

Abstract
Methylation of flavivirus RNA is vital for its stability and translation in the infected host cell. This methylation is mediated by the flavivirus methyltransferase (MTase), which methylates the N7 and 2′-O positions of the viral RNA cap by using S-adenosyl-l-methionine (SAM) as a methyl donor. In this report, we demonstrate that SAM, in contrast to the reaction by-product S-adenosyl-l-homocysteine, which was assumed previously, is copurified with the Dengue (DNV) and West Nile virus MTases produced in Escherichia coli (E. coli). This endogenous SAM can be removed by denaturation and refolding of the MTase protein. The refolded MTase of DNV serotype 3 (DNV3) displays methylation activity comparable to native enzyme, and its crystal structure at 2.1 Å is almost identical to that of native MTase. We characterized the binding of Sinefungin (SIN), a previously described SAM-analog inhibitor of MTase function, to the native and refolded DNV3 MTase by isothermal titration calorimetry, and found that SIN binds to refolded MTase with more than 16 times the affinity of SIN binding to the MTase purified natively. Moreover, we show that SAM is also copurified with other flavivirus MTases, indicating that purification by refolding may be a generally applicable tool for studying flavivirus MTase inhibition.
Keywords: flavivirus, methyltransferase, refolding, S-adenosyl methionine
Introduction
The virus genus Flavivirus consists of many arthropod-borne viruses, some of which cause significant disease burden and mortality worldwide. Of these viruses, West Nile virus (WNV), DNV, tick-borne encephalitis virus (TBEV), Japanese encephalitis virus (JEV), and Yellow fever virus (YFV) are among the most deadly; DNV is the most prolific, resulting in ∼50–100 million cases, 500,000 severe cases, and 22,000 deaths per year.1 Despite the existence of effective vaccines for TBEV, JEV, and YFV, challenges in vaccinating large at-risk populations and difficulties in developing a vaccine against DNV's multiple serotypes2–4 highlight the importance of developing therapeutics against proteins essential to flavivirus functions.
The flavivirus genome consists of an ∼11 kb ss-RNA (+) containing one large open reading frame encoding a single polyprotein, which is cleaved during and immediately after translation by cellular and a virally encoded proteases.5 These cleavages produce the viral structural proteins: capsid, premembrane or membrane, and envelope, as well as seven nonstructural proteins (NSs) required for genome replication: NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5. The NS5 protein functions as an RNA dependent-RNA polymerase and, directly relevant to this study, a methyltransferase (MTase)/guanylyltransferase that adds a GMP cap to the newly produced viral RNA.6–9 The MTase subsequently methylates this cap at the N7 position of the Guanine and the ribose 2′-OH of the first replicated nucleotide as well as internal adenosines.7–10 The methyl donor for these reactions is S-adenosyl-l-methionine (SAM), which becomes S-adenosyl-l-homocysteine (SAH) as a byproduct of the methylation reaction.
Prior work,11,12 has identified sinefungin (SIN), an analogue of the methyl-donor SAM, as a potent inhibitor of both flavivirus MTase N7 and 2′-O function as well as viral replication. The MTase-SIN cocrystal structure11 confirmed that SIN binds in place of SAM in the SAM-binding site. In addition, SAH was assumed to be copurified with flavivirus MTases in previous structural studies,7,13–15 although the identity of the copurified compounds were never verified. In this study, we demonstrate that SAM, but not SAH, is copurified with flavivirus MTases produced in Escherichia coli (E. coli). This endogenous SAM, a potential obstacle to in vitro compound characterization, can be removed by denaturation and subsequent refolding of the MTase protein. This refolded MTase is fully functional and we subsequently measure its physical binding properties to SAM and SIN by isothermal titration calorimetry (ITC). Additionally, we solve the crystal structure of the refolded MTase at a resolution of 2.1 Å and show that it is nearly identical to that of the native DNV3 MTase, validating the use of refolded MTase for further in vitro studies.
Results
Refolding of DNV3 MTase
Flavivirus MTase has been an attractive drug target in recent years since it is essential for the virus life cycle.8,13,16,17 Because the cofactor SAM-binding site is conserved across flavivirus families, it has been heavily investigated for inhibitor development.11,12,17–22 However, to date, only one crystal structure of the MTase in complex with an SAH-derived inhibitor has been determined.20 It is generally believed that the recombinant flavivirus MTase proteins, except for that from the Modoc virus,23,24 were copurified with SAH, the by-product of the MTase reaction preoccupying the SAM-binding site.7,13–15 The pre-bound by-product may increase the difficulty for inhibitors to bind to the SAM-binding site.
To produce flavivirus MTase with an empty SAM pocket, we explored a refolding strategy. We lysed the cells expressing the DNV3 MTase with a buffer containing 8M urea, and extensively washed the denatured protein to ensure that residue bound “putative” SAH would be removed. We employed an on-column refolding strategy in which the entire MTase-bound resin was transferred to a dialysis bag and subjected to refolding by dialysis against the refolding buffer. The DNV3 MTase was successfully refolded. The purification profile of the refolded MTase was very similar to that of the MTase purified under the native condition (data not shown). As expected, the refolded protein ran identically to native protein on SDS-PAGE [ Fig. 1(A)].
Figure 1.
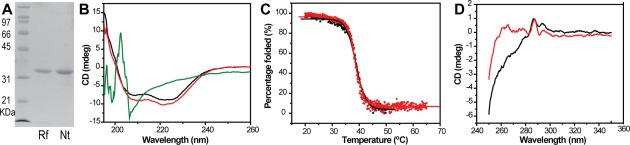
Purification and CD spectrum analysis of refolded DNV3 MTase. A: SDS-PAGE gel of the purification of refolded (Rf) and native (Nt) DNV3 MTase. B: Far UV spectrum CD of native (black line), refolded (red line), and denatured (green line) DNV3 MTase. C: Temperature dependence of molar ellipticity at 222 nm for native (black) and refolded (red) DNV3 MTase. D: Near UV Spectrum CD of native (black line) and refolded (red line) DNV3 MTase.
Circular dichroism spectra analysis of refolded DNV3 MTase
To characterize the refolded MTase and to see if it behaves similarly as the native MTase, we measured the Circular dichroism (CD) spectra for the DNV MTase purified under both native and refolding conditions (Fig. 1). Clearly the MTase under denaturing condition (in a buffer containing 8M urea) does not display any ordered secondary structures typically observed for a correctly folded protein, indicating that the denatured protein is totally unfolded [ Fig. 1(B), green]. In contrast, the shapes and amplitudes of the far-UV CD spectra for the MTase proteins purified under both native (black) and refolding (red) conditions are characteristic of proteins with a typical αβ structure [ Fig. 1(B)]. Small differences in the spectral range between 205 and 225 are observed, which could result from small differences in the loading concentrations between refolded and native MTase proteins. Consistent with this finding, we found that the secondary structural content of the refolded MTase, estimated using the CDPro suite,25 is nearly identical (within 1%) to that of native MTase, despite the differences in the measured CD spectra.
