

Abstract
Pain in chronic pancreatitis (CP) shows similarities with other visceral pain syndromes (i.e., inflammatory bowel disease and esophagitis), which should thus be managed in a similar fashion. Typical causes of CP pain include increased intrapancreatic pressure, pancreatic inflammation and pancreatic/extrapancreatic complications. Unfortunately, CP pain continues to be a major clinical challenge. It is recognized that ongoing pain may induce altered central pain processing, e.g., central sensitization or pro-nociceptive pain modulation. When this is present conventional pain treatment targeting the nociceptive focus, e.g., opioid analgesia or surgical/endoscopic intervention, often fails even if technically successful. If central nervous system pain processing is altered, specific treatment targeting these changes should be instituted (e.g., gabapentinoids, ketamine or tricyclic antidepressants). Suitable tools are now available to make altered central processing visible, including quantitative sensory testing, electroencephalograpy and (functional) magnetic resonance imaging. These techniques are potentially clinically useful diagnostic tools to analyze central pain processing and thus define optimum management approaches for pain in CP and other visceral pain syndromes. The present review proposes a systematic mechanism-orientated approach to pain management in CP based on a holistic view of the mechanisms involved. Future research should address the circumstances under which central nervous system pain processing changes in CP, and how this is influenced by ongoing nociceptive input and therapies. Thus we hope to predict which patients are at risk for developing chronic pain or not responding to therapy, leading to improved treatment of chronic pain in CP and other visceral pain disorders.
Keywords: Chronic pancreatitis, Pain, Pain treatment, Central sensitization, Quantitative sensory testing, Electroencephalograpy, Magnetic resonance imaging
Core tip: Pain in chronic pancreatitis (CP) shows many similarities with other visceral pain syndromes. CP pain frequently leads to peripheral and central sensitization. When the latter is present, treating the nociceptive focus, with i.e., analgesic therapy, surgical or endoscopic procedures for local complications may fail even after technically successful procedures. In this case, treatment must be aimed at the central nervous system (CNS). Suitable tools to visualize altered central processing include quantitative sensory testing, electroencephalograpy and magnetic resonance imaging. Future research should be aimed at the circumstances under which CNS processing changes and how this is influenced by pain and therapies.
INTRODUCTION
Chronic pancreatitis (CP) involves progressive inflammatory changes of the pancreas resulting in morphological alterations and loss of pancreatic endocrine and exocrine function[1]. Quality of life is impaired and life expectancy is reduced[2,3]. The two main clinical manifestations of CP are pancreatic insufficiency and (chronic) abdominal pain. Pancreatic insufficiency is marked by exocrine dysfunction resulting in impaired food digestion and absorption, and endocrine dysfunction which results in diabetes mellitus[1]. Pain in CP is considered to be of visceral origin. When compared to other (chronic) visceral pain syndromes there are many similarities with the pain presentation of CP patients. The pain of CP is typically present as chronic epigastric pain, often radiating to the back, severe, dull, worse after eating and exhibiting episodic flares. This conforms to typical clinical characteristics of visceral pain which are: (1) the pain is not always simply or directly linked to morphological changes of the diseased organ; (2) pain is diffuse and poorly localized; (3) the pain may be referred to other locations; and (4) the pain is accompanied by motor and autonomic reflexes (vomiting, nausea and muscle tension)[4]. These parallels suggest that CP pain provides a useful model for the diagnosis and treatment of visceral pain syndromes with an identifiable nociceptive source in general.
Pain management in CP is at present mostly aimed at the nociceptive source, the pancreas. General recommendations include correction of pancreatic insufficiency and management of local complications, flanked by dietary modifications and cessation of alcohol use and smoking[1]. Currently a conservative step-up approach is advocated for pain treatment in CP, consisting of symptomatic pain relief and dealing with the pancreas as nociceptive source. For symptomatic pain relief, patients are treated with analgesics based on the “pain relief ladder” provided by the World Health Organization[5]. When such analgesic therapy is not successful, patients usually are referred for endoscopic interventions to attempt to reduce nociceptive input from the diseased pancreas. Eventually, patients may be referred for invasive surgical intervention if pain still persists despite prolonged analgesic (usually opioid) use and multiple endoscopic interventions (up to 75% of all patients).
