadenylase responsible for the Gln signalled effect (Morozov et al., 2010a).
Figure 3.
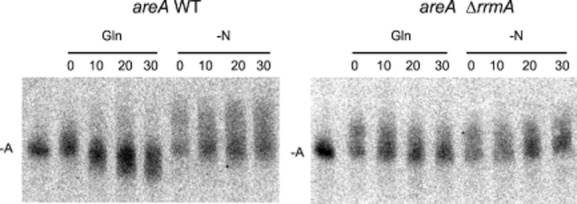
Analysis of deadenylation of areA mRNA using RNase H assay. Total RNA was subjected to RNase H analysis using oligonucleotides specific to the areA 3′ UTR (Morozov et al., 2001). Total RNA was extracted from both wild type and ΔrrmA strains over a 30 min. time-course after the inhibition of transcription. Cultures were incubated either in the absence of a nitrogen source (−N) or in the presence of Gln, as indicted. A deadenylated control was included [-A], providing a reference point (A0). Cycloheximide was added to the culture before the beginning of the time-course to prevent decapping and 5′-3′ degradation of the mRNA. The deadenylation and 3′-5′ degradation observed for the wild type in the presence of Gln is lost in the ΔrrmA strain.
(4) RrmA modulates the stability of both agaA and otaA mRNA
RrmA was originally identified via random transposon mutagenesis and selection to obtain suppressors of proline auxotrophy in a proA1 strain (Olszewska et al., 2007), proA being required for proline biosynthesis. We have confirmed that deletion of rrmA, as with the insertional mutation, results in a slower growth phenotype (Fig. 4) and suppression of proA1 with respect to proline auxotrophy (data not shown). It was previously shown that the suppression mechanism is neither allele nor gene specific as the rrmA insertional mutation suppresses not only proA1 but also proA7, proB3 and proB4 mutations. The suppression mechanism was also shown to require arginine catabolism enzymes arginase and ornithine aminotransferase (OAT) activities (Olszewska et al., 2007). Exogenous arginine is catabolized by arginase to urea and ornithine. In a transamination reaction OAT converts ornithine to glutamate-5-semialdehyde which is a proline precursor. Previous analysis suggested that suppression of proA− and proB− alleles might be due to changes in the expression of arginine catabolism genes agaA and otaA encoding arginase and OAT respectively. Additionally a homologue of RrmA from Schizosaccharomyces pombe, Csx1, has been implicated in modulating the oxidative stress response, with gene disruption leading to destabilization of atf1 mRNA, which encodes a bZIP transcription factor essential for stress responses, and decreased resistance to H2O2 (Rodriguez-Gabriel et al., 2003).
Figure 4.
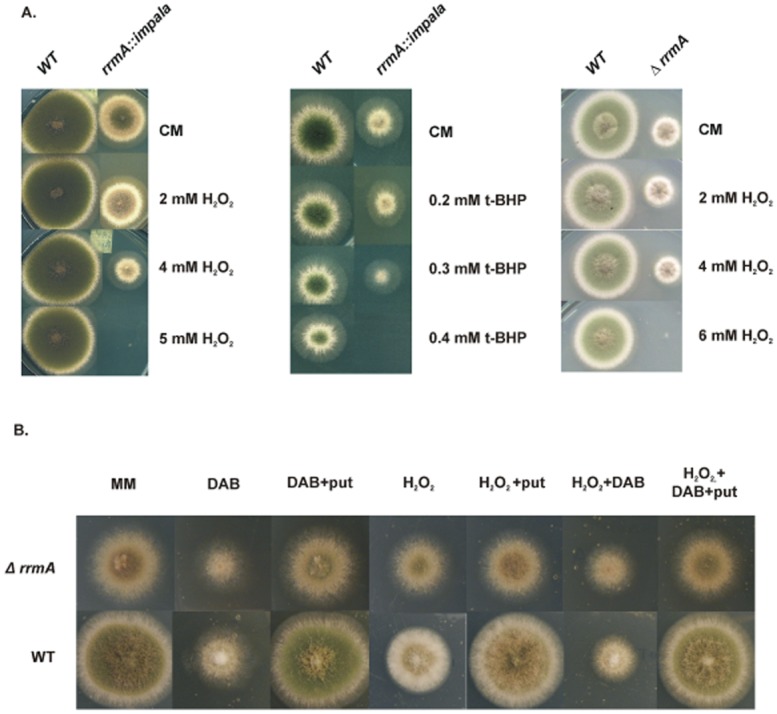
Loss-of-function mutations in rrmA result in hypersensitivity to oxidative stress.A. The rrmA insertional mutant (rrmA::impala), the deletion strain (ΔrrmA) and the wild type (WT) were grown on complete medium with H2O2or tert-butyl hydroperoxide (t-BHP). This demonstrates increased sensitivity of rrmA− mutants to oxidative stress.B. The involvement of polyamines in the oxidative stress response was shown by comparing wild type and ΔrrmA on minimal medium with H2O2, putrescine (put) and/or the ornithine decarboxylase inhibitor 1,4-diamino-2-butanone (DAB). In order to compare the response of the wild type and ΔrrmA strains to DAB and putrescine, H2O2 was used at the maximum concentrations which allowed growth; 12 mM for the wild type and 4 mM for the ΔrrmA strain.
We therefore analysed the effect of ΔrrmA on agaA and otaA transcript stability under conditions including nitrogen repression and oxidative stress. In order to induce expression of both agaA and otaA in a wild type background it is necessary to grow the fungus in the presence of arginine. Northern analysis of transcript stability in ΔrrmA and wild type strains revealed some differences in transcript stability but these were generally subtle. The strongest effect was the stabilization of otaA mRNA in the ΔrrmA strain compared with the wild type grown in the presence of arginine or after transfer to arginine and glutamine (Arg→Arg+Gln) (Table S4A). For agaA mRNA the only significant effect was a slight destabilization of the transcript with glutamine (Arg→Arg+Gln) (Table S5A). It has been shown previously that the agaA transcript binds arginine in vitro through an aptamer-like element within the 5′UTR (Borsuk et al., 2007). To test whether this direct binding of arginine influences the transcript's stability, masking the ΔrrmA phenotype, we utilized an arcAd47 strain. This bears a mutation in arcA, the pathway-specific activator for arginine catabolism, resulting in induction of the pathway in the absence of arginine (Empel et al., 2001). From these assays, clear differences were observed between rrmA+ and ΔrrmA strains for agaA and otaA transcript stability. In particular, otaA mRNA was stabilized by deletion of rrmA under all conditions that we tested, including nitrogen repression and oxidative stress (point estimates for half-life medians being 2.7 and 3.3 times higher in the mutant respectively) (Table 1A and Table S4B). Potentially this could lead to higher OAT activity which may explain the suppression of both proA− and proB− in rrmA− mutants. For agaA mRNA, more than sevenfold increase in stability was observed in the ΔrrmA strain under oxidative stress (Table 1A and Table S5B). To determine if the role of RrmA in mediating altered transcript stability in response to oxidative stress extended to other transcripts whose stability is regulated by RrmA, we monitored areA mRNA in the presence of H2O2. From this analysis there was no evidence that RrmA affected the areA transcript stability under oxidative stress (Table S7).