To examine the thermal stability of the refolded and native DNV3 MTase proteins, we performed thermal denaturation experiments, by monitoring secondary structural changes at 222 nm using CD spectra [ Fig. 1(C)]. The denaturation experiment indicated that the thermal denaturations of the MTase molecules purified under both conditions are irreversible processes. Using the JASCO Spectra Analysis software, we calculated thermal denaturation (melting) midpoint values (Tm) as 38.7 ± 0.03°C and 38.8 ± 0.06°C for native and refolded MTase proteins, respectively. Obviously, the Tms for native and refolded MTase proteins are statistically identical. These results imply that removal of the co-factor does not destabilize the DNV3 MTase structure.
In addition to the far-UV CD spectra, which monitor a protein's secondary structural content, we also measured the near-UV (250–350 nm) CD spectra, which can be used to monitor a protein's tertiary structure in the vicinity of the aromatic amino acid residues Tyr, Trp, and Phe.26 The near-UV CD spectrum can be sensitive to small changes in tertiary structure due to protein-protein interactions and/or changes in solvent conditions. Signal changes in the near-UV range can be attributed to alterations to Phe (250–270 nm), Tyr (270–290 nm), and Trp (280–300 nm). As shown in Figure 1(D), the near-UV CD spectra of apo-native and refolded MTase proteins are very similar from 280 to 350 nm, indicating that no overall conformational alterations to aromatic residues are associated with the cofactor removal and refolding procedure. In contrast, significant differences can be seen for the near-UV spectra between native and refolded MTase proteins. These differences may be due to the presence of SAH bound to the native protein. In the crystal structure of the flavivirus MTase proteins, the adenosine base of the copurified putative cofactor SAM (or SAH) is at a close proximity to the residues Phe133 and Tyr134 of the DNV3 MTase.7,11,13,20 Therefore, removal of the cofactor, leading to the loss of the interactions between the base and Phe133 and Tyr134, may affect the conformations of these residues, resulting in a near UV spectra different from that with the bound cofactor.
ITC analysis
Previous work has confirmed that SIN, a broad-spectrum inhibitor of MTase proteins, also inhibits flavivirus MTases by competing with the methyl-donor, S-adenosyl-l-methionine (SAM), for binding in a pocket adjacent to the active site.11 To characterize the binding of this inhibitor, we carried out ITC on DNV3 MTase purified under both native and denaturing conditions with SIN as the injectant. As shown in Figure 2, the downward projecting peaks indicate an exothermic heat of binding, in which the heat of binding for that particular addition of ligand is the area under the peak. After a number of injections, the peaks no longer decrease in magnitude because all binding sites are now saturated; these peaks of constant magnitude represent background heat from buffer addition. We observed the binding of SIN to native DNV3 MTase was exothermic (ΔH = −17.6 kcal/mol) with a KD = 2.15 µM, but to our surprise, saturated very rapidly, yielding a much lower stoichiometry of binding than expected (N = 0.08) [ Fig. 2(A)]; we predicted a stoichiometry of ∼1.0 based on the available MTase structures,7,11,13–15,23,24,27,28 which exhibit a single binding pocket per molecule of protein.
Figure 2.
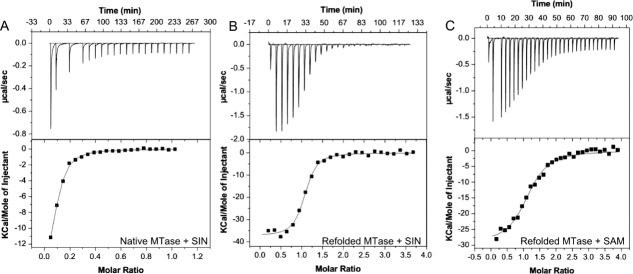
ITC Analysis of SAM or SIN binding to DNV3 MTase. Heat flow versus time (upper panels) and reaction enthalpy (lower panels) at 25°C of (A) SIN injected into ITC buffer containing native DNV3 MTase, (B) SIN injected into ITC buffer containing refolded DNV3 MTase, and (C) SAM injected into ITC buffer containing refolded DNV3 MTase. The negative peaks in the upper panels represent exothermic heat of binding at each injection of SIN or SAM.
One possible explanation for the binding stoichiometry of SIN to DNV3 MTase being much lower than predicted is a lack of available binding sites. Since the MTase methyl donor, SAM or its by-product SAH, is ubiquitous in living cells, it was formally possible that SAM or SAH endogenous to E. Coli was being copurified with DNV3 MTase entrenched within the MTase binding site, as demonstrated by a number of crystal structures of the flavivirus MTase proteins in which an SAH was present in the SAM cofactor binding site, although SAH was not added into the cell culture media during any protein production or purification steps.7,11,13–15,23,24,27,28
To determine if this was the case, we proceeded to examine the binding of SIN to the refolded DNV3 MTase by ITC. The enthalpy of binding of was exothermic and roughly twice that of the native protein (ΔH = −37.4 kcal/mol). This binding (KD = 136 nM) was stronger than that of the native protein, and unlike the native MTase, saturated over the course of more than ten injections of SIN. The stoichiometry of binding (N = 1.02) determined is in perfect agreement with the concept that the flavivirus MTase only contains one SAM binding site [ Fig. 2(B)]. We next assayed the binding of the refolded MTase to its natural methyl donor, SAM. The stoichiometry and enthalpy of binding were comparable to that of SIN, (N = 1.15, ΔH = −29.6 kcal/mol), but was approximately six times weaker (KD = 826 nM) than the inhibitor [ Fig. 2(C)] (See Table I for comparison of physical constants). These data are consistent with the finding that SIN is an effective inhibitor for the flavivirus MTase.11
Table I.
Physical Constants from ITC
| DENV3 MTase | Compound | N | KD (nM) | ΔH (KCal/mol) | ΔS cal/mol/deg |
|---|---|---|---|---|---|
| Native | SIN | 0.08 | 2150 | −17.6 | −33.2 |
| Refolded | SIN | 1.02 | 136 | −37.4 | −94.1 |
| Refolded | SAM | 1.15 | 826 | −29.6 | −71.5 |
Functional analysis of refolded DNV3 MTase
To determine whether the refolded DNV3 MTase protein functions as an MTase as expected, we performed the N7 and 2′-O methylation assays for both native and refolded MTase proteins in the presence or absence of exogenously added SAM. As expected, the native DNV3 MTase was fully active in the presence of the exogenously added SAM [ Fig. 3(A,B)]. However, to our surprise, the native DNV3 MTase was also fully active without addition of exogenous SAM [ Fig. 3(A,B)]. In contrast, the refolded DNV3 MTase's activity was reduced ∼80% in the absence of exogenous SAM; addition of SAM restored the activity of the refolded protein to native levels [ Fig. 3(A,B)]. These data demonstrated (1) that it is SAM, but not SAH, that is copurified with the DNV3 MTase as SAH will not support methylation reactions and (2) that refolded DNV3 MTase has full MTase activity in the presence of exogenously added SAM.