Usually endoscopic interventions are performed for pancreatic duct strictures (stenting) and pancreatic duct stones (extracorporal shockwave therapy). Multiple surgical procedures have been described in the literature, all with different indications and success rates[6]. Drainage procedures like the pancreaticojejunostomy are performed for an enlarged pancreatic duct. When an enlarged pancreatic duct with an inflammatory mass in the pancreatic head is present, usually a Frey or Beger procedure is performed. Indications for (partial) pancreatic resections, i.e., pancreaticoduodenectomy, distal pancreatectomy and total pancreatectomy, are inflammatory masses in the head or tail of the pancreas, or failure of other therapies. Alternative approaches for dealing with the pancreas as a nociceptive source include deafferentation techniques such as nerve blocks and denervation procedures like bilateral thoracoscopic sphlanchnicectomy, which have shown to be beneficial for pain reduction in CP patients[7]. The success rate in terms of pain reduction after endoscopy or surgery is highly variable[6]. The optimal timing of interventions and which patients should be treated endoscopically or surgically continues to be intensively debated[6]. Despite these many management options, a significant number of chronic pancreatitis patients continue to experience pain even after conventional successful treatments, resulting in recurrent hospitalization, opioid dependence and severely impaired quality of life[8,9].
It is increasingly accepted that in many patients with refractory chronic pain, the pain may be the result of abnormal central pain processing which should be taken into account and targeted when pain management is planned[10]. This is in line with the key new insight of the last two to three decades of pain research, demonstrating that the central nervous system is not hard-wired, but rather highly plastic in the face of ongoing nociceptive input, exhibited as extensive alterations in central pain processing[10]. These changes typically involve increased pain sensitivity and facilitatory changes in modulation of painful inputs[11-13]. Further support for this view comes from recent successful studies with non-classical analgesic medication, i.e., S-ketamine and pregabalin, which targets mainly the central nervous system, and which has been shown to be effective in both visceral and somatic chronic pain syndromes[11,14,15].
To optimize (pain) treatment in CP, it is thus evident that we need to move away from approaches exclusively based on dealing with peripheral nociceptive input from the pancreas towards more holistic strategies taking into account alterations in central pain processing due to ongoing nociceptive inputs. The aim of this review is to highlight the recent progress in understanding the central mechanisms underlying chronic pain in CP and its impact on pain management. We present the evidence presently available that such central changes take place and operate in the human clinical context. Next, we focus on the diagnostics that are currently available to measure/visualize changes in central pain processing and how these are related to chronic pain in CP and other chronic abdominal visceral pain syndromes. Finally, based on these diagnostics we propose a new systematic mechanism-orientated approach to diagnosing and treating pain in CP as an example of an abdominal visceral pain syndrome.
A SYSTEMATIC MECHANISM-ORIENTATED APPROACH TO CHRONIC PAIN
Even after tissue healing, pain may persist as chronic pain with a major impact on quality of life. To date, the majority of publications on chronic pain adopt an empirical approach to the treatment of such pain, primarily based on dealing with the putative peripheral nociceptive source of the pain. At present, a holistic systematic mechanism-orientated approach to the prevention and treatment of chronic pain is lacking.
Key: Altered pain processing
A key insight has been that nervous system processing of pain is not hard-wired: sensory processing in the central nervous system typically changes as a result of noxious sensory inputs[16]. Acute nociception initially results in increased pain sensitivity (hyperalgesia) affecting the peripheral and central nervous system. When ongoing nociception (due to ongoing damage to tissues and nerves) is present, it initially sensitizes the peripheral nervous system. Subsequently, such ongoing nociceptive barrage will excite the spinal cord, brainstem and brain leading to central sensitization. In the end the whole nervous system may become sensitized, leading to exaggerated pain with minor stimuli (hyperalgesia) or even pain without nociceptive input (allodynia)[16-18]. Counteracting modulatory responses to nociceptive input like descending inhibition may fail as well, or even become facilitatory, resulting in more pain[9].
