

Abstract
Spinal and bulbar muscular atrophy, or Kennedy's disease, is an X-linked motor neuron disease caused by polyglutamine repeat expansion in the androgen receptor. The disease is characterised by weakness, atrophy and fasciculations in the limb and bulbar muscles. Affected males may have signs of androgen insensitivity, such as gynaecomastia and reduced fertility. Neurophysiological studies are typically consistent with diffuse denervation atrophy, and serum creatine kinase is usually elevated 2–5 times above normal. Progression of the disease is slow, and the focus of spinal and bulbar muscular atrophy (SBMA) management is to prevent complications.
Keywords: genetics, craniofacial, motor neuron disease, spinal and bulbar muscular atrophy, Kennedy's disease, weakness
Introduction
Spinal and bulbar muscular atrophy (SBMA), also known as Kennedy's disease (Kennedy et al, 1968), is a slowly progressive disease with lower motor neuron loss and muscle weakness, atrophy and fasciculations. The disease is caused by a trinucleotide (CAG) repeat expansion in the androgen receptor (AR) gene on the X chromosome (La Spada et al, 1991). This mutation results in an expanded polyglutamine tract and an androgen-dependent toxic gain of function in the mutant protein. The disease has been reported across many different ethnic groups, with an estimated prevalence of about 1 in 40 000. Normal individuals will have repeat lengths between 11 and 36 CAGs, and the disease will occur when the repeat length increases to between 38 and 62 CAGs (Atsuta et al, 2006; Rhodes et al, 2009). Although there is no treatment available to alter the progression of this disease, patients may have benefit from symptomatic therapy.
The toxic gain of function of the AR is dependent on the androgens testosterone and dihydrotestosterone. In addition to these toxic effects of the AR, loss of normal receptor function also contributes to the disease phenotype, with gynaecomastia and reduced fertility in some patients. The mutant AR is prone to aggregation (Li et al, 1998; Walcott and Merry, 2002), and its effects on cellular signal transduction, mitochondrial function and axonal transport likely contribute to the disease (McCampbell et al, 2000; Lieberman et al, 2002; Katsuno et al, 2006; Ranganathan et al, 2009).
From altered AR function to motor neuron disease: pathophysiology of SBMA
Similar CAG repeat expansion mutations have been found in Huntington's disease, dentatorubral–pallidoluysian atrophy (DRPLA) and several spinocerebellar ataxias (Gatchel and Zoghbi, 2005). In each case, the CAG repeat is translated to a polyglutamine tract; thus, these disorders are known as polyglutamine diseases. In SBMA, as in the other polyglutamine diseases, there is an inverse correlation between the CAG repeat length and the age of onset, and a direct correlation with the disease severity, as adjusted by the age of examination (Doyu et al, 1992; Atsuta et al, 2006).
Although the mutant proteins are expressed widely in the CNS, specific populations of neurons are vulnerable in each disease, resulting in characteristic patterns of neurode-generation and clinical features (Zoghbi and Orr, 2000).
In SBMA, anterior horn cells degenerate in the spinal cord of affected individuals (Kennedy et al, 1968; Ogata et al, 1994). Patients also have degeneration of the dorsal root ganglia, leading to mild abnormalities in sensory function in the distal extremities (Sobue et al, 1981; Nagashima et al, 1988). The mutant AR may also have a direct toxic effect on skeletal muscle: histological and molecular signs of muscle pathology are detectable before the appearance of pathological abnormalities in the spinal cord in a knock-in mouse model of SBMA (Yu et al, 2006), and muscle biopsy samples from SBMA patients may have a mixed pathology with both myopathic and neurogenic features (Soraru et al, 2008).
Unlike other polyglutamine diseases in which the normal function of the disease protein may not be clear, in SBMA, the disease protein has a well-characterised role as a ligand-dependent transcription factor. The AR is a nuclear hormone receptor and is located in the cytoplasm when inactive, in a complex with heat-shock proteins. Upon binding with its natural ligands, testosterone and its more potent derivative dihydrotestosterone, a number of events occur, including AR post-translational modification, nuclear translocation and DNA binding. These changes ultimately result in AR-mediated activation or repression of target genes. These occur in concert with conformational changes that result in the exposure of two co-regulator interaction surfaces, activation function-1 (AF-1) and activation function-2 (AF-2) (Nedelsky et al, 2010).
