
Fig 3.
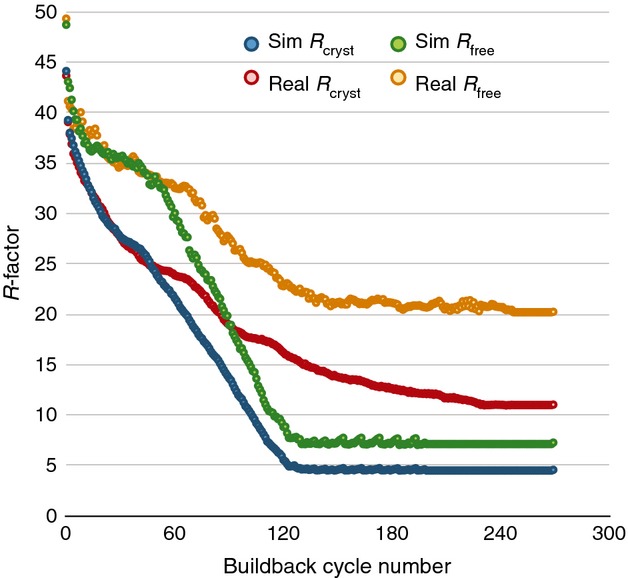
Reduction in R-factors as individual atoms are automatically built into difference maps. The starting coordinate model used to compute Fstart for the simulated data (blue and green) was a single-conformer model of 100% occupied atoms with isotropic B factors. In both cases, the processed data were provided to phenix.autobuild with anomalous differences, the sequence of lysozyme and no other sources of phase information. The resulting model was trimmed of all water atoms and subjected to a simple five-step rebuilding procedure using data truncated to 2.0 Å: (1) refine in REFMAC for 500 cycles, or until atoms and B factors stop moving, (2) add dummy atoms to the five highest peaks in the difference map, (3) assign new atoms the proper atom name if they are within 0.5 Å of an atom in the reference model, (4) remove atoms with B > 100 Å2 or that fall on negative difference features < −6σ, and (5) repeat until convergence. Models built into the simulated data converge to very low Rfree values (Rcryst = 4.57% and Rfree = 7.27%), roughly the same magnitude as the Rmerge from the XDS/MOSFLM-processed data, whereas the Rfree for the real data never goes below 20%.