Four key questions
To achieve a holistic and systematic mechanism-orientated approach to chronic pain four key questions need to be answered[18].
What is the source of nociception? The majority of chronic pain disorders start off with a nociceptive source. Knowledge of the source enables us to aim our therapy at it and provides us with information regarding the type and intensity of nociception (e.g., visceral vs somatic pain).
Is nociceptive transmission altered? A common reason for altered nociceptive transmission by peripheral nerves to the central nervous system is peripheral nerve sensitization and damage. Nerve damage is a strong predictor for pain that is difficult to control or treat and can become a source of nociceptive input in itself. Nerve damage is associated with extensive and aggressive alteration in central nervous system function[19]. In addition, cytokines, hormones and other acute phase proteins may be released due to pathological processes and may facilitate sensitization of the central nervous system, e.g., via humoral pathways[9,20].
Is central pain processing altered? The first alteration in central nervous system processing to be taken into account is central sensitization, defined as an increased responsiveness of central pain transmitting neurons[9]. The presence and persistence of central sensitization affects both disease prognosis and effectiveness of therapy in chronic pain conditions. More extensive spread of central sensitization (generalized hyperalgesia) is associated with more pain. When central sensitization is present, therapy targeting only the source of nociception (the disease site) will be relatively ineffective. Thus drug treatment modulating the sensitization of the central nervous system need to be instituted. Examples of agents achieving this are gabapentinoids and antidepressants. Secondly, the state of descending central pain modulation must also be taken into account. If there is a pro-nociceptive (facilitatory) shift in central pain modulation, this has a negative effect on prognosis and requires specific treatment strategies[21].
Is altered central processing (still) dependent on peripheral nociceptive drive? If altered central processing becomes independent of peripheral nociceptive drive this further worsens the prognosis for controlling pain, and therapies aimed at controlling the nociceptive input from the source of disease are highly prone to failure. In this context, specific treatment dealing with altered central pain processing is mandatory e.g., gabapentinoids and antidepressants[18].
Implications
In summary, increasing evidence shows that (ongoing) nociceptive input results in altered central pain processing and should be taken into account in the management of chronic pain. However knowledge is lacking on how chronic painful inputs leads to altered central pain processing, and how this is influenced by disease progression and therapeutic interventions. Hence, the key to better treatment of chronic pain is measuring or visualizing the changes in the central nervous system - or neuroplasticity - that accompany the development and existence of chronic pain conditions. Together with measurements before and after treatment, the introduction of such systematic mechanism-orientated diagnostics will provide the basis for optimization of treatment indications and schedules.
A SYSTEMATIC MECHANISM-ORIENTATED APPROACH TO DIAGNOSING ALTERED PAIN PROCESSING IN CHRONIC PAIN
Quantitative sensory testing (QST), electroencephalograpy (EEG) and (functional) magnetic resonance imaging [(f)MRI] have increasingly been used in chronic pain disorders to describe changes in structure and function of the central nervous system. In the next paragraphs we will give a short introduction to QST, EEG and (f)MRI and their use in chronic pain conditions.
QST
The basis for QST was laid by Ulf Lindblom in the 1950s[22]. He was one of the first to describe the use of physiologic stimulation of the peripheral afferent unit in animals to test sensory processing. Later on he applied his experience in patients with sensory abnormalities i.e., chronic pain, which was the start of the use of QST in humans[23].
QST gives clinicians and researchers the opportunity to study abnormalities in the sensory system and characterize mechanisms underlying pathologic pain disorders. Compared to bedside clinical tests, QST is reliable and quantifies both the test stimulus (i.e., heat or pressure) and the patient’s response (i.e., pain)[24,25]. Somatosensory evoked responses to electrical, mechanical, thermal or chemical test modalities are involved in QST[26]. The stimulus is applied in a systematic fashion to an anatomical site (skin, muscle, joint or viscera like the esophagus or sigmoid). Stimulus intensity is gradually increased until the subject reaches a predefined sensory threshold (e.g., sensation or pain). By using multiple stimuli with differing intensities it is possible to construct a stimulus-response relationship (or curve) characterizing the subjects’ state of pain processing. This stimulus-response relationship is particularly useful as it also involves suprathreshold stimulation, particularly relevant to clinical pain. Measurements at the affected site or sites more distant are used to differentiate between signs of peripheral and (spinal or supraspinal) central sensitization.