Finally, the effects of nitrogen and carbon starvation on both agaA and otaA mRNAs were assayed in the wild type and ΔrrmA backgrounds. From this it was shown that disruption of rrmA leads to a slight but significant stabilization of the otaA transcript under nitrogen starvation conditions (Table 1B and Table S4C), while agaA mRNA is destabilized in carbon starvation conditions (Table 1B and Table S5C).
(5). Disruption of rrmA results in hypersensitivity to oxidative stress
We used growth tests to determine whether the disruption of rrmA impacts on the phenotype during oxidative stress induced with H2O2 or tert-butyl hydroperoxide. As can be seen from Fig. 4A, disruption of rrmA leads to hypersensitivity to oxidative stress in both deletion and insertional mutants.
Based on these data, we hypothesized that RrmA may play a key role in modulating the stability of transcripts directly involved in the oxidative stress response, so we assayed the stability of three transcripts required in this process, SrrA (AN3688), the response regulator of a two component signal transduction system (Hagiwara et al., 2007) and two bZIP transcription factors, AtfA (AN2911) and NapA (AN7513) (Asano et al., 2007; Hagiwara et al., 2008). We did not observe any significant differences in mRNA stability between the rrmA deletion strain and the wild type under stress conditions (data not shown). Similarly, no difference was observed in stability of catB mRNA, encoding catalase B (AN9339). An alternative possibility was that the effect was indirect, with enzyme levels and not mRNA stability being altered. We therefore assayed the activity of catalase, superoxide dismutase (SOD) and glutathione peroxidase (GPX) under oxidative stress conditions. Analysis of these enzyme activities did not reveal any consistent significant differences between the ΔrrmA and wild type strains (data not shown).
(6). RrmA modulates stability of eIF5A and deoxyhypusine synthase mRNAs
In addition to proline biosynthesis, ornithine is utilized for the production of polyamines, including putrescine and spermidine, which are important during oxidative stress as free radical scavengers (Ha et al., 1998; Rider et al., 2007). They have also been implicated in a variety of distinct cellular processes including translation, RNA binding and chromatin modification (Igarashi and Kashiwagi, 2000). Specifically, spermidine is necessary for a post-translational modification of eIF5A, a translation factor implicated in polysome disassembly, translational repression and stress granule assembly (Li et al., 2010; Ohn and Anderson, 2010). eIF5A is the only protein known to contain hypusine, a post-translationally modified lysine. In the first step of modification, a specific lysine residue in eIF5A is converted to deoxyhypusine by deoxyhypusine synthase using spermidine as donor of a 4-amino-butyl moiety. Then in the second step, deoxyhypusine is converted to hypusine. This modification is essential for the activity of eIF5A (Park, 2006).
As rrmA is implicated in both ornithine metabolism and the oxidative stress response we undertook growth tests to determine whether polyamines are implicated in the A. nidulans oxidative stress response and if rrmA is involved. As can be seen from Fig. 4B, the ornithine decarboxylase inhibitor DAB, which prevents the conversion of ornithine to putrescine, inhibits the growth of both wild type and ΔrrmA strains and extracellular putrescine reverses this effect. In the presence of DAB and H2O2, addition of putrescine reverses the growth inhibition in both the wild type and the mutant. This confirms that in A. nidulans polyamines play an important role in the oxidative stress response, which is consistent with other organisms. However, putrescine did not reverse the sensitivity of the ΔrrmA mutant to oxidative stress, showing only a marginal effect, unlike the wild type (Fig. 4B). This suggests that the sensitivity of rrmA− strains to oxidative stress is not primarily due to reduced intracellular levels of polyamines.
To determine if the ΔrrmA mutation directly alters eIF5A or deoxyhypusine synthase expression we assayed the stability of their respective transcripts. The orthologue for eIF5A was identified as AN4015, and designated eifE. Its transcript stability under oxidative stress conditions was analysed by quantitative northern analysis (Fig. 5A and Table S6) and from these data it is clear that the eifE mRNA is significantly less stable in the ΔrrmA strain (point estimates for half-life medians three times lower in the mutant). In the case of the deoxyhypusine synthase mRNA (dhsA, AN8075), qRT-PCR was used since transcript levels were too low for northern analysis (Fig. 5B and Table S6) and stability was again significantly reduced in the ΔrrmA background (point estimates for half-life medians 1.8 times lower in the mutant). These results show that deletion of rrmA results in destabilization of both eifE and dhsA transcripts and might potentially lead to lower eIF5A activity. This may in part explain the sensitivity of the rrmA mutants to the oxidative stress.
Figure 5.
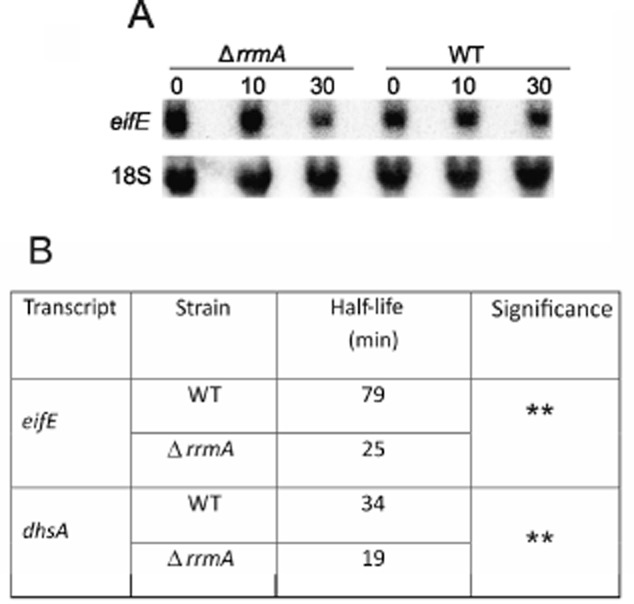
RrmA influences the stability of eifE and dhsA mRNAs under oxidative stress conditions. Stability of the eifE transcript was determined by quantitative northern analysis with 18S rRNA as a control (A). For dhsA, qPCR was utilized due to the low level of expression (data not shown). In this case tubC, which encodes β-tubulin (May and Morris, 1988), was used as the reference. Stability was monitored in both wild type (WT) and ΔrrmA strains over a 30 min time-course after transcription was inhibited. The estimated median half-life for each transcript is given (B). In both cases there is a significant difference between the two strains (P < 0.01; **). Full statistical analysis of these data is presented in Table S6.