Figure 3.
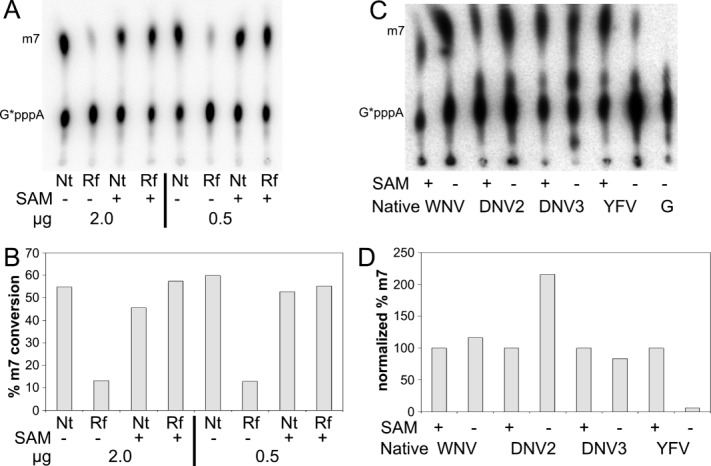
Endogenous SAM activity is present in native Flavivirus MTases. A: Methylation reactions were performed on WNV 32P-labeled G*pppA-capped RNA (0.25 µM) with the indicated amount of either native (Nt) or refolded (Rf) DNV3 MTase in the presence or absence of exogenously added SAM. The methylated RNA cap (m7) migrates higher on the TLC plate than the nonmethylated cap (G*pppA). B: Quantification of the methylation reaction in (A). C: Methylation reactions performed as in (A) using 2 µg of native MTases (3 µM) from either WNV, DNV2, DNV3, or YFV in the presence or absence of exogenously added SAM. Untreated G*pppA (G) is included for comparison. D: Quantification of the methylation reaction in (C). The percentage of conversion of G*pppA to m7G*pppA was normalized in such a way that the conversion rate for each native flavivirus MTase with exogenous SAM addition was set to 100%, and the conversion rate without exogenous SAM was normalized to the one with SAM.
Flavivirus MTases were copurified with SAM
To determine whether other flavivirus MTase proteins were copurified with SAM, we queried native MTase proteins from other flaviviruses with this assay. As shown in Figure 3(C,D), in the absence of exogenous SAM, the purified MTase enzymes from WNV and DNV serotype 2 (DNV2) were fully active. The addition of SAM did not further increase the activity of the MTase proteins from WNV, DNV2, or DNV3. These results confirmed that it was SAM, but not SAH, that was co-purified along with the flavivirus MTase enzymes. The MTase from YFV, however, only displayed full activity when SAM was added exogenously [ Fig. 3(C,D)], indicating that SAM is removed (or simply not present) from native YFV MTase during the purification process. In addition, one interesting observation from this experiment is that the native DNV2 MTase displayed higher activity in the absence of exogenous SAM than that in the presence of exogenous SAM; we are currently unsure as to what would account for this increase in activity.
Mass spectrometry analysis of copurified cofactor
To further verify that it was endogenous SAM occupying the available binding sites of the native flavivirus MTase, we extracted small molecules from our native WNV MTase by ethanol denaturation and extraction. The extracted small molecules were subjected to LC-MS/MS analysis [ Fig. 4(A)]. The MRM transitions used for SAM were automatically optimized using the Analyst 1.62 software. Detection of SAM was achieved by using MRM trigged MS/MS experiment on a hybrid QTRAP 6500 mass spectrometer. The sample extracted from the native WNV MTase was eluted at 2.3 min, a retention time the same as a commercial available SAM (data not shown). Furthermore, the product ion spectrum of the sample is identical to that of control SAM (Fig. 4). In conclusion, we identified the unknown extracted compound as SAM using the combination of chromatographic separation, MRM transitions, and the product ion spectrum. These results undoubtedly confirmed that SAM was copurified with the flavivirus MTase.
Figure 4.
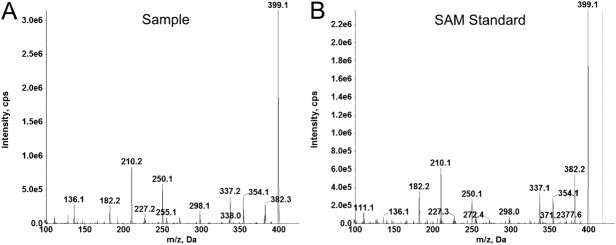
Mass Spectrometry confirms that SAM copurifies with native DNV3 MTase. A: LC-MS/MS of ETOH extraction from native WNV MTase. B: LC-MS/MS of commercial SAM standard.
Crystal structure of the refolded DNV3 MTase
We crystallized the refolded DNV3 MTase and determined its structure at 2.1 Å with an Rfactor of 24.9% and Rfree of 30.7%. When the refolded MTase structure was aligned with the previously solved native DNV3 MTase structure (3P8Z),20 the two were nearly indistinguishable, with a root mean square deviation (RMSD) of 0.34 Å when the A-chains were aligned and 0.3 Å when the B-chains were aligned. Alignment of native A-chain to the refolded B-chain yielded an RMSD of 0.32 Å and alignment of native B-chain with refolded A-chain yielded an RMSD of 0.29 Å. The A- and B-chains of 3P8Z differ in that an SAH-derived compound is bound in the cofactor-binding site of the A-chain, but SAH is bound in the active site of the B-chain. While the B-chains of the native and refolded structures were almost identical, when the A-chains of the two structures are compared [ Fig. 5(A)], a small shift is observed in two loops near the cofactor-binding site. In the first loop [residues 104–111, red box in Fig. 5(A)], the main-chain of the natively purified MTase is shifted an average of 1.2 Å closer to the co-factor site relative to that of the refolded protein, with the largest Cα shifts between G107 (1.61 Å), P108 (1.89 Å), and G109 (1.73 Å). The RMSD of the 104–111 loop is 1.28 Å. Similarly, the loop consisting of residues 80–84 [blue circle in Fig. 5(A)] is also shifted a small distance closer in the native A-chain structure (average Cα shift 0.53 Å) with an RMSD of 0.54 Å. A third loop, distant from the cofactor-binding site [residues 243–247, green box in Fig. 5(A)], is also shifted between the native and refolded structures. The average distance between these residues is 1.77 Å, with an RMSD of 2.07 Å. Interestingly, it was reported that structural differences at loops 104–111 and 243–247 could be observed between the structures of the flavivirus MTases.13 Alternatively, crystal packing could account for the structure differences, as both loops participate in either non-crystallographic symmetry-related dimer contacts or crystallographic contacts in crystals of the refolded DNV3 MTase.
Figure 5.