Descending pain modulation (“pain inhibits pain”, a response to a noxious stimulus is inhibited by another noxious stimulus) is measured using the conditioned pain modulation paradigm (CPM, formerly known as diffuse noxious inhibitory controls or DNIC). In the case of CPM a test stimulation is applied (e.g., pain threshold, pain score), afterwards a conditioning stimulus is applied (e.g., cold pressor task via ice water bucket immersion) and then again the test stimulation is applied. The difference between the two test stimuli signals the size of inhibitory or facilitatory descending modulation. When central sensitization is present descending modulatory mechanisms often fail, due to a decreased activity in the inhibitory pathway of the spinal cord and an increase in facilitatory pathway activity, resulting in a further increase in pain (Figure 1)[27,28]. QST is increasingly used to compare pain sensitivity before and after interventions for patients and healthy controls in acute and chronic pain disorders.
Figure 1.
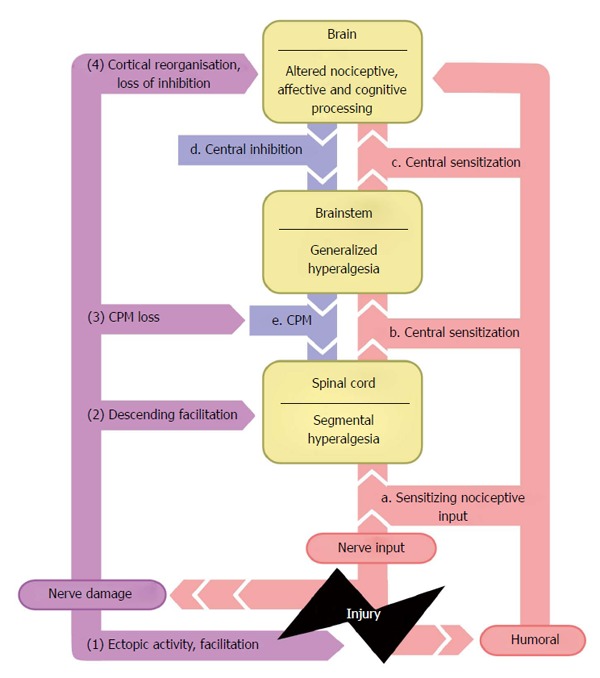
Summarizes the views presented above regarding the mechanisms underlying pain. This figure illustrates the concept of spread of altered central pain processing (progression marked via letters) following ongoing nociceptive input due to tissue and nerve damage (progression marked by numbers). This figure is based on the original figure of ref [18]. CPM: Conditioned pain modulation.
EEG
EEG is the recording of electrical brain activity, generated by synchronous activity of thousands of millions of neurons in the cortex. Neural networks are usually randomly active at any given time in a resting state, and can be synchronized in response to an external stimulus. Therefore, EEG can be used in chronic pain conditions to study the brains’ default state reflected by the resting state EEG (static element) and brain activity due to external stimuli reflected by event related or evoked brain potentials (dynamic element). As early as 1953, the EEG was already being studied in patients with pain due to peptic ulcers and functional gastric disorders by Kirschbaum et al[29]. Their study is an early example of the recognition of the brain-gut axis as a possible substrate for visceral pain syndromes. Although the use of EEG can be demanding and complex, this technique is a potentially useful non-invasive method for clinical practice. EEG has a poor spatial resolution, but superior millisecond-range temporal resolution compared to other neurodiagnostic instruments such as positron emission tomography or fMRI, enabling direct measurements of neuronal processing[30].