(7). RrmA subcellular localization and role in P-body function
In order to determine the subcellular localization of RrmA we constructed a C-terminal rrmA::GFP fusion construct and transformed this into A. nidulans replacing the wild-type allele. The fusion is functional in vivo as demonstrated by the resistance of the tagged strain to oxidative stress being similar to wild type (Fig. 6A). The resulting strain was analysed using confocal fluorescence microscopy and the protein appeared to be located in the cytoplasm (Fig. 6B). Considering the apparent role of RrmA in mRNA degradation, and in particular the alteration of specific differential responses, we investigated the effect of ΔrrmA on processing (P) body formation. P-bodies are implicated in mRNA degradation, being highly dynamic RNA-protein complexes, which include translationally repressed transcripts and key factors involved in RNA decapping and degradation. To undertake these experiments we introduced the ΔrrmA allele into a strain expressing Dcp1::GFP which we have previously shown to localize to P-bodies in A. nidulans (Morozov et al., 2010b). Confocal microscopy revealed that P-bodies are still formed in the ΔrrmA strain (Fig. 6C and D). However, unlike the wild type, perturbations, including oxidative stress or altered nitrogen regime lead to relatively minor shifts in P-body number (Fig. 6E–I). Additionally, treatment with cycloheximide, which leads to rapid loss of P-bodies in the wild type showed a significantly delayed response. A similar phenotype has previously been observed in caf1− mutants, in which differential deadenylation and subsequent transcript degradation appears to be lost (Morozov et al., 2010b).The general effects on P-bodies would be consistent with RrmA playing a key role in the regulated degradation of a wide array of transcripts.
Figure 6.
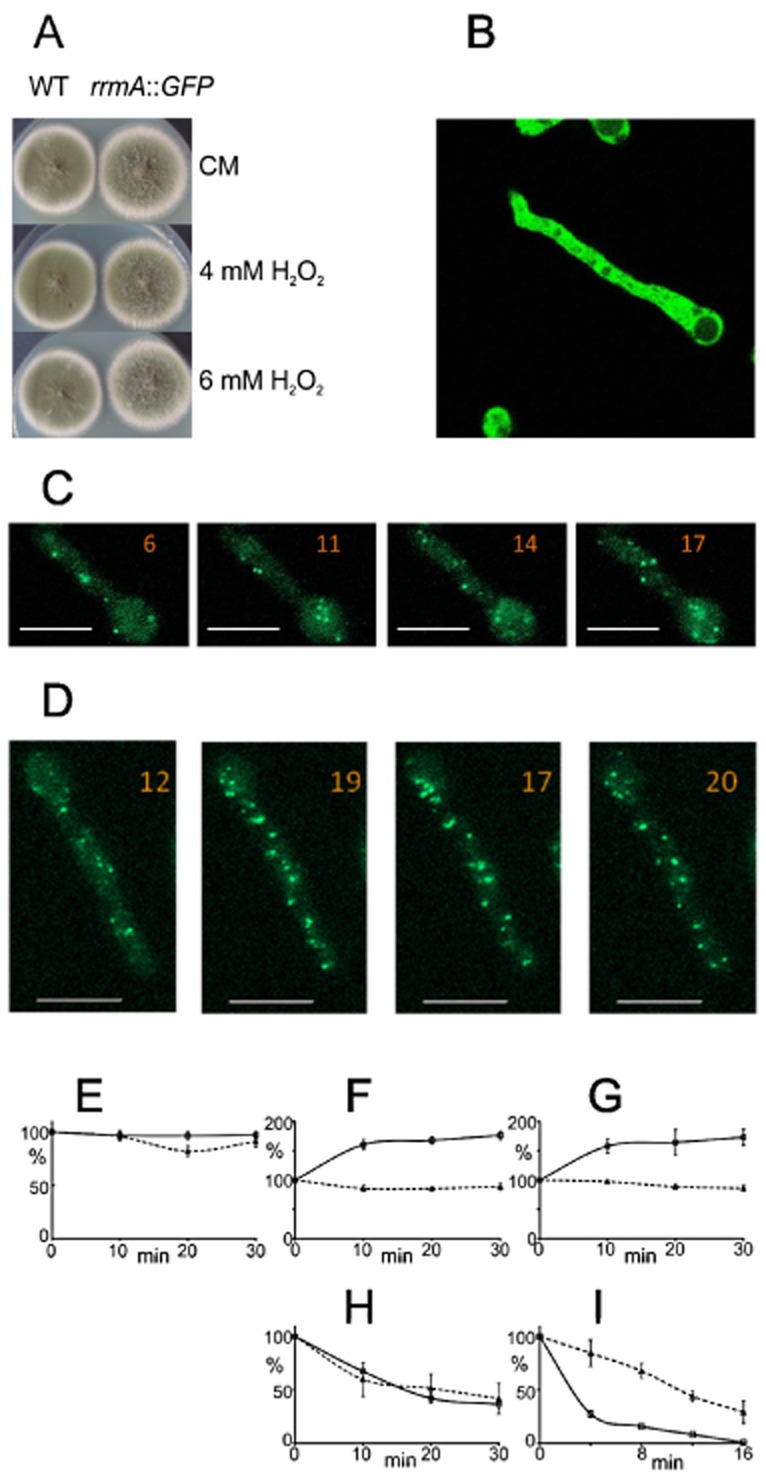
RrmA localization and role in P-body formation.A. Functionality of an RrmA::GFP fusion was assessed utilizing growth tests, with a near wild type growth phenotype and sensitivity to oxidative stress.B. Fluorescence microscopy revealed that RrmA::GFP is evenly distributed throughout the cytoplasm.C and D. P-body formation, as observed using Dpc1::GFP (Morozov et al., 2010b) in rrmA+ (C) or ΔrrmA (D) strains. In the ΔrrmA strain the P-bodies appeared normal, being evenly distributed and highly dynamic, as can be seen by comparing the four images taken at 10 min intervals. The number of P-bodies in each image is indicated and a 10 μm scale included.E–I. Quantitative analysis of the number of the P-bodies, normalized by cell volume, based on a minimum of four independent experiments with five to seven cells in each. The numbers given (± SE) were those observed in the rrmA+ (solid line) and ΔrrmA mutant (broken line) after different growth shifts. As a control, P-body number was monitored without shifting conditions (NH4+) for 30 min (E). Compared with wild type no shift in numbers was observed on transfer to carbon free media (F) or after the addition of H2O2 to the media (G) suggesting the loss of a regulatory response. On transfer from ammonium to nitrate (NH4+ to NO3−), the number of P-bodies did change but this was less dramatic than in the wild type background (H). Cycloheximide treatment which leads to loss of P-bodies, probably due to disruption of RNA decapping and degradation (Beelman and Parker, 1994), was observed and they were lost at a greatly reduced rate in the ΔrrmA strain compared with wild type (I). This would be consistent with delayed degradation of mRNA associated with P-bodies.