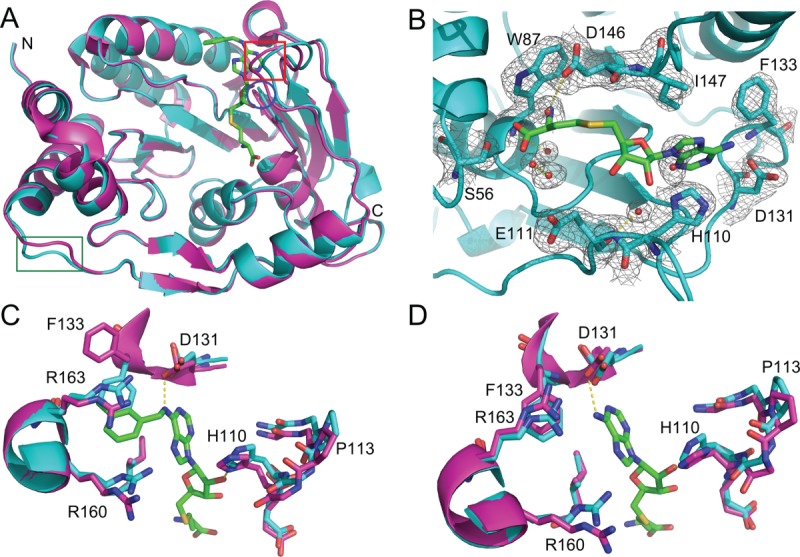
Refolded and native DNV3 MTase have similar crystal structures. A: Alignment of the A-chains of native (magenta, PDB 3P8Z) and refolded (cyan) DNV3 MTase crystal structures. Loops where the structures differ (residues 80–84, 104–111, and 243–247) are indicated by a blue circle, red box, and green box, respectively. B: The 2Fo-Fc electron density map of the refolded DNV3 MTase SAM-binding site at 2.1 Å resolution, contoured at 1.08 σ level (0.2701 e/Å3) above the mean density. SAH from the native structure B-chain is added to the SAM-binding site for reference and is colored by element, carbon, green; nitrogen, blue; oxygen, red; sulfur, yellow. C: Active sites from the aligned native (magenta) and refolded (cyan) DNV3 MTase A-chains. The SAH-derived compound present in the 3P8Z A-chain is colored as SAH in (B). D: As in (C), but SAM-binding sites are from the aligned B-chains. SAH is present in the 3P8Z B-chain and is colored by element as in (B).
The SAM-binding sites of the native and refolded MTase proteins are structurally very similar, but also contain several differences. Unlike the native MTase,20 only water molecules appear to be present in the density map of the SAM-binding site of the refolded MTase [ Fig. 5(B)]; it appears to lack any density corresponding to SAM [ Fig. 5(B)]. This is consistent with the fact that denaturation of the MTase prior to refolding removed the co-factor SAM from its binding site. Second, when an SAH-derived compound is present in the SAM-binding site of the native MTase, F133 is “flipped” ∼120° toward the active site relative to the refolded protein [ Fig. 5(C)]. The F133 “flip” does not occur in the empty SAM-binding site of the refolded MTase [ Fig. 5(D)]. The conformation of F133 is almost identical to that when SAH is bound in the SAM-binding site [ Fig. 5(D)].20 Other than the F133 “flip” the SAM-binding site residues align very closely with very little variations for conformations at the side chain level [ Fig. 5(C,D)]. Altogether, this data provides validation that the refolded DNV3 MTase faithfully replicates the structure and function of the native protein.
Discussion
We have previously shown that SIN potently inhibits N7 and 2′-O methylation in vitro as well inhibiting infection by WNV, DNV2, and YFV.11,12 Because SIN is a SAM analogue, chemically identical except for the substitution of C–NH2 amine for an S–CH3 sulfonium moiety (the transferred methyl group), we would expect that SIN would inhibit all MTase proteins, including host cell proteins, that use SAM as a methyl group donor. Fortunately, host RNA cap MTase enzymes and flaviviral MTase enzymes display several key differences that can be exploited to design potential inhibitors specific for flavivirus MTase proteins.
In the host cell, the N7 and 2′-O methylations of the RNA cap are carried out by separate enzymes, guanine N-7 and 2′-O-ribose MTases, respectively.29,30 Flaviviral MTase proteins differ in that the same enzyme carries out both types of methylations,7,8 as well as formation of the initial cap structure.6 Mutagenesis experiments8,10,12,17 strongly suggests that the capped RNA binds to flavivirus MTase proteins in two different positions, one for N7 methylation and one for 2′-O methylation. If the transition from the N7 RNA position to the 2′-O position occurs within the same molecule,17 then it may be possible for a small molecule to block this transition and specifically inhibit flavivirus RNA cap methylation.
A second key difference present in flavivirus MTase proteins is an ∼5 Å deep extension of the SAM binding pocket that appears to be conserved in all flavivirus MTase sequences.11 This extended pocket, bounded by F133, I147, G148, E149, R160, R163, V164, and L182, forms a cavity adjacent to the adenine base of SAM. This cavity, not present in cellular MTase proteins, accommodates the binding of SAM analogues modified with additional chemical groups projecting into the extended pocket. These compounds specifically inhibit DNV3 MTase in vitro and induce conformational change in the surrounding amino acid residues.20
Despite of great efforts in development of inhibitors targeting the flavivirus MTases,11,12,18,20–22,31–33 currently only one crystal structure of the MTase-inhibitor complex was available,20 besides the WNV MTase-SIN complexes and the DNV2 MTase in complex with ribavirin that targets the GTP-binding site.11,31 We reasoned that the lack of cocrystal structures of the MTase in complex with inhibitors, particularly those targeting the SAM-binding site, is due to the preoccupation of SAH, the by-product of the methyl transfer reactions.