Resting state EEG: The resting state EEG is commonly analyzed by transforming data from the time domain into the frequency domain. Spontaneous brain activity in the frequency domain is divided into different frequency bands (delta = 1-3.5 Hz, theta = 3.5-7.5 Hz, alpha = 7.5-13 Hz, and beta = 13-32 Hz). The awake human brain activity recorded during rest is typically dominated by oscillations in the alpha frequency band. This dominant alpha activity is most prominent over parietal and occipital cortices, and is largest when the eyes are closed[31]. Recent developments in cognitive neuroscience suggests that alpha activity reflects selective cortical inhibition, rather than neural idling[32].
Alterations in the brains’ default state as reflected by resting state EEG, particularly in the alpha band, have been observed in multiple studies in various chronic pain conditions. Typically these changes consist of a shift of peak alpha or theta frequency to lower frequencies and/or a reduction in alpha or theta power[33-35]. It seems unlikely that alpha activity is directly related to the pain experience, as a correlation between pain intensity and alpha power is absent[35].
Evoked brain potentials: Event-related potentials or evoked potentials (EPs) are voltage polarity changes in the EEG time-locked to the onset of an external stimulus. They reflect the summed activity of postsynaptic potentials produced when a large number of similarly oriented neurons fire in synchrony while processing information[36]. EPs are traditionally extracted from the EEG by averaging similar repetitive stimuli within a stimulus block. Human EPs can be divided into two parts. The early components peaking roughly within the first 100 milliseconds after stimulus presentation are termed “sensory” or “exogenous” as they depend largely on the physical parameters of the stimulus. In contrast, later components of EPs reflect the manner in which the subject evaluates the stimulus and are termed “cognitive” or “endogenous” EPs as they examine information processing[37]. Alterations in evoked potentials are traditionally studied in the amplitudes and latencies of the (positive and negative) potential peaks, and can also be studied in the time frequency domain[38].
In order to obtain evoked potentials that are specific to nociceptive input, such input should be the result of physiological processing of nociceptive stimuli, i.e., involving selective activation of nociceptive Aδ/C-fibers in the periphery and recording resultant EPs generated in the cortex[39]. Brain mapping studies have established a positive relationship between the intensity of pain reported to nociceptive selective laser stimuli and EP amplitude[40]. In the context of evoked EEG studies, it must be noted that the experimental visceral electrical stimulation of large and small peripheral afferents that is generally applied to different gut segments is painful but not nociception specific[41]. Whether EPs resulting from stimuli entirely selective for nociceptive peripheral afferents represent the experience of pain or a more generalized response of heightened attention or arousal to afferent stimuli is current topic of debate[40,42,43]. Mouraux and Iannetti demonstrated that laser-evoked EEG responses reflect neural activities equally involved in processing nociceptive and non-nociceptive sensory inputs[43]. Thus, a stimulus entirely selective for nociceptive peripheral afferents does not imply that the elicited brain activity is nociception specific. However, even if EPs reflect neuronal activities that are unspecific for the nociceptive system, their generation still relies on the consequences of nociceptive activation and resultant changes in CNS state at both peripheral and central levels[43].
(f)MRI: (f)MRI has been increasingly used to describe brain activity and structural changes in chronic pain disorders. (f)MRI uses different techniques to measure functional brain activity. Changes in oxygenated and deoxygenated hemoglobin can be measured by the blood oxygenation level dependent technique[44]. By this technique the change in oxygenation (reflecting neuronal activity) in different areas of the brain can be estimated. Recently diffusion tensor imaging (DTI) has been used to measure changes in gray and white matter microstructure, and connectivity between brain areas[45]. Other functional techniques are signal enhancement by extravascular water protons and arterial spin labeling which allows the measurement of whole brain cerebral blood flow[46,47].