Discussion
The regulatory response to the level and quality of available nitrogen has been studied extensively in filamentous fungi (Platt et al., 1996; Morozov et al., 2000; Caddick et al., 2006a). The intracellular glutamine levels represent a key regulatory signal, and this is known to modulate transcript stability for a number of genes involved in nitrogen metabolism, including structural genes such as niaD and meaA as well as areA, which encodes the key transcription factor (Caddick et al., 2006b). In the case of areA mRNA, a 218 nucleotide region of the 3′ UTR, which is highly conserved between the Aspergilli orthologues, is required and sufficient for regulated transcript stability (Platt et al., 1996; Morozov et al., 2000). In order to identify proteins with the potential to regulate the stability of areA mRNA we undertook RNA pull down analysis to identify proteins which bind to the 3′ UTR of the transcript. This analysis led to the identification of RrmA, which had previously been implicated in the regulation of arginine/proline metabolism (Olszewska et al., 2007). Consistent with its role in modulating RNA stability, RrmA has three RRM domains (RNA Recognition Motif; Pfam Accession No. PF00076; SMART Accession No. SM00360) that are likely to mediate RNA binding. Disruption of RrmA leads to significant stabilization of the areA transcript, both in the presence of Gln and in nitrogen starvation conditions. However, in the case of meaA, which encodes an ammonium transporter, transcript stability was not significantly different in the ΔrrmA background compared with wild type, although differential stability was observed between nitrogen starvation and Gln sufficiency, as in the wild type. This suggests that more than one mechanism is involved in regulating transcript stability in response to nitrogen availability.
In the case of niaD, which encodes nitrate reductase, it has previously been shown that the transcript is rapidly degraded in the presence of Gln but that nitrate stabilizes the transcript and that this stabilization overrides Gln signalled destabilization (Caddick et al., 2006b). From the data presented it is apparent that both Gln signalled destabilization and nitrate signalled stabilization are lost in ΔrrmA strains. However, the stabilization in response to nitrogen starvation is still observed in the mutant and appears to be accentuated – the half-life being equivalent to the wild type in the presence of nitrate. Combining these data the simplest model is that RrmA mediates Gln signalled destabilization of specific transcripts. However, there is a separate mechanism mediating stabilization in response to nitrogen starvation which is independent of RrmA. Additionally, RrmA mediates a nitrate signal which stabilizes specific transcripts (e.g. niaD and niiA). The specificity of the nitrate signal may involve other RNA binding proteins which could interact with a limited subset of RrmA regulated transcripts. Importantly this also demonstrates that RrmA can have both a role in stabilization and destabilization, mediating distinct signals to specific transcripts.
Gln signalled transcript degradation has previously been associated with accelerated deadenylation and this is mediated primarily by Caf1, an active deadenylase which appears to effect regulated deadenylation as part of the Ccr4-Caf1-Not complex (Morozov et al., 2010a,b). Consistent with this we found that deadenylation of the areA transcript in the response to Gln signalling was lost in the rrmA deletion strain. It is therefore possible that RrmA interacts directly with Caf1, as has been shown to be the case for other RNA binding proteins that modulate transcript stability (Goldstrohm et al., 2006; Hook et al., 2007; Kadyrova et al., 2007).
RrmA was originally identified via transposon mutagenesis, being selected on the basis of suppression of proline auxotrophy in a proA1 strain and shown to suppress other proA and proB alleles (Olszewska et al., 2007). It was proposed that the basis for rrmA− suppression of these mutations may relate to overexpression of the arginine catabolism genes agaA and otaA. One possibility is that RrmA modulates the stability of certain transcripts involved in amino acid metabolism, with agaA and otaA being prime candidates. Our data confirms that under various growth regimes disruption of rrmA leads to increased stability of otaA mRNA. This could explain the mechanism of proline suppression as the elevated level of OAT should result in a higher concentration of glutamic-5-semialdehyde which is a proline precursor. However, with regards to agaA, both stabilization and destabilization were observed suggesting that RrmA mediates distinct signals at this transcript, as for niaD.
The specific regimes that displayed differential transcript stability between the wild type and the rrmA deletion strains include oxidative stress. This was of particular interest as an RrmA homologue in S. pombe, Csx1, had previously been implicated in the oxidative stress response (Rodriguez-Gabriel et al., 2003). In the presence of hydrogen peroxide both the agaA and otaA transcripts are significantly more stable in the ΔrrmA strain, although in both cases the presence of arginine masked the effect. Interestingly this role in mediating the oxidative stress response was not extended to areA mRNA, again emphasizing the transcript specificity of particular responses. As with the effect of nitrogen regime on areA, meaA and niaD, the implication is that further factors are involved and the specificity is potentially defined by the specific combination of proteins able to bind particular transcripts. Consistent with this, the oxidative stress response mediated by Csx1 in S. pombe is complex and involves at least two other RRM proteins, Cip1 and Cip2, which potentially bind the same transcripts and counter the effect of Csx1 (Martin et al., 2006). However, although structurally related RNA binding proteins from A. nidulans and S. pombe are implicated in oxidative stress response, disruption of Csx1 has a major effect on the expression of a key transcription factor, Atf1, by destabilizing its transcript. This results in csx1 mutants having a major defect in their response to oxidative stress at the transcriptional level. Csx1 is also implicated in modulation of the sexual cycle by modulating the stability of ste11 mRNA, which encodes a key transcription factor that regulates sexual development (Matia-Gonzalez et al., 2012). Intriguingly, a key regulatory signal for sexual development in S. pombe is nitrogen availability, which suggests conservation of a second function between Csx1 and RrmA. We have observed no effects on sexual development in rrmA− strains (data not shown).
The role of RrmA in the oxidative stress response was explored further and we have shown that rrmA− strains are hypersensitive to both H2O2 and tert-butyl hydroperoxide. In order to determine the primary basis for this phenotype we assayed several key transcripts and activities known to be involved in the oxidative stress response, including superoxide dismutase and catalase activity but these appeared unaltered by disruption of RrmA. The role of RrmA in the catabolism of ornithine, which is a precursor for putrescine and subsequently spermidine biosynthesis, suggested a possible link with polyamine biosynthesis, polyamines having diverse biological roles including the binding of oxygen radicals (Ha et al., 1998; Rider et al., 2007). It has been shown that modulation of expression of arginase coding genes results in change in tolerance to multiple abiotic stresses in Arabidopsis and this correlates with changes in concentration of polyamines and ROS in the cell (Shi et al., 2013). In yeast arginine synthesis from proline confers resistance to oxidative stress (Nishimura et al., 2010).