Here, we described the successful refolding of the DNV3 MTase and characterization of the refolded MTase. The DNV3 MTase was purified under a denaturing condition, and refolded through an on-column dialysis strategy. The refolded DNV3 MTase displays CD spectra characteristic for a correctly folded protein with αβ structures [ Fig. 1(B)]. More importantly, the far-UV CD spectra and thermal denaturation behavior of the refolded MTase are indistinguishable from those of the MTase purified under the native condition, suggesting that the MTase was correctly folded. Small differences were observed for the CD spectra at near-UV region between the native and refolded MTases, which could be attributed to the removal of the adenosine base of SAH/SAM in the refolded MTase that interacts with near-UV contributing residues Phe133 and Tyr134 of the MTase at the SAM-binding site.11,13,34
Binding studies using ITC suggest that SIN binds the refolded DNV3 MTase with 6-fold higher affinity (136 nM) than does the cofactor SAM (826 nM), which is consistent with the fact that SIN is an effective inhibitor for both the MTase in vitro and virus growth in vivo.11,12 In contrast, SIN binds the native MTase with a much lower affinity (2.15 µM), suggesting that the presence of pre-bound SAH/SAM reduces the binding affinity of SIN to the MTase. A very low stoichiometry (N = 0.08) of the binding between SIN and the native MTase also suggests that only limited sites of the native MTase protein are available in solution. In contrast, stoichiometric ratios obtained for SIN and SAM binding to the refolded DNV3 MTase (Fig. 2 and Table I) are consistent with previously obtained structural data, which predicts a single SAM binding site per MTase molecule. Likewise, successful in vitro and in vivo inhibition of flavivirus MTase proteins by SIN implies that it binds to flavivirus MTase proteins with higher affinity.11,12,19 Although ∼20% of methylation activity remained in the refolded MTase when no SAM was added to the methylation reaction [ Fig. 3(A,B)], the stoichiometry observed by ITC suggested that virtually all SAM binding sites were vacant [ Fig. 2(B) and Table I]. There are several nonexclusive explanations for this discrepancy. First, the stoichiometry measured by ITC is calculated directly from the input protein concentration; variability in the measurement of input protein concentration (often on the order of +/− 10%) will directly impact the calculated stoichiometry to the same degree. Second, MTase samples were centrifuged immediately before ITC to remove any aggregates; MTase protein associated with residual SAM may be disproportionately represented in these aggregates. Third, it is possible that a small amount of residual SAM could have a larger than proportional effect when measuring methylation activity at low concentrations. Despite the residual methylation activity present, the denaturation and refolding process has removed the vast majority of SAM from the DNV3 MTase, which will facilitate the in vitro characterization of potential inhibitor compounds.
Functional studies indicated that the refolded DNV3 MTase has full MTase function equivalent to the native MTase in the presence of exogenously added SAM, the methyl donor; whereas the MTase activity was barely detectable in the absence of SAM (Fig. 3). In the presence of SAM, the MTase activity of the refolded protein is comparable with that of the native protein, indicating again that the denatured MTase is correctly refolded and fully functional. Interestingly, when we evaluated the biological function of the refolded DNV3 MTase, to our surprise we found that the DNV3 MTase purified under native condition is fully active in N7 methylation activity in the absence of exogenously added SAM, indicating that possibly a SAM molecule is present in the SAM-binding pocket of the native MTase protein. This possibility raised a question of whether SAM or SAH was co-purified with the flavivirus MTases under native conditions, as several previous structural studies of recombinant flavivirus MTase proteins assumed that SAH was copurified with these MTase proteins.7,13–15 Since SAM has a higher affinity for the MTase proteins than SAH 19 and both are chemically identical except for the donated methyl group, it is possible that SAM, and not SAH, was copurified with these MTase proteins. In fact, the ability of our purified MTases of DNV2, DNV3, and WNV to support N7 cap methylation in the absence of additional SAM (Fig. 3) supports the idea that endogenous SAM, but not reaction byproduct SAH, was copurified with these proteins. We further verified that endogenous SAM, but not SAH, was present in the WNV MTase by mass spectrometry (Fig. 4). It was reported that the intracellular concentration of SAM (400 µM) in E. coli is 300-fold higher than that of SAH (1.3 µM).35,36 In addition, SAM was known to bind flavivirus MTase with an affinity 28-fold more than SAH.19 Therefore, it is unlikely that the MTases expressed in E. coli copurify with SAH, although it cannot be completely ruled out that some flavivirus MTases may copurify with SAH or a mixture of SAM and SAH.
Curiously, native YFV MTase did not appear to retain a significant degree of endogenous SAM relative to the other MTase proteins we tested [ Fig. 3(C,D)], suggesting that these binding sites are either unoccupied or are bound with SAH, instead. Structurally, YFV MTase is almost identical to that of DNV3; the only difference of note in the residues surrounding the SAM binding pocket is that F133 is replaced with a Histidine residue in YFV (PDB # 3EVA14). Since a satisfying explanation for this difference in endogenous SAM retention does not arise from the solved crystal structures, we can only speculate that the loss of endogenous SAM from YFV MTase occurs because of differences in protein dynamics not captured by crystal structures and/or conditions of the protein purification. Furthermore, a significant difference in SIN sensitivity between the two MTase proteins11 suggests that the two SAM binding sites are not functionally identical.
Nevertheless, the crystal structure of the refolded DNV3 MTase clearly demonstrated that the MTase was successfully and correctly refolded into the native MTase fold, and that the co-factor SAM was successfully removed from its binding site. The structure of the refolded MTase is almost identical with that of the native protein, although a few small differences were observed at three loop regions including two at the SAM-binding site. These small differences could be attributed to the removal of the cofactor SAM from its binding site. Consequently, the SAM-binding site is more open in the refolded MTase than that in the native protein [ Fig. 5(A)]. Since flavivirus N7 methylation is vital for replication and an important target for antiviral therapy,8,13,17 a more open SAM-binding site and the removal of endogenous SAM may facilitate the characterization of candidate inhibitor compounds.
Experimental Procedures
Cloning, expression, denaturation, refolding, and purification of the DNV3 MTase
To construct the His-tag version of the DNV3 MTase, we grafted the insert containing the DNV3 MTase from the pGEX-6P-1 vector19 into a home-made pET28a-3C vector, in which the thrombin protease recognition sequence was replaced with the 3C protease recognition sequence. The DNV3 MTase was expressed normally in E. coli strain Rosetta 2(DE3) (EMD Biosciences). The cells were lysed in a denaturing buffer containing 50 mM Tris, pH 8.0, 500 mM NaCl, 10 mM β-Me, 10% glycerol, and 8M urea. The denatured MTase cell lysate was loaded to the Ni-NTA affinity column under denaturing condition, and extensively washed (>30 column volume) with the lysis buffer in the presence of 10 mM imidazole. The MTase-bound Ni-NTA beads were transferred to a dialysis bag, and dialyzed overnight at 4°C against a buffer containing 25 mM Tris-HCl, pH 8.0, 500 mM NaCl, 10 mM β–mercaptoethanol (β-Me), and 10% Glycerol. Home-made 3C protease was added to the dialysis bag, and the mixture was continued to dialyze for overnight at 4°C. The protease treated mixture was collected in an empty Bio-Rad Econo column. Flow-through was collected, and the beads were washed with the dialysis buffer for 3–6 column volumes or until the OD280 <0.1. The wash fractions and the flow-through were combined and concentrated to 5 mL, and was subjected to gel filtration chromatography in a buffer containing 25 mM Tris-HCl, pH 7.5, 200 mM NaCl, 10% glycerol, 2 mM DTT, using a Superdex S-200 column (GE HealthCare). The MTase fractions were collected and concentrated to 10 mg/mL, and flash-frozen in liquid nitrogen for crystallization and functional analysis.