Taken together, the (f)MRI techniques allow assessment of the neural activation induced by stimuli like pain, and the structural neuroplastic changes induced by a long-lasting pain input. Compared to QST and EEG the advantage of (f)MRI is that it can take into account anatomy and can quantify the area of neuronal activity. The downside of the technique is that it is difficult to assess whether neural activity has a facilitatory or inhibitory effect on the pain processing. The main use for fMRI lies in anatomical resting state and activation studies[48]. Increasing evidence from studies using these tools has provided us with more information on central pain processing and how it can be influenced by disease progression and treatments.
Clinical diagnostics of pain processing
For implementation in the clinical context, a suitable tool to diagnose altered pain processing in chronic pain should fulfill the following criteria[18].
The tool should be validated and suitable for a clinical setting with a minimal burden for the patient. Measurements should be easy to reproduce and stimuli should be standardized so data can be compared between patients and populations. A tool that is easy to use can be used in an outpatient setting and has a low burden, increases patient compliance and makes the method more practical for clinical use.
The tool should reveal altered pain processing for both superficial and deep tissue stimulation. Differences in deep and superficial tissue stimulation may help discriminate between somatic and visceral origin of pain and the extent of central sensitization (e.g., somato-somatic, viscero-visceral and viscera-somatic spread of hyperalgesia).
The tool should contain static (pain sensitivity) and dynamic (pain modulation) elements. Static measurements provide insights into basal pain sensitivity (e.g., central sensitization) and dynamic measurements test how the body actively modulates nociceptive input.
The tool should sensitively assess changes in sensitization of pain processing as well as alterations in state of cortical/descending modulation. In the context of sensitized signal processing by the central nervous system, this will help differentiate e.g., between a situation of ongoing nociceptive input directly sensitizing central processing and pro-nociceptive alterations of descending nociceptive control by brainstem and brain (Figure 1).
Application of such a holistic approach to chronic pain is the basis for systematic mechanism-orientated pain management enabling: (1) diagnosis and prognosis of chronic pain; (2) rationale for treatment choice and responder identification; and (3) monitoring of chronic pain and its treatment[18].
EVIDENCE FOR A SYSTEMATIC MECHANISM-ORIENTATED APPROACH TO CHRONIC PAIN
In the next paragraphs we will focus on QST, EEG and (f)MRI research documenting the reality of altered pain processing in chronic visceral pain disorders such as chronic pancreatitis and thus providing further evidence for the feasibility of achieving a systematic mechanism-orientated approach in clinical practice.
What is the source of nociception?
In the literature the following pathophysiological mechanisms have most commonly been suggested as causes of pain in CP: (1) increased intrapancreatic pressure within the parenchyma and/or pancreatic duct causing tissue ischemia (due to pancreatic duct strictures and stones); (2) inflammation of the pancreas; and (3) pancreatic and extrapancreatic complications (i.e., pseudocysts, bile duct/duodenal strictures and peptic ulcers)[49-53]. The exact pathophysiology of chronic pancreatitis is still unknown and which mechanisms starts first are still subject to debate i.e., are duct strictures caused by tissue ischemia or inflammation or both?
Is nociceptive transmission altered?
In the past years, increasing evidence has been published regarding altered nociception transmission (e.g., nerve damage, peripheral sensitization) in chronic pain patients like CP[12,16,27,54]. In CP transmission of nociceptive input from the pancreas to the spinal cord can be altered and influenced by lesions in intrapancreatic and peripheral nerves, as described in histological studies[55,56]. These changes are comparable with other neuropathic pain disorders[9,57]. Not only an increase of excitability of nerves innervating the pancreas, but also structural changes of nerves in the pancreas may be a part of the problem. Hence, hypertrophy, increased neural density and neuritis of intrapancreatic nerves have been reported to be associated with pain in CP patients[58,59]. Ongoing nociceptive input due to the inflammation of the pancreas and its local complications may lead to nociceptors becoming more sensitive to further stimulation. This peripheral sensitization may be caused by upregulation of nerve growth factors, brain-derived neurotrophic factors and proinflammatory cytokines, and lead to increased pain intensity[60,61]. Pancreatic neuroplasticity (remodelling) and peripheral sensitization (increased excitability) will increase the nociceptive drive to the central nervous system resulting in an increased reaction of pain transmitting neurons (increase of pain)[59]. Finally, this process may result in spontaneous nociceptive activity without the presence of nociceptive inputs and to an aggressive increase of pain signals to the spinal cord[16,62].