This postulated link between polyamine biosynthesis and resistance to oxidative stress in A. nidulans was supported by plate tests which show that disruption of putrescine biosynthesis with an ornithine decarboxylase inhibitor (DAB) exacerbates sensitivity to oxidative stress in wild type A. nidulans. Furthermore, this was reversible on addition of extracellular putrescine.
One specific role for spermidine is the post-translational modification of a specific Lys residue to hypusine in eIF5A. There is strong evidence that both eIF5A expression and its post-translational modification are altered in ΔrrmA strains, with both the eifE and dhsA transcripts being significantly less stable in the ΔrrmA background. eIF5A is a highly conserved translation factor involved in initiation and/or elongation (Saini et al., 2009; Henderson and Hershey, 2011). It has also been implicated as having a role in eukaryotic stress responses (Ohn and Anderson, 2010; Wang et al., 2012) and nonsense mediated decay (Kang and Hershey, 1994; Schrader et al., 2006) and as a key determinant of cell proliferation making it a key factor in pathogenicity of various diseases including cancer (Kaiser, 2012). However, from our data we cannot exclude the possibility that the effect of the ΔrrmA mutation on eifE and dhsA transcript stability is indirect, for example as a consequence of an altered concentration of a specific polyamine within the cells.
The function of RrmA in modulating specific transcripts at the level of stability would be consistent with it acting in the cytoplasm. The subcellular location of its GFP fusion shows this appears to be the case, with abundant RrmA::GFP evenly distributed. There is no obvious association with P-bodies suggesting that it does not specifically associated with translationally repressed or degrading mRNAs. This is consistent with RrmA being associated with functional transcripts and having a role in co-ordinating different signals by modulating their deadenylation. Interestingly, there appears to be a marked effect of ΔrrmA on P-bodies themselves, these being generally less responsive to specific environmental shifts, consistently with partial loss of regulation at the level of RNA stability. Additionally, the P-bodies show a delayed dissociation in response to cycloheximide treatment – both of which would be consistent with a general delay to mRNA degradation.
In conclusion, RrmA is an intriguing RNA binding protein implicated in the regulation of specific transcripts in response to distinct signals. Its role varies depending on both the signal and the transcript, implicating other factors, possibly additional RNA binding proteins, which combine to exert appropriate regulation. In this respect RrmA appears to be the equivalent of a DNA binding protein which can act at different enhancers and promoters through its interaction with co-activators and co-repressors. It will be interesting to determine the full spectrum of proteins that RrmA interacts with and the transcripts it regulates. Although our study has focused on mRNA stability, closely coupled with this is translational efficiency (Hoshino, 2012) and it is possible that RrmA may also influence this. Recent global analysis of the full spectrum of proteins associated with mammalian mRNA has revealed a diverse array of proteins (Castello et al., 2012) suggesting a level of complexity not dissimilar to that observed for transcriptional regulation and emphasizing the biological importance of the differential regulation of both mRNA stability and translational efficiency.
Experimental procedures
A. nidulans strains, genetic techniques and imaging
Aspergillus nidulans strains carried markers in standard use (Clutterbuck, 1974; 1993). Standard genetic techniques were used (Clutterbuck, 1974). Growth media were as described by Cove (1966). The ΔrrmA and rrmA:GFP strains were constructed by direct transformation of recombinant PCR constructs utilizing Aspergillus fumigatus pyrG as the selectable marker, as described by Szewczyk et al. (2006). The recipient strain was pyrG89 sE15 wA3 fwA1 chaA1 pyroA4 nirA− ΔnkuA::argB.
The A. nidulans strains employed for growth tests, transcript analysis and protein production were: (1) biA1 (wild type), (2) ΔrrmA, (3) rrmA::(impala::yA) proA6 pabaA1 ΔyA::pyr4 (4) arcAd47proA6 pabaA9 yA (5) ΔrrmA arcAd47 proA6 pyroA4 (6) gpd::crnA fwA1 biA1 (7) ΔrrmA gpd::crnA pabaA1 pyroA4 sE15.
Growth tests of rrmAΔ and WT (biA1) were performed on complete medium (CM) with different concentrations of H2O2, tert-butyl hydroperoxide (t-BHP), 1 mM putrescine and/or 1.5 mM 1,4-diamino-2-butanone (DAB), at 37°C for three days. Confocal fluorescence imaging of P-bodies utilized strains expressing Dcp1::GFP as described previously (Morozov et al., 2010b).
Preparation of protein extracts
Cells were grown overnight in fully supplemented minimal medium at 30°C in the presence of 10 mM NH4+ and transferred into fresh fully supplemented minimal medium lacking any nitrogen source (−N). After incubation for 1 h the cells were either harvested for the −N samples or 10 mM of Gln was added for 5 min to prior harvesting. Cells were washed with cold wash buffer, containing 20 mM HEPES pH 7.6, 100 mM KCl, 0.1 mM EDTA and 10 % glycerol. The mycelium was blotted dry, ground in liquid nitrogen and immediately transferred into cold wash buffer supplemented with 2 mM DTT, 0.5 mM PMSF and a protease inhibitor cocktail (Sigma, P8215), in the ratio 5 ml buffer per 1 g frozen cells. The extracts were agitated in ice for 30 min followed by two high speed centrifugation stages at 30 000 g for 1 h and 100 000 g (Ti-1284 rotor) for 90 min. The resulting S100 cell-free extracts were frozen in liquid nitrogen and stored at −80°C.
Electrophoretic mobility shift assay (EMSA)
The EMSA was performed as described previously (Baker et al., 2002). Briefly, 1 nM of gel-purified 5′ end-labelled RNA [200 nucleotide region of the conserved 3′ UTR of the areA mRNA (Morozov et al., 2000)] was incubated with 10 μg protein (according the Bradford concentration assay) from S100 extracts of mycelia grown with Gln, −N or without any protein extract. The volume of the reaction mixture was adjusted to 20 μl with the extraction buffer, and then incubated for 30 min at 37°C. The competition assay contained 100× excess (0.1 μM) of the same but unlabelled RNA. Samples were then fractionated on native 10% polyacrylamide gels and visualized using a Storm phosphorimager.