In SBMA, the disease likely results from a toxic gain of function, because AR gene deletion in humans does not lead to motor neuron degeneration, and mouse models with expanded polyglutamine AR protein accurately reflect the human disease, with lower motor neuron specificity and gender specificity (Katsuno et al, 2002). Nevertheless, a loss of normal AR function may also play a role in the pathogenesis, as suggested by the fact that SBMA patients often show mild signs of partial androgen insensitivity like gynaecomastia and decreased fertility.
A pathological hallmark of the polyglutamine diseases is the presence of intranuclear and cytoplasmic inclusions of the disease proteins that are ubiquitinated and associated with various transcription factors, chaperones and proteasome components (Li et al, 1998). As human AR is widely expressed, nuclear inclusions of the pathogenic AR protein are detected not only in the brain and spinal cord, but also in non-neuronal tissues such as kidney, skeletal muscle, adrenal gland and scrotal skin. Although nuclear inclusions are a disease-specific histopathological finding, they may not be pathogenic per se. Monomers and soluble oligomers of the expanded protein may constitute the toxic species in polyglutamine diseases, and the intranuclear inclusions likely represent a protective response to the abnormal polyglutamine protein (Arrasate et al, 2004; Miller et al, 2011).
Although the expansion may interfere with the normal function of the host protein, it is not yet clear how this contributes to pathogenesis. Studies have indicated that transcriptional dysregulation may underlie the molecular mechanism of neuronal dysfunction in SBMA and other polyglutamine diseases (McCampbell et al, 2000; Minamiyama et al, 2004). CREB-binding protein (CBP), a transcriptional co-activator that mediates the nuclear response to a variety of cell signalling cascades, is incorporated into nuclear inclusions formed by polyglutamine-containing proteins in cultured cells, transgenic mice and tissue from patients with SBMA (McCampbell et al, 2000). Histone acetylation is decreased in a mouse model of SBMA, and this may be a consequence of altered CBP localisation (Minamiyama et al, 2004). Mitochondrial impairment and consequent oxidative stress have also been suggested as a causative molecular event in SBMA (Ranganathan et al, 2009). Moreover, the pathogenic AR protein represses the transcription of the subunits of peroxisome proliferators– activated receptor gamma co-activator (PGC-1), a transcriptional co-activator that regulates the expression of nuclear-encoded mitochondrial proteins (Ranganathan et al, 2009). In the muscle of both SBMA model mice and SBMA patients, neurofilaments and synaptophysin accumulate at the distal motor axon, suggesting an impairment of retrograde axonal transport (Katsuno et al, 2006).
Diagnostic studies and evaluation
The diagnosis of SBMA is confirmed by genetic testing, because all affected individuals have a trinucleotide repeat expansion with at least 38 CAGs in the first exon of the AR gene (La Spada et al, 1991).
Serum creatine kinase is elevated in most subjects to the 900–1400 U/l range, and aminotransferases may be mildly elevated (Atsuta et al, 2006; Rhodes et al, 2009). SBMA subjects may also have impaired glucose tolerance or elevation of the low-density lipoprotein levels above normal. Although most patients have elevations in total testosterone, free testosterone and dihydrotestosterone, the levels of free testosterone and dihydrotestosterone may be reduced in some individuals.
The characteristic features on electromyography and nerve conduction study are low sensory nerve amplitudes, decreased compound motor action potentials and evidence of diffuse denervation. Motor unit nerve estimation (MUNE) is reduced to about half of healthy control values (Lehky et al, 2009). A muscle biopsy is not needed to make a diagnosis, but if carried out may show evidence of neurogenic and myogenic atrophy (Kennedy et al, 1968; Soraru et al, 2008).
A majority of SBMA patients have a positive family history, but about one-third of diagnosed patients are unaware of other affected family members (Rhodes et al, 2009). As SBMA is an X-linked recessive disease, the children of affected patients are asymptomatic. The disease may be passed through asymptomatic females, for example mothers, sisters and daughters of affected patients. Female carriers do not usually develop weakness, although a minority may have muscle cramps (Ishihara et al, 2001). The CAG repeat length may change between generations, with longer repeats correlating with earlier age of disease onset (La Spada et al, 1992). Carriers and those who are at risk should have access to genetic counselling.