Circular dichroism
CD spectra of the DNV3 MTases (WT and refolded) at 1 mg/mL in PBS buffer were measured on a J-720 Jasco spectropolarimeter (Japan Spectroscopic Co.) equipped with a temperature-controlled cell holder. Static measurements were recorded at 20°C. For the far-UV (190–260 nm), CD spectra were measured at a bandwidth of 1 nm using a quartz cell of path length 0.2 mm; and for the near-UV (250–350 nm) using a 1-cm quartz cell. Data from four scans were averaged using the J-720 operating software. Far-UV CD spectra were smoothed by using the Jasco noise reduction routine. The extent of secondary structure of the MTases was calculated from the CD spectra in the far-UV spectral range, using the CDPro program suite.25
The thermal denaturations were carried out for the DNV3 MTases (WT and refolded) at 1 mg/mL in PBS buffer by measurement of the CD ellipticity at 222 nm, with a quartz cell of path length 5 mm. A scan rate of 1°C/min and a response time of 2 s were used. The midpoint (Tm) of the heat denaturation was calculated by simulating the experimental melting curve using the protein denaturation routine in the Spectra Analysis software of the Jasco Spectra Manager suite. All the CD experiments were repeated several times, similar results were obtained.
Sample preparation for mass spectrometry
The WNV MTase purified under native condition (50 µL) at 10 mg/mL was precipitated by addition of methanol (1 mL) followed by incubation for 30 min. The mixture was spun down; the supernatant (900 µL) was removed and the remaining mixture was evaporated to dryness under a speed vacuum.
Mass spectrometry analysis
LC-MS/MS analysis was performed on a QTRAP 6500 (ABSCIEX) configured with a Shimadzu UHPLC Nexera LC-30AD pumping system, a SIL-30AC auto sampler, and CTO-30A column oven. The system was operated under Analyst 1.62 control. The chromatographic separation of products was achieved on a Waters hydrophilic interaction liquid chromatography analytical column (3.0 ID 100 mm packed with 1.6 μm CORTEC particles). The mobile phase consisted of solvent A (100 mM ammonium formate in water, pH 3.0) and solvent B (100% acetonitrile). The samples were eluted, at a flow rate of 0.6 mL/min, with 90% A for 1 min, followed by 3 min gradient of 90% B to 10% B, and then 100% B for a further 1.8 min. The total run time was 6 min. The compound identification was conducted through multiple reaction monitoring (MRM) triggered enhanced product ion (EPI) scan using information dependent acquisition (IDA). The utilization of chromatographic separation, MRM transitions, and EPI scan allows accurate compound identification and confirmation. Two MRM transitions including (m/z 398.99 > 150.1) and (m/z 398.99 > 136.1) for SAM were used to triggered the EPI experiment. The instrument was operated in a positive ion mode with a turbo V ion drive electrospray source. The parameters for the operation were as follows: curtain gas, 30 psi; heated nebulizer temperature 350°C, ion spray voltage, 5300 V; gas 1, 50 psi; gas 2, 70 psi, declustering potential, 65V, EP, 10 V, and CAD gas, medium.
N7 Methylation Assay
The production of capped WNV RNA and measurement of N7 methylation activity were performed essentially as described in Refs.18 and 19 Briefly, the first 90 nucleotides of the WNV genome were produced from the A12 WNV replicon in vitro using the Ambion Megascript T7 transcription kit (AM1334), purified over Micro-G25 spin columns (Santa Cruz Biotechnology, #202390) and ethanol precipitated overnight. The purified RNA was treated with Vaccinia virus Guanylyl transferase (0.3 mg/mL) in the presence of α-32P [GTP] (0.25 µCi/µL; MPBio) at 37°C for 1 h to produce 32P-labeled G*pppA WNV RNA, which was purified as the initial RNA transcript (the asterisk indicates that the following phosphate is 32P labeled). Conversion of G*pppA RNA to m7G*pppA RNA was performed essentially as described in refs18,19 in the presence of the indicated MTase (2 µg (3 µM final concentration) or 0.5 µg (0.75 µM) where indicated), 80 µM SAM, and 5 pmol G*pppA RNA (or m7G*pppA) RNA (0.25 µM final concentration) with a total reaction volume of 20 µL. The methylated RNA was treated with nuclease P1 (US Biological N7000) to release the 32P-labeled cap moieties, which were separated by thin-layer chromatography (TLC) and imaged on a Typhoon 9400 imager. The resolved bands were quantified using Imagequant software and percent conversion of G*pppA → m7G*pppA was calculated as (m7G*pppA signal)/(m7G*pppA signal + G*pppA signal) × 100%.
Isothermal titration calorimetry
ITC experiments were performed at the Analytical Biochemistry Core Facility in the Center for Biotechnology and Interdisciplinary Studies (CBIS) at Rensselaer Polytechnic Institute in Troy, NY. Native or refolded DNV3 MTase was dialyzed into ITC buffer (20 mM Hepes 7.4, 250 mM NaCl, 1 mM TCEP) and centrifuged at 12,000g for 30 min to remove any debris or precipitates, quantified by UV spectrum, and loaded into the 1.4 mL cell of a Microcal VP-ITC machine (GE Life Sciences) at a concentration of 27 µM (native MTase) or 9 µM (refolded MTase), respectively. SAM or SIN (500 nM) was initially injected at a volume of 1 µL, followed by 3 µL injections at 5 min intervals. Control injections of SAM or SIN into ITC buffer were performed after each experiment to account for signal from dilution. All ITC reactions were carried out at 25°C. Data was processed and physical constants calculated using the Microcal Origin software.
Crystallization, X-ray data collection, structure determination, and refinement
Initial crystallization condition of the refolded DNV3 MTase was established as described previously.20 Large crystals were grown in an optimized condition with a reservoir solution containing 50 mM sodium citrate, pH 5.6, 24–30% PEG 4,000, 5% saturated ammonium sulfate, 10% glycerol, 0–20% DMSO, and 5 mM DTT. The crystals belong to space group P1 with two molecules per asymmetric unit (Table II). Prior to data collection, all crystals were transferred to a reservoir solution containing 25% glycerol, and then flash-cooled under a nitrogen stream at 100 K, and stored in liquid nitrogen. Diffraction data were collected to 2.1 Å resolution at 100 K using an R-AXIS IV++ detector and an in-house Rigaku microfocus MicroMax-007 X-ray generator. Diffraction data were processed and scaled using CrystalClear 1.3.6 (Rigaku Corp.).
Table II.
Diffraction data collection and structure refinement statistics
| Data collection | ||
| Space group | P1 | |
| Cell parameters | ||
| a = 44.69 Å, b = 48.39 Å, c = 68.07 Å | ||
| α = 79.11°, β = 78.75°, γ = 69.71° | ||
| Resolution (Å) | 45–2.1 (2.18–2.1)a | |
| Redundancy | 2.1 (2.1) | |
| Completeness (%) | 94.9 (92.7) | |
| Average I/σ(I) | 3.9 (1.3) | |
| Rsym (%) | 12.3 (45.0) | |
| Refinement | ||
| Resolution limits (Å) | 45–2.1 | |
| No. reflections | 28706 | |
| Rwork (%) | 24.9 | |
| Rfree (%) | 30.7 | |
| Non-H atoms | ||
| Protein | 4094 | |
| Water | 248 | |
| Average B (Å2) | 40 | |
| Geometry | ||
| rmsd bond length (Å) | 0.004 | |
| rmsd bond angle (°) | 0.821 | |
Values in parentheses are those for the highest-resolution shell.