RNA pull down assay
Ten micrograms polyadenylated 3′ end areA RNA (see above) was incubated with 1 mg S100 cell free extract (Gln or nitrate) for 30 min at 37°C, as for the gel shift assay. The RNA and associated proteins were then isolated utilizing oligo-dT immobilized on magnetic beads (Dynabeads, no. 610.05) following the supplier's protocol. After washing, the associated proteins were eluted and analysed by 2D gel electrophoresis, following Bio-Rad protocols, utilizing 11 cm ReadyStrip IPG strips with linear 3–10 pH gradient followed by Laemmli 10% PAGE. Spots, after detection according to Heukeshoven and Dernick method (Heukeshoven and Dernick, 1988) were extracted from the gel and in-gel trypsin digests were performed (Shevchenko et al., 1996). The extracted peptides were used directly for MALDI-TOF mass spectrometry using a Waters Q-Tof micro coupled to an LC-Packings nano LC-MS. This was operated in the positive ion electrospray mode, and data acquisition was done using the data directed analysis mode. The data files are Waters MassLynx raw files. Local MASCOT searches were conducted using the resulting peptide mass fingerprints.
RNA isolation and transcript analysis
Growth of mycelia, transcriptional inhibition, RNA extraction, Northern blot and RNase H analysis was conducted as described previously (Platt et al., 1996; Morozov et al., 2000; Macios et al., 2012). Cultures were grown in fully supplemented minimal media for 14 h at 30°C. Appropriate nitrogen sources were added at a final concentration of 10 mM nitrate or arginine. Mycelia were harvested, washed and transferred to the same medium or to nitrogen/carbon free media and incubated for a further 2 h. To inhibit transcription, proflavin (15 μg ml−1) was added 10 min prior the time-course commencing. As indicated, glutamine (5 mM), nitrate (5 mM) or H2O2 (1 mM) were added at t0. For quantitative northern analysis, probes for specific transcripts were produced by PCR and signals were quantified using a phosphorimager (Storm). The full list of primers used is given in the supplementary data (Table S8). For dhsA transcript analysis, qRT-PCR was performed. cDNA was synthesized using High-Capacity cDNA Reverse transcription Kit (Applied Biosystems) and dhsA specific primer deox3end. qPCR was performed using SYBRGreen JumpStart Taq Ready Mix for Quantitative PCR (Sigma-Aldrich) and a LightCycler 480 (Roche). For qPCR primers deox3P and deox3L were used for dhsA, and tub1L/tub1P for a reference tubC transcript. For both northern and qRT-PCR at least three independent experiments were conducted and the data subjected to statistical analysis. RNase H analysis was performed as described previously (Caddick et al., 2006b).
Statistical analysis
For each biological experiment, point estimate of transcript half-life time (HLT) was obtained using linear regression fitted to log-transformed data by the ordinary least squares method (SAS/REG procedure; SAS Institute Inc. 2010). Then, assuming approximate log-normality of HLTs, comparisons were made by computing Satterthwaite confidence intervals for the mean difference of log-transformed HLTs (SAS/TTEST procedure; SAS Institute 2010. SAS/STAT® 9.22 User's Guide. Cary, NC: SAS Institute). This yielded also a P-value for the test of no difference. Confidence intervals for the ratio of median HLTs, to avoid over-interpretation of median HLTs, were found by applying inverse transformation to confidence limits for the mean difference of log-transformed values. Conventionally, confidence coefficient was set at 1 − α = 0.95.
Acknowledgments
The project was supported by the BBSRC (P20143 and BB/E017657/1), EU Marie Curie Early Stage Training Host Fellowship (Sensible: MEST-CT-2005–020526), the Polish Ministry of Science and Higher Education Grants (0292/B/P01/2008/34 and 1855/B/P01/2010/38) and partly by intramural grants BW175531, BW179129 and BW183114 from the Faculty of Biology, University of Warsaw, and with the support of the project financing agreements POIG.02.02.00–14-024/08-00 project (CePT). Jean Wood provided technical support and mass spectrometry was conducted by Mark Prescott. AspGD provided invaluable resources for genome analysis.
Supporting Information
Supporting Information
References
- Asano Y, Hagiwara D, Yamashino T. Mizuno T. Characterization of the bZip-type transcription factor NapA with reference to oxidative stress response in Aspergillus nidulans. Biosci Biotechnol Biochem. 2007;71:1800–1803. doi: 10.1271/bbb.70133. [DOI] [PubMed] [Google Scholar]
- Baker CS, Morozov I, Suzuki K, Romeo T. Babitzke P. CsrA regulates glycogen biosynthesis by preventing translation of glgC in Escherichia coli. Mol Microbiol. 2002;44:1599–1610. doi: 10.1046/j.1365-2958.2002.02982.x. [DOI] [PubMed] [Google Scholar]
- Beelman CA. Parker R. Differential effects of translational inhibition in cis and in trans on the decay of the unstable yeast MFA2 mRNA. J Biol Chem. 1994;269:9687–9692. [PubMed] [Google Scholar]
- Bevilacqua A, Ceriani MC, Capaccioli S. Nicolin A. Post-transcriptional regulation of gene expression by degradation of messenger RNAs. J Cell Physiol. 2003;195:356–372. doi: 10.1002/jcp.10272. [DOI] [PubMed] [Google Scholar]
- Borsuk P, Przykorska A, Blachnio K, Koper M, Pawlowicz JM, Pekala M. Weglenski P. L-arginine influences the structure and function of arginase mRNA in Aspergillus nidulans. Biol Chem. 2007;388:135–144. doi: 10.1515/BC.2007.015. [DOI] [PubMed] [Google Scholar]
- Caddick MX, Dobson C, Morozov IY. Jones MG. Gene regulation in Aspergillus: from genetics to genomics. Med Mycol. 2006a;44:S13–S16. doi: 10.1080/13693780600835781. [DOI] [PubMed] [Google Scholar]
- Caddick MX, Jones MG, van Tonder JM, Le Cordier H, Narendja F, Strauss J. Morozov IY. Opposing signals differentially regulate transcript stability in Aspergillus nidulans. Mol Microbiol. 2006b;62:509–519. doi: 10.1111/j.1365-2958.2006.05383.x. [DOI] [PubMed] [Google Scholar]