Is central pain processing altered?
QST-CP: Increasing evidence has been published on segmental and generalized hyperalgesia and referred pain as a sign of spinal and supraspinal central sensitization in CP. Accordingly, decreased pain thresholds (i.e., hyperalgesia) for somatic stimulation in dermatomes near and distant to the pancreas in chronic pancreatitis patients are evident[7,11,13,27,54]. In agreement with this, other studies report increased areas of referred pain to electrical stimulation of viscera of upper gastrointestinal organs and decreased pain thresholds to visceral stimulation of the rectosigmoid[28,63]. These results suggest that peripheral visceral and somatic nerves converge at spinal levels in the central nervous system to elicit (somatic) referred pain as a sign of spinal central sensitization[64,65]. Failure of descending inhibitory pain modulation has also been observed in CP patients[11,25,27,28,54]. Probably this is due to a decreased activity in descending inhibitory pathways to the spinal cord as well as an increase in facilitatory activity projecting to the posterior spinal horn.
QST-visceral pain conditions: Similar to CP, sensitization of the central nervous system is seen in other inflammatory visceral pain conditions e.g., esophagitis and inflammatory bowel disorders, where it can be local in the viscera, spreading in the surrounding area or more distant in the case of referred pain. Drewes et al[66] showed segmental sensitization to thermal stimulation of the distal esophagus in esophagitis patients, together with a larger referred somatic pain area to mechanical stimulation, both reflecting central sensitization. Comparable results were found in ulcerative colitis and Crohn’s disease patients, who showed decreased pain thresholds to balloon dilation of the colon or rectal stimulationagain suggesting visceral hypersensitivity as a sign of central sensitization[67-69]. Evidence for descending counter-regulatory mechanisms has been described for patients with peptic ulcer and Crohn’s disease, both of whom showed hypoalgesia to visceral stimulation as a sign of effective tonic descending inhibition[70-72].
Clinical application of QST: In addition to characterization of the pain mechanisms underlying visceral pain disorders, QST has been used to study the effects of pain treatment on pain processing. In a study of S-ketamine, a noncompetitive NMDA receptor antagonist whose activity is related to central sensitization, infusions in CP patients were associated with a short-lasting increase in pain pressure thresholds, without a reduction in clinical pain. However, this study was not powered on clinical endpoints and had a short infusion time[11]. Another study showed that pregabalin reduced clinical pain in CP and was associated with a moderate anti-hyperalgesic effect. Interestingly patients treated with placebo also showed a reduction in clinical pain, but this effect came without changes in pain thresholds measured by QST[12,15].
The role of disease progression in CP and how it is influenced by interventions has not been well studied. Just one exploratory study in CP patients showed a relation between a more severe disease stage and lower pain thresholds (more hyperalgesia) compared to a moderate disease stage and healthy controls[27]. Interestingly, a study in CP patients after pain-relieving pancreatic surgery showed that patients with a poor pain outcome after surgery showed more central sensitization and more pronociceptive descending pain modulation compared to patients with a good pain outcome and healthy controls[73].
To summarize: CP and other abdominal visceral pain syndromes show similarities in pain mechanisms and physiology. In the area of tissue damage and its surrounding tissue there is typically hypersensitivity to all kinds of different stimuli as signs of segmental hyperalgesia. When pain is ongoing, tissues more distant of the area of injury also become sensitized as (generalized hyperalgesia) as a sign of spreading central sensitisation. Failure of counter-regulatory mechanisms such as DNIC, measured via e.g., CPM, also leads to hyperalgesia and pain increases. Treatments aimed at central pain mechanisms may reduce pain and hyperalgesia in such patients. Evidence regarding the role of disease progression and treatments aimed at reducing pain and central sensitization is still scarce. However, it is evident that QST can play a useful role in quantifying pain processing and its impact on clinical pain before and after pain treatment[74,75].