Clinical presentation
The onset ranges from about ages 18–64, with most patients presenting in the fourth or fifth decade of life (Rhodes et al, 2009). The initial symptoms include tremor, cramping and weakness (Rhodes et al, 2009; Dias et al, 2011). The characteristic signs of lower motor neuron disease are seen, including muscle atrophy, fasciculations in the face and extremities, and reduced deep tendon reflexes. Weakness may present as difficulty climbing stairs and walking, particularly long distances. The weakness is both proximal and distal, with some asymmetry and more weakness on the dominant side (Rhodes et al, 2009). Degeneration of the dorsal root ganglia can result in a loss of sensation in the distal extremities. Patients may also have androgen insensitivity, which can manifest as gynaecomastia, oligospermia and erectile dysfunction. The decrease in normal androgen receptor function may decrease the risk of androgenetic alopecia in SBMA (Sinclair et al, 2007).
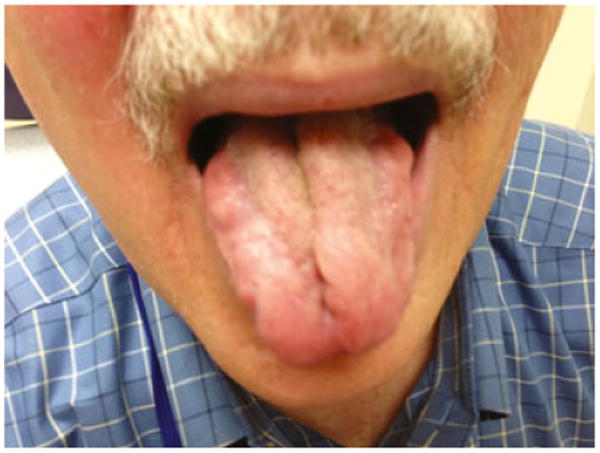
Most individuals have involvement of the bulbar muscles and difficulty with speech articulation and swallowing (Figure 1). The dysarthria and dysphagia have median onset in the early 50 s (Atsuta et al, 2006). Other findings include weakness and atrophy of the face and tongue muscles, with perioral fasciculations (Figure 2; Video S1), and hypernasality of the voice, with decreased range of pitch and loudness. SBMA patients may present with fatigue when chewing and occasionally with jaw drop, caused by weakness of the jaw-closure muscles (temporalis and mas-seter muscles) with preservation of jaw-opening muscles (pterygoid muscles) (Sumner and Fischbeck, 2002).
Figure 1.
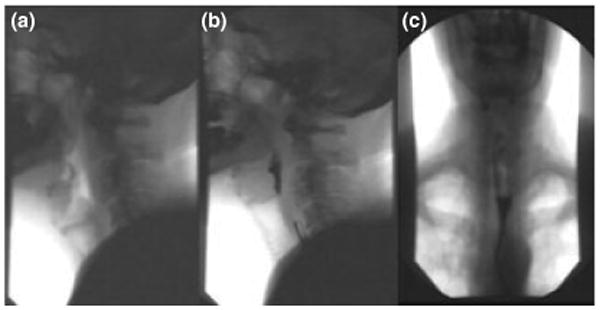
Video swallow study in a patient with Kennedy's disease spinal and bulbar muscular atrophy (SBMA), with images before (a) and after (b) swallow, showing retention of barium at the base of the tongue and in the postcricoid region. There is also slow propulsion of liquid in the upper oesophagus above the aortic arch (c)
Figure 2. Tongue atrophy in a patient with spinal and bulbar muscular atrophy (SBMA).
The disease progresses slowly; in a recent clinical trial, muscle strength as measured by quantitative muscle assessment declined by 2% per year in the placebo group (Fernández-Rhodes et al, 2011).