- Castello A, Fischer B, Eichelbaum K, Horos R, Beckmann BM, Strein C, et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell. 2012;149:1393–1406. doi: 10.1016/j.cell.2012.04.031. [DOI] [PubMed] [Google Scholar]
- Clutterbuck AJ. Aspergillus Nidulans. New York: Plenum Press; 1974. [Google Scholar]
- Clutterbuck AJ. Aspergillus Nidulans. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1993. [Google Scholar]
- Conlon H, Zadra I, Haas H, Arst HN, Jr, Jones MG. Caddick MX. The Aspergillus nidulans GATA transcription factor gene areB encodes at least three proteins and features three classes of mutation. Mol Microbiol. 2001;40:361–375. doi: 10.1046/j.1365-2958.2001.02399.x. [DOI] [PubMed] [Google Scholar]
- Cove DJ. The induction and repression of nitrate reductase in the fungus Aspergillus nidulans. Biochim Biophys Acta. 1966;113:51–56. doi: 10.1016/s0926-6593(66)80120-0. [DOI] [PubMed] [Google Scholar]
- Empel J, Sitkiewicz I, Andrukiewicz A, Lasocki K, Borsuk P. Weglenski P. arcA, the regulatory gene for the arginine catabolic pathway in Aspergillus nidulans. Mol Genet Genomics. 2001;266:591–597. doi: 10.1007/s004380100575. [DOI] [PubMed] [Google Scholar]
- Goldstrohm AC, Hook BA, Seay DJ. Wickens M. PUF proteins bind Pop2p to regulate messenger RNAs. Nat Struct Mol Biol. 2006;13:533–539. doi: 10.1038/nsmb1100. [DOI] [PubMed] [Google Scholar]
- Ha HC, Sirisoma NS, Kuppusamy P, Zweier JL, Woster PM. Casero RA., Jr The natural polyamine spermine functions directly as a free radical scavenger. Proc Natl Acad Sci USA. 1998;95:11140–11145. doi: 10.1073/pnas.95.19.11140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagiwara D, Asano Y, Marui J, Furukawa K, Kanamaru K, Kato M, et al. The SskA and SrrA response regulators are implicated in oxidative stress responses of hyphae and asexual spores in the phosphorelay signaling network of Aspergillus nidulans. Biosci Biotechnol Biochem. 2007;71:1003–1014. doi: 10.1271/bbb.60665. [DOI] [PubMed] [Google Scholar]
- Hagiwara D, Asano Y, Yamashino T. Mizuno T. Characterization of bZip-type transcription factor AtfA with reference to stress responses of conidia of Aspergillus nidulans. Biosci Biotechnol Biochem. 2008;72:2756–2760. doi: 10.1271/bbb.80001. [DOI] [PubMed] [Google Scholar]
- Henderson A. Hershey JW. The role of eIF5A in protein synthesis. Cell Cycle. 2011;10:3617–3618. doi: 10.4161/cc.10.21.17850. [DOI] [PubMed] [Google Scholar]
- Heukeshoven J. Dernick R. Improved silver staining procedure for fast staining in PhastSystem Development Unit. I. Staining of sodium dodecyl sulfate gels. Electrophoresis. 1988;9:28–32. doi: 10.1002/elps.1150090106. [DOI] [PubMed] [Google Scholar]
- Hook BA, Goldstrohm AC, Seay DJ. Wickens M. Two yeast PUF proteins negatively regulate a single mRNA. J Biol Chem. 2007;282:15430–15438. doi: 10.1074/jbc.M611253200. [DOI] [PubMed] [Google Scholar]
- Hoshino S. Mechanism of the initiation of mRNA decay: role of eRF3 family G proteins. Wiley Interdiscip Rev RNA. 2012;3:743–757. doi: 10.1002/wrna.1133. [DOI] [PubMed] [Google Scholar]
- Igarashi K. Kashiwagi K. Polyamines: mysterious modulators of cellular functions. Biochem Biophys Res Commun. 2000;271:559–564. doi: 10.1006/bbrc.2000.2601. [DOI] [PubMed] [Google Scholar]
- Iwai Y, Akahane K, Pluznik DH. Cohen RB. Ca2+ ionophore A23187-dependent stabilization of granulocyte-macrophage colony-stimulating factor messenger RNA in murine thymoma EL-4 cells is mediated through two distinct regions in the 3′-untranslated region. J Immunol. 1993;150:4386–4394. [PubMed] [Google Scholar]
- Kadyrova LY, Habara Y, Lee TH. Wharton RP. Translational control of maternal Cyclin B mRNA by Nanos in the Drosophila germline. Development. 2007;134:1519–1527. doi: 10.1242/dev.002212. [DOI] [PubMed] [Google Scholar]
- Kaiser A. Translational control of eIF5A in various diseases. Amino Acids. 2012;42:679–684. doi: 10.1007/s00726-011-1042-8. [DOI] [PubMed] [Google Scholar]
- Kang HA. Hershey JW. Effect of initiation factor eIF-5A depletion on protein synthesis and proliferation of Saccharomyces cerevisiae. J Biol Chem. 1994;269:3934–3940. [PubMed] [Google Scholar]
- Li CH, Ohn T, Ivanov P, Tisdale S. Anderson P. eIF5A promotes translation elongation, polysome disassembly and stress granule assembly. PLoS ONE. 2010;5:e9942. doi: 10.1371/journal.pone.0009942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macios M, Caddick MX, Weglenski P, Scazzocchio C. Dzikowska A. The GATA factors AREA and AREB together with the co-repressor NMRA, negatively regulate arginine catabolism in Aspergillus nidulans in response to nitrogen and carbon source. Fungal Genet Biol. 2012;49:189–198. doi: 10.1016/j.fgb.2012.01.004. [DOI] [PubMed] [Google Scholar]
- Martin V, Rodriguez-Gabriel MA, McDonald WH, Watt S, Yates JR, 3rd, Bahler J. Russell P. Cip1 and Cip2 are novel RNA-recognition-motif proteins that counteract Csx1 function during oxidative stress. Mol Cell Biol. 2006;17:1176–1183. doi: 10.1091/mbc.E05-09-0847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matia-Gonzalez AM, Sotelo J. Rodriguez-Gabriel MA. The RNA binding protein Csx1 promotes sexual differentiation in Schizosaccharomyces pombe. PLoS ONE. 2012;7:e30067. doi: 10.1371/journal.pone.0030067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May GS. Morris NR. Developmental regulation of a conidiation specific beta-tubulin in Aspergillus nidulans. Dev Biol. 1988;128:406–414. doi: 10.1016/0012-1606(88)90302-8. [DOI] [PubMed] [Google Scholar]
- Morozov IY, Martinez MG, Jones MG. Caddick MX. A defined sequence within the 3′ UTR of the areA transcript is sufficient to mediate nitrogen metabolite signalling via accelerated deadenylation. Mol Microbiol. 2000;37:1248–1257. doi: 10.1046/j.1365-2958.2000.02085.x. [DOI] [PubMed] [Google Scholar]
- Morozov IY, Galbis-Martinez M, Jones MG. Caddick MX. Characterization of nitrogen metabolite signalling in Aspergillus via the regulated degradation of areA mRNA. Mol Microbiol. 2001;42:269–277. doi: 10.1046/j.1365-2958.2001.02636.x. [DOI] [PubMed] [Google Scholar]