The structure of the refolded DNV3 MTase was determined by the Phaser molecular-replacement program within the PHENIX suite,37 with the native structure of the DNV3 MTase (3P8Z20) used as a starting model. Prior to molecular replacement calculations, the bound SAH was removed from the SAM-binding site of the MTase structure. Structural refinement was carried out using PHENIX. Model rebuilding was carried out using Coot.38 The final refinement statistics are summarized in Table II.
Acknowledgments
The authors thank the core facilities at the Wadsworth Center, including the Molecular Genetics Core for DNA sequencing, the Biochemistry Core for assistance in CD measurement, and the Macromolecular Crystallography Core for facility usage. They thank M. Lopez at the Rensselaer Polytechnic Institute for assistance in ITC measurement.
Glossary
- CD
circular dichroism
- DNV2
DNV serotype 2
- DNV3
DNV serotype 3
- DNV
dengue virus
- E. coli
Escherichia coli
- ITC
isothermal titration calorimetry
- JEV
Japanese encephalitis virus
- min
minute
- MTase
Methyltransferase
- NS
nonstructural protein
- RMSD
root mean square deviation
- SAH
S-adenosyl-l-homocysteine
- SAM
S-adenosyl-l-methionine
- SIN
sinefungin
- TBEV
tick-borne encephalitis virus
- WNV
West Nile virus
- YFV
yellow fever virus
- β-Me
β–mercaptoethanol
References
- 1.Global Dengue. 2014. Available at: http://www.cdc.gov/dengue/epidemiology/index.html. Last accessed on November 16, 2014.
- 2.Sabchareon A, Wallace D, Sirivichayakul C, Limkittikul K, Chanthavanich P, Suvannadabba S, Jiwariyavej V, Dulyachai W, Pengsaa K, Wartel TA, Moureau A, Saville M, Bouckenooghe A, Viviani S, Tornieporth NG, Lang J. Protective efficacy of the recombinant, live-attenuated, CYD tetravalent dengue vaccine in Thai schoolchildren: a randomised, controlled phase 2b trial. Lancet. 2012;380:1559–1567. doi: 10.1016/S0140-6736(12)61428-7. [DOI] [PubMed] [Google Scholar]
- 3.Burke DS, Monath TP. Flaviviruses. In: Knipe DM, Howley PM, editors. Fields Virology. Philadelphia, PA: Lippincott-Williams & Wilkins; 2001. pp. 1043–1125. [Google Scholar]
- 4.Fink K, Shi PY. Live attenuated vaccine: the first clinically approved dengue vaccine? Expert Rev Vaccines. 2014;13:185–188. doi: 10.1586/14760584.2014.870888. [DOI] [PubMed] [Google Scholar]
- 5.Chambers TJ, Hahn CS, Galler R, Rice CM. Flavivirus genome organization, expression, and replication. Annu Rev Microbiol. 1990;44:649–688. doi: 10.1146/annurev.mi.44.100190.003245. [DOI] [PubMed] [Google Scholar]
- 6.Issur M, Geiss BJ, Bougie I, Picard-Jean F, Despins S, Mayette J, Hobdey SE, Bisaillon M. The flavivirus NS5 protein is a true RNA guanylyltransferase that catalyzes a two-step reaction to form the RNA cap structure. RNA. 2009;15:2340–2350. doi: 10.1261/rna.1609709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Egloff MP, Benarroch D, Selisko B, Romette JL, Canard B. An RNA cap (nucleoside-2'-O-)-methyltransferase in the flavivirus RNA polymerase NS5: crystal structure and functional characterization. EMBO J. 2002;21:2757–2768. doi: 10.1093/emboj/21.11.2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ray D, Shah A, Tilgner M, Guo Y, Zhao Y, Dong H, Deas TS, Zhou Y, Li H, Shi PY. West Nile virus 5'-cap structure is formed by sequential guanine N-7 and ribose 2'-O methylations by nonstructural protein 5. J Virol. 2006;80:8362–8370. doi: 10.1128/JVI.00814-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dong H, Chang DC, Hua MH, Lim SP, Chionh YH, Hia F, Lee YH, Kukkaro P, Lok SM, Dedon PC, Shi PY. 2'-O methylation of internal adenosine by flavivirus NS5 methyltransferase. PLoS Pathog. 2012;8:e1002642. doi: 10.1371/journal.ppat.1002642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dong H, Ren S, Li H, Shi PY. Separate molecules of West Nile virus methyltransferase can independently catalyze the N7 and 2'-O methylations of viral RNA cap. Virology. 2008;377:1–6. doi: 10.1016/j.virol.2008.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dong H, Liu L, Zou G, Zhao Y, Li Z, Lim SP, Shi PY, Li H. Structural and functional analyses of a conserved hydrophobic pocket of flavivirus methyltransferase. J Biol Chem. 2010;285:32586–32595. doi: 10.1074/jbc.M110.129197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dong H, Ren S, Zhang B, Zhou Y, Puig-Basagoiti F, Li H, Shi PY. West Nile virus methyltransferase catalyzes two methylations of the viral RNA cap through a substrate-repositioning mechanism. J Virol. 2008;82:4295–4307. doi: 10.1128/JVI.02202-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou Y, Ray D, Zhao Y, Dong H, Ren S, Li Z, Guo Y, Bernard KA, Shi PY, Li H. Structure and function of flavivirus NS5 methyltransferase. J Virol. 2007;81:3891–3903. doi: 10.1128/JVI.02704-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Geiss BJ, Thompson AA, Andrews AJ, Sons RL, Gari HH, Keenan SM, Peersen OB. Analysis of flavivirus NS5 methyltransferase cap binding. J Mol Biol. 2009;385:1643–1654. doi: 10.1016/j.jmb.2008.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bollati M, Milani M, Mastrangelo E, Ricagno S, Tedeschi G, Nonnis S, Decroly E, Selisko B, de Lamballerie X, Coutard B, Canard B, Bolognesi M. Recognition of RNA cap in the Wesselsbron virus NS5 methyltransferase domain: implications for RNA-capping mechanisms in Flavivirus. J Mol Biol. 2009;385:140–152. doi: 10.1016/j.jmb.2008.10.028. [DOI] [PubMed] [Google Scholar]
- 16.Kroschewski H, Lim SP, Butcher RE, Yap TL, Lescar J, Wright PJ, Vasudevan SG, Davidson AD. Mutagenesis of the dengue virus type 2 NS5 methyltransferase domain. J Biol Chem. 2008;283:19410–19421. doi: 10.1074/jbc.M800613200. [DOI] [PubMed] [Google Scholar]