The majority of individuals with SBMA have a normal life expectancy and do not die from direct complications of the disease. Affected individuals are at risk of choking on food and aspiration pneumonia because of weakness of the bulbar muscles (Kennedy et al, 1968; Atsuta et al, 2006). Occasional patients have difficulty breathing due to respiratory muscle weakness. Dysarthria, dysphagia and the onset of pneumonia correlate with the length of the CAG repeat (Atsuta et al, 2006). As bulbar symptoms affect the prognosis, videofluorography-assessed barium swallow is clinically indicated and has been used as an outcome measure in SBMA clinical trials (Katsuno et al, 2010; Fernández-Rhodes et al, 2011).
Differential diagnosis
The diagnosis of SBMA is often delayed due to limited awareness of the disease. With recognition of the characteristic clinical features and the availability of confirmatory genetic testing, a diagnosis can be relatively straightforward. In a recent study, the time to diagnosis after onset of weakness averaged 5.5 years, and the time from first medical evaluation to diagnosis averaged more than 3 years (Rhodes et al, 2009). SBMA patients are often misdiagnosed with amyotrophic lateral sclerosis (ALS). Dysphagia, dysarthria and progressive weakness are common findings in ALS; nevertheless, differentiation of ALS from SBMA can usually be made based on history and physical examination. Patients with ALS usually have upper and lower motor neuron signs and more rapid disease progression. The bulbar and extremity weakness of SBMA can lead to the misdiagnosis of myasthenia gravis, and, although the manifestations usually differ, SBMA should be considered in patients who are thought to have myasthenia but have negative anti-acetylcholine receptor antibody testing and lack of improvement with myasthenia treatment (pyridostigmine, corticosteroids or other immunosuppressants). Other misdiagnoses in SBMA patients include polymyositis, metabolic myopathy and chronic inflammatory neuropathy, given the myopathic and neuropathic features.
Management
There is currently no treatment available to alter the course of the disease. Animal models recapitulate the disease phenotype, and androgen reduction therapy in these animals is effective (Katsuno et al, 2003). However, testing of the androgen-reducing agents leuprorelin and dutasteride in randomised, placebo-controlled clinical trials has not shown significant effects on the primary outcome measures of swallow function and muscle strength (Banno et al, 2009; Katsuno et al, 2010; Fernández-Rhodes et al, 2011). SBMA management should focus on enhancing mobility and function and preventing complications of the disease. Subjects who have choking spells or dysphagia should undergo a speech and swallow evaluation. Behavioural measures can be instituted to reduce the risk of aspiration. While dyspnoea is not often encountered in SBMA, pulmonary function testing should be carried out in subjects with shortness of breath. Mobility and ambulation can be enhanced with assistive devices.
The effect of exercise on function in SBMA remains uncertain. A study of moderate-intensity aerobic exercise in eight SBMA subjects by Preisler et al (2009) showed mixed results, with an increase in maximal work capacity but no effect on maximal oxygen uptake. Given the variability in function across the SBMA population, exercise needs to be tailored to the patient's level of function. Although we would expect exercise to help maintain and improve mobility, more studies are needed to assess whether functional exercise is beneficial.
Supplementary Material
Video S1. Tongue fibrillations and chin fasciculations in a patient with SBMA. (Patient data were collected with the understanding and written consent of each subject and according to ethical principles, including the World Medical Association Declaration of Helsinki (version, 2002)).
Footnotes
Author contributions: Christopher Grunseich, Carlo Rinaldi, and Kenneth Fischbeck all contributed to drafting the paper.