- Morozov IY, Jones MG, Razak AA, Rigden DJ. Caddick MX. CUCU modification of mRNA promotes decapping and transcript degradation in Aspergillus nidulans. Mol Cell Biol. 2010a;30:460–469. doi: 10.1128/MCB.00997-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morozov IY, Jones MG, Spiller DG, Rigden DJ, Dattenbock C, Novotny R, et al. Distinct roles for Caf1, Ccr4, Edc3 and CutA in the co-ordination of transcript deadenylation, decapping and P-body formation in Aspergillus nidulans. Mol Microbiol. 2010b;76:503–516. doi: 10.1111/j.1365-2958.2010.07118.x. [DOI] [PubMed] [Google Scholar]
- Morozov IY, Jones MG, Gould PD, Crome V, Wilson JB, Hall AJ, et al. mRNA 3′ tagging is induced by nonsense-mediated decay and promotes ribosome dissociation. Mol Cell Biol. 2012;32:2585–2595. doi: 10.1128/MCB.00316-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura A, Kotani T, Sasano Y. Takagi H. An antioxidative mechanism mediated by the yeast N-acetyltransferase Mpr1: oxidative stress-induced arginine synthesis and its physiological role. FEMS Yeast Res. 2010;10:687–698. doi: 10.1111/j.1567-1364.2010.00650.x. [DOI] [PubMed] [Google Scholar]
- Ohn T. Anderson P. The role of posttranslational modifications in the assembly of stress granules. Wiley Interdiscip Rev RNA. 2010;1:486–493. doi: 10.1002/wrna.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olszewska A, Krol K, Weglenski P. Dzikowska A. Arginine catabolism in Aspergillus nidulans is regulated by the rrmA gene coding for the RNA-binding protein. Fungal Genet Biol. 2007;44:1285–1297. doi: 10.1016/j.fgb.2007.07.001. [DOI] [PubMed] [Google Scholar]
- Park MH. The post-translational synthesis of a polyamine-derived amino acid, hypusine, in the eukaryotic translation initiation factor 5A (eIF5A) J Biochem. 2006;139:161–169. doi: 10.1093/jb/mvj034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker R. Song H. The enzymes and control of eukaryotic mRNA turnover. Nat Struct Mol Biol. 2004;11:121–127. doi: 10.1038/nsmb724. [DOI] [PubMed] [Google Scholar]
- Platt A, Langdon T, Arst HN, Jr, Kirk D, Tollervey D, Sanchez JM. Caddick MX. Nitrogen metabolite signalling involves the C-terminus and the GATA domain of the Aspergillus transcription factor AREA and the 3′ untranslated region of its mRNA. EMBO J. 1996;15:2791–2801. [PMC free article] [PubMed] [Google Scholar]
- Puig S, Askeland E. Thiele DJ. Coordinated remodeling of cellular metabolism during iron deficiency through targeted mRNA degradation. Cell. 2005;120:99–110. doi: 10.1016/j.cell.2004.11.032. [DOI] [PubMed] [Google Scholar]
- Rider JE, Hacker A, Mackintosh CA, Pegg AE, Woster PM. Casero RA., Jr Spermine and spermidine mediate protection against oxidative damage caused by hydrogen peroxide. Amino Acids. 2007;33:231–240. doi: 10.1007/s00726-007-0513-4. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Gabriel MA, Burns G, McDonald WH, Martin V, Bahler JR, Yates J., 3rd Russell P. RNA-binding protein Csx1 mediates global control of gene expression in response to oxidative stress. EMBO J. 2003;22:6256–6266. doi: 10.1093/emboj/cdg597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs AB, Sarnow P. Hentze MW. Starting at the beginning, middle, and end: translation initiation in eukaryotes. Cell. 1997;89:831–838. doi: 10.1016/s0092-8674(00)80268-8. [DOI] [PubMed] [Google Scholar]
- Saini P, Eyler DE, Green R. Dever TE. Hypusine-containing protein eIF5A promotes translation elongation. Nature. 2009;459:118–121. doi: 10.1038/nature08034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAS Institute Inc. SAS/STAT® 9.22 User's Guide. Cary, NC: SAS Institute Inc; 2010. [Google Scholar]
- Schrader R, Young C, Kozian D, Hoffmann R. Lottspeich F. Temperature-sensitive eIF5A mutant accumulates transcripts targeted to the nonsense-mediated decay pathway. J Biol Chem. 2006;281:35336–35346. doi: 10.1074/jbc.M601460200. [DOI] [PubMed] [Google Scholar]
- Shevchenko A, Wilm M, Vorm O. Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- Shi H, Ye T, Chen F, Cheng Z, Wang Y, Yang P, et al. Manipulation of arginase expression modulates abiotic stress tolerance in Arabidopsis: effect on arginine metabolism and ROS accumulation. J Exp Bot. 2013;64:1367–1379. doi: 10.1093/jxb/ers400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim J. Karin M. The control of mRNA stability in response to extracellular stimuli. Mol Cells. 2002;14:323–331. [PubMed] [Google Scholar]
- Stoecklin G, Hahn S. Moroni C. Functional hierarchy of AUUUA motifs in mediating rapid interleukin-3 mRNA decay. J Biol Chem. 1994;269:28591–28597. [PubMed] [Google Scholar]
- Szewczyk E, Nayak T, Oakley CE, Edgerton H, Xiong Y, Taheri-Talesh N, et al. Fusion PCR and gene targeting in Aspergillus nidulans. Nat Protoc. 2006;1:3111–3120. doi: 10.1038/nprot.2006.405. [DOI] [PubMed] [Google Scholar]
- Tucker M, Valencia-Sanchez MA, Staples RR, Chen J, Denis CL. Parker R. The transcription factor associated Ccr4 and Caf1 proteins are components of the major cytoplasmic mRNA deadenylase in Saccharomyces cerevisiae. Cell. 2001;104:377–386. doi: 10.1016/s0092-8674(01)00225-2. [DOI] [PubMed] [Google Scholar]
- Wang L, Xu C, Wang C. Wang Y. Characterization of a eukaryotic translation initiation factor 5A homolog from Tamarix androssowii involved in plant abiotic stress tolerance. BMC Plant Biol. 2012;12:118. doi: 10.1186/1471-2229-12-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Liu CL, Storey JD, Tibshirani RJ, Herschlag D. Brown PO. Precision and functional specificity in mRNA decay. Proc Natl Acad Sci USA. 2002;99:5860–5865. doi: 10.1073/pnas.092538799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang E, van Nimwegen E, Zavolan M, Rajewsky N, Schroeder M, Magnasco M. Darnell JE., Jr Decay rates of human mRNAs: correlation with functional characteristics and sequence attributes. Genome Res. 2003;13:1863–1872. doi: 10.1101/gr.1272403. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information