- 17.Liu L, Dong H, Chen H, Zhang J, Ling H, Li Z, Shi PY, Li H. Flavivirus RNA cap methyltransferase: structure, function, and inhibition. Front Biol. 2010;5:286–303. doi: 10.1007/s11515-010-0660-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen H, Liu L, Jones SA, Banavali N, Kass J, Li Z, Zhang J, Kramer LD, Ghosh AK, Li H. Selective inhibition of the West Nile virus methyltransferase by nucleoside analogs. Antiviral Res. 2013;97:232–239. doi: 10.1016/j.antiviral.2012.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen H, Zhou B, Brecher M, Banavali N, Jones SA, Li Z, Zhang J, Nag D, Kramer LD, Ghosh AK, Li H. S-adenosyl-homocysteine is a weakly bound inhibitor for a flaviviral methyltransferase. PLoS One. 2013;8:e76900. doi: 10.1371/journal.pone.0076900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lim SP, Sonntag LS, Noble C, Nilar SH, Ng RH, Zou G, Monaghan P, Chung KY, Dong H, Liu B, Bodenreider C, Lee G, Ding M, Chan WL, Wang G, Jian YL, Chao AT, Lescar J, Yin Z, Vedananda TR, Keller TH, Shi PY. Small molecule inhibitors that selectively block dengue virus methyltransferase. J Biol Chem. 2011;286:6233–6240. doi: 10.1074/jbc.M110.179184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luzhkov VB, Selisko B, Nordqvist A, Peyrane F, Decroly E, Alvarez K, Karlen A, Canard B, Qvist J. Virtual screening and bioassay study of novel inhibitors for dengue virus mRNA cap (nucleoside-2'O)-methyltransferase. Bioorg Med Chem. 2007;15:7795–7802. doi: 10.1016/j.bmc.2007.08.049. [DOI] [PubMed] [Google Scholar]
- 22.Podvinec M, Lim SP, Schmidt T, Scarsi M, Wen D, Sonntag LS, Sanschagrin P, Shenkin PS, Schwede T. Novel inhibitors of dengue virus methyltransferase: discovery by in vitro-driven virtual screening on a desktop computer grid. J Med Chem. 2010;53:1483–1495. doi: 10.1021/jm900776m. [DOI] [PubMed] [Google Scholar]
- 23.Jansson AM, Jakobsson E, Johansson P, Lantez V, Coutard B, de Lamballerie X, Unge T, Jones TA. Structure of the methyltransferase domain from the Modoc virus, a flavivirus with no known vector. Acta Cryst. 2009;D65:796–803. doi: 10.1107/S0907444909017260. [DOI] [PubMed] [Google Scholar]
- 24.Bollati M, Milani M, Mastrangelo E, de Lamballerie X, Canard B, Bolognesi M. Crystal structure of a methyltransferase from a no-known-vector Flavivirus. Biochem Biophys Res Commun. 2009;382:200–204. doi: 10.1016/j.bbrc.2009.03.008. [DOI] [PubMed] [Google Scholar]
- 25.Sreerama N, Woody RW. Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal Biochem. 2000;287:252–260. doi: 10.1006/abio.2000.4880. [DOI] [PubMed] [Google Scholar]
- 26.Adler AJ, Greenfield NJ, Fasman GD. Circular dichroism and optical rotatory dispersion of proteins and polypeptides. Methods Enzymol. 1973;27:675–735. doi: 10.1016/s0076-6879(73)27030-1. [DOI] [PubMed] [Google Scholar]
- 27.Mastrangelo E, Bollati M, Milani M, Selisko B, Peyrane F, Canard B, Grard G, de Lamballerie X, Bolognesi M. Structural bases for substrate recognition and activity in Meaban virus nucleoside-2'-O-methyltransferase. Protein Sci. 2007;16:1133–1145. doi: 10.1110/ps.072758107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Assenberg R, Ren J, Verma A, Walter TS, Alderton D, Hurrelbrink RJ, Fuller SD, Bressanelli S, Owens RJ, Stuart DI, Grimes JM. Crystal structure of the Murray Valley encephalitis virus NS5 methyltransferase domain in complex with cap analogues. J Gen Virol. 2007;88:2228–2236. doi: 10.1099/vir.0.82757-0. [DOI] [PubMed] [Google Scholar]
- 29.Fabrega C, Hausmann S, Shen V, Shuman S, Lima CD. Structure and mechanism of mRNA cap (guanine-N7) methyltransferase. Mol Cell. 2004;13:77–89. doi: 10.1016/s1097-2765(03)00522-7. [DOI] [PubMed] [Google Scholar]
- 30.Smietanski M, Werner M, Purta E, Kaminska KH, Stepinski J, Darzynkiewicz E, Nowotny M, Bujnicki JM. Structural analysis of human 2'-O-ribose methyltransferases involved in mRNA cap structure formation. Nat Commun. 2014;5:3004. doi: 10.1038/ncomms4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Benarroch D, Egloff MP, Mulard L, Guerreiro C, Romette JL, Canard B. A structural basis for the inhibition of the NS5 dengue virus mRNA 2'-O-methyltransferase domain by ribavirin 5'-triphosphate. J Biol Chem. 2004;279:35638–35643. doi: 10.1074/jbc.M400460200. [DOI] [PubMed] [Google Scholar]
- 32.Milani M, Mastrangelo E, Bollati M, Selisko B, Decroly E, Bouvet M, Canard B, Bolognesi M. Flaviviral methyltransferase/RNA interaction: structural basis for enzyme inhibition. Antiviral Res. 2009;83:28–34. doi: 10.1016/j.antiviral.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Puig-Basagoiti F, Qing M, Dong H, Zhang B, Zou G, Yuan Z, Shi PY. Identification and characterization of inhibitors of West Nile virus. Antiviral Res. 2009;83:71–79. doi: 10.1016/j.antiviral.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li H, Zhao Y, Guo Y, Vanvranken SJ, Li Z, Eisele L, Mourad W. Mutagenesis, biochemical, and biophysical characterization of Mycoplasma arthritidis-derived mitogen. Mol Immunol. 2007;44:763–773. doi: 10.1016/j.molimm.2006.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Javor GT. Depression of adenosylmethionine content of Escherichia coli by thioglycerol. Antimicrob Agents Chemother. 1983;24:860–867. doi: 10.1128/aac.24.6.860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Halliday NM, Hardie KR, Williams P, Winzer K, Barrett DA. Quantitative liquid chromatography-tandem mass spectrometry profiling of activated methyl cycle metabolites involved in LuxS-dependent quorum sensing in Escherichia coli. Anal Biochem. 2010;403:20–29. doi: 10.1016/j.ab.2010.04.021. [DOI] [PubMed] [Google Scholar]
- 37.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Cryst. 2010;D66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Cryst. 2010;D66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]