Supporting Information: Additional Supporting Information may be found in the online version of this article:
References
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant hun-tingtin and the risk of neuronal death. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- Atsuta N, Watanabe H, Ito M, et al. Natural history of spinal and bulbar muscular atrophy (SBMA): a study of 223 Japanese patients. Brain. 2006;129:1446–1455. doi: 10.1093/brain/awl096. [DOI] [PubMed] [Google Scholar]
- Banno H, Katsuno M, Suzuki K, et al. Phase 2 trial of leuprorelin in patients with spinal and bulbar muscular atrophy. Ann Neurol. 2009;65:140–150. doi: 10.1002/ana.21540. [DOI] [PubMed] [Google Scholar]
- Dias FA, Munhoz RP, Raskin S, Werneck LC, Teive HAG. Tremor in X-linked recessive spinal and bulbar muscular atrophy (Kennedy's disease) Clinics. 2011;66:955–957. doi: 10.1590/S1807-59322011000600006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyu M, Sobue G, Mukai E, et al. Severity of X-linked recessive bulbospinal neuronopathy correlates with size of the tandem CAG repeat in androgen receptor gene. Ann Neurol. 1992;32:707–710. doi: 10.1002/ana.410320517. [DOI] [PubMed] [Google Scholar]
- Fernández-Rhodes LE, Kokkinis AD, White MJ, et al. Efficacy and safety of dutasteride in patients with spinal and bulbar muscular atrophy: a randomised placebo-controlled trial. Lancet Neurol. 2011;10:140–147. doi: 10.1016/S1474-4422(10)70321-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatchel JR, Zoghbi HY. Diseases of unstable repeat expansion: mechanisms and common principles. Nat Rev Genet. 2005;10:743–755. doi: 10.1038/nrg1691. [DOI] [PubMed] [Google Scholar]
- Ishihara H, Kanda F, Nishio H, Sumino K, Chihara K. Clinical features and skewed X-chromosome inactivation in female carriers of X-linked recessive spinal and bulbar muscular atrophy. J Neurol. 2001;248:856–860. doi: 10.1007/s004150170069. [DOI] [PubMed] [Google Scholar]
- Katsuno M, Adachi H, Kume A, et al. Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron. 2002;35:843–854. doi: 10.1016/s0896-6273(02)00834-6. [DOI] [PubMed] [Google Scholar]
- Katsuno M, Adachi H, Doyu M, et al. Leuprorelin rescues polyglutamine-dependent phenotypes in a transgenic mouse model of spinal and bulbar muscular atrophy. Nat Med. 2003;9:768–773. doi: 10.1038/nm878. [DOI] [PubMed] [Google Scholar]
- Katsuno M, Adachi H, Minamiyama M, et al. Reversible disruption of dynactin 1-mediated retrograde axonal transport in polyglutamine-induced motor neuron degeneration. J Neurosci. 2006;26:12106–12117. doi: 10.1523/JNEUROSCI.3032-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuno M, Banno H, Suzuki K, et al. Efficacy and safety of leuprorelin in patients with spinal and bulbar muscular atrophy (JASMITT study): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2010;9:875–884. doi: 10.1016/S1474-4422(10)70182-4. [DOI] [PubMed] [Google Scholar]
- Kennedy WR, Alter M, Sung JH. Progressive proximal spinal and bulbar muscular atrophy of late onset: a sex-linked recessive trait. Neurology. 1968;18:671–680. doi: 10.1212/wnl.18.7.671. [DOI] [PubMed] [Google Scholar]
- La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature. 1991;352:77–79. doi: 10.1038/352077a0. [DOI] [PubMed] [Google Scholar]
- La Spada AR, Roling D, Harding AE, et al. Meiotic stability and genotype-phenotype correlation of the trinucleotide repeat in X-linked spinal and bulbar muscular atrophy. Nat Genet. 1992;2:301–304. doi: 10.1038/ng1292-301. [DOI] [PubMed] [Google Scholar]
- Lehky TJ, Chen CJ, Di Prospero NA, Rhodes LE, Fischbeck K, Floeter MK. Standard and modified statistical MUNE evaluations in spinal-bulbar muscular atrophy. Muscle Nerve. 2009;40:809–814. doi: 10.1002/mus.21399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Miwa S, Kobayashi Y, et al. Nuclear inclusions of the androgen receptor protein in spinal and bulbar muscular atrophy. Ann Neurol. 1998;44:249–254. doi: 10.1002/ana.410440216. [DOI] [PubMed] [Google Scholar]
- Lieberman AP, Harmison G, Strand AD, Olson JM, Fischbeck KH. Altered transcriptional regulation in cells expressing the expanded polyglutamine androgen receptor. Hum Mol Genet. 2002;11:1967–1976. doi: 10.1093/hmg/11.17.1967. [DOI] [PubMed] [Google Scholar]
- McCampbell A, Taylor JP, Taye AA, et al. CREB-binding protein sequestration by expanded polyglutamine. Hum Mol Genet. 2000;9:2197–2202. doi: 10.1093/hmg/9.14.2197. [DOI] [PubMed] [Google Scholar]
- Miller J, Arrasate M, Brooks E, et al. Identifying polyglutamine protein species in situ that best predict neurodegenera-tion. Nat Chem Biol. 2011;7:925–934. doi: 10.1038/nchembio.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamiyama M, Katsuno M, Adachi H, et al. Sodium butyrate ameliorates phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Hum Mol Genet. 2004;13:1183–1192. doi: 10.1093/hmg/ddh131. [DOI] [PubMed] [Google Scholar]
- Nagashima T, Seko K, Hirose K, et al. Familial bulbospinal muscular atrophy associated with testicular atrophy and sensory neuropathy (Kennedy-Alter-Sung syndrome). Autopsy case report of two brothers. J Neurol Sci. 1988;87:141–152. doi: 10.1016/0022-510x(88)90240-7. [DOI] [PubMed] [Google Scholar]
- Nedelsky NB, Pennuto M, Smith RB, et al. Native functions of the androgen receptor are essential to pathogenesis in a Drosophila model of spinobulbar muscular atrophy. Neuron. 2010;67:936–952. doi: 10.1016/j.neuron.2010.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata A, Matsuura T, Tashiro K, et al. Expression of androgen receptor in X-linked spinal and bulbar muscular atrophy and amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 1994;57:1274–1275. doi: 10.1136/jnnp.57.10.1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preisler N, Andersen G, Thogersen F, et al. Effect of aerobic training in patients with spinal and bulbar muscular atrophy (Kennedy disease) Neurology. 2009;72:317–323. doi: 10.1212/01.wnl.0000341274.61236.02. [DOI] [PubMed] [Google Scholar]
- Ranganathan S, Harmison GG, Meyertholen K, Pennuto M, Burnett BG, Fischbeck KH. Mitochondrial abnormalities in spinal and bulbar muscular atrophy. Hum Mol Genet. 2009;18:27–42. doi: 10.1093/hmg/ddn310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes LE, Freeman BK, Auh S, et al. Clinical features of spinal and bulbar muscular atrophy. Brain. 2009;132:3242–3251. doi: 10.1093/brain/awp258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair R, Greenland KJ, Egmond Sv, Hoedemaker C, Chapman A, Zajac JD. Men with Kennedy disease have a reduced risk of androgenetic alopecia. Br J Dermatol. 2007;157:290–294. doi: 10.1111/j.1365-2133.2007.08026.x. [DOI] [PubMed] [Google Scholar]
- Sobue G, Matsuoka Y, Mukai E, Takayanagi T, Sobue I. Pathology of myelinated fibers in cervical and lumbar ventral spinal roots in amyotrophic lateral sclerosis. J Neurol Sci. 1981;50:413–421. doi: 10.1016/0022-510x(81)90153-2. [DOI] [PubMed] [Google Scholar]
- Soraru G, D'Ascenzo C, Polo A, et al. Spinal and bulbar muscular atrophy: skeletal muscle pathology in male patients and heterozygous females. J Neurol Sci. 2008;264:100–105. doi: 10.1016/j.jns.2007.08.012. [DOI] [PubMed] [Google Scholar]
- Sumner CJ, Fischbeck KH. Jaw drop in Kennedy's disease. Neurology. 2002;59:1471–1472. doi: 10.1212/01.wnl.0000033325.01878.13. [DOI] [PubMed] [Google Scholar]
- Walcott JL, Merry DE. Ligand promotes intranuclear inclusions in a novel cell model of spinal and bulbar muscular atrophy. J Biol Chem. 2002;277:50855–50859. doi: 10.1074/jbc.M209466200. [DOI] [PubMed] [Google Scholar]
- Yu Z, Dadgar N, Albertelli M, et al. Androgen-dependent pathology demonstrates myopathic contribution to the Kennedy disease phenotype in a mouse knock-in model. J Clin Invest. 2006;116:2663–2672. doi: 10.1172/JCI28773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi HY, Orr HT. Glutamine repeats and neurodegeneration. Annu Rev Neurosci. 2000;23:217–247. doi: 10.1146/annurev.neuro.23.1.217. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. Tongue fibrillations and chin fasciculations in a patient with SBMA. (Patient data were collected with the understanding and written consent of each subject and according to ethical principles, including the World Medical Association Declaration of Helsinki (version, 2002)).