Resting state electroencephalography - CP: Olesen et al[76] reported an increase in amplitude strength in the theta and alpha band in patients with CP compared to healthy controls, reflecting slowed EEG rhythmicity in patients with CP compared to controls. Another study demonstrated a significant shift toward lower frequencies in patients with CP compared with healthy controls[33]. This was observed as a decrease in peak alpha frequency over all scalp electrodes. Interestingly, these changes correlated with duration of pain, further supporting alterations in resting state EEG as a potential biomarker in chronic pain conditions.
The mechanisms underlying these observations are still poorly understood. One hypothesis is that of thalamocortical dysrythmia (TCD), where damage or lesions to afferent neural pathways results in deafferentation and a decrease in excitatory input to the thalamic relay cells. This results in disfacilitation and cell membrane hyperpolarization due to activation of T-type calcium channels. In this hyper-excitatory state thalamic relay neurons fire low threshold spike bursts and the normal thalamo-cortical rhythmicity is disturbed[30]. Application of drugs that interfere with T-type calcium channel function may prevent low frequency bursting, reverse TCD, and alleviate pain in conditions with underlying TCD. Thus resting state EEG may be of value not only as a potential biomarker for chronic pain progression via shifts in oscillatory activity, but also in treatment decisions and evaluation via identification of TCD. Another hypothesis is based on recent experiments indicating that the phase of alpha activity modulates perception and that alpha oscillations are produced by periodic pulses of inhibition. It was suggested that posterior alpha oscillations provide a mechanism for prioritizing and ordering unattended visual input according to “relevance” or saliency[32]. However, it is unclear whether the proposed role of alpha activity can be generalized to other modalities, such as the somatosensory and nociceptive system.
Evoked brain potentials EEG - CP: Dimcevski et al[63] recorded EPs after stimuli given with a constant current electric stimulator at the three different sites of the upper gastrointestinal tract. Patients with CP had a significantly decreased latency for the N1 and P1, while N2 latency was borderline significant compared to healthy subjects. No differences were found in the amplitudes of the N1, P1, and N2 potentials. In another study using evoked visceral pain of the upper gastrointestinal tract, patients showed higher activity than controls in the theta band, with prolonged persistence of the signal and at lower frequency (4.4 Hz in patients compared to 5.5 Hz in controls)[10]. In a second study, patients with CP showed hyperalgesia to electrical stimulation and prolonged latencies of early visceral EPs components in the frontal region of the cortex compared to healthy controls. Additionally, scalp distributions of EP amplitudes were more scattered and more posteriorly located in the patient group[28]. As the changes in cortical processing were correlated to the pain this further validates the findings. To date, no comparable data are available for other types of abdominal focus-related chronic pain.
Clinical application of EEG: Studies using EEG to identify patients who may benefit from treatment strategies targeting central pain mechanisms are limited. Graversen et al[77] studied the resting state EEG after a three week regimen of pregabalin or matching placebo in patients with CP. Patients in the pregabalin group showed a significant increase in theta activity after pregabalin treatment, while no changes were observed for the other frequency bands, nor were any changes found in the placebo group. The authors concluded that quantitative pharmaco-EEG can be used to monitor central analgesic mechanisms of pregabalin and may in the future be used to predict treatment effects[77].
To summarize: Studies in chronic visceral pain have investigated both the resting state as well as the evoked EEG. The use of multiple analysis techniques and different stimulation methods makes these results difficult to compare. Alpha activity in the resting state EEG has been shown to be affected in multiple chronic pain states including CP, suggesting a change in the default state of the brain as a result of chronic pain. Pain-evoked EEG studies in CP patients demonstrate alterations in dynamic pain processing reflected by prolonged latencies of visceral EPs and higher theta activity with prolonged persistence of the signal at a lower frequency during experimental visceral pain. Taken together, these EEG findings further support the concept that chronic visceral pain conditions such as chronic pancreatitis are associated with significant and ubiquitous alterations in resting state and evoked CNS processing, both nociceptive and non-nociceptive.
(f)MRI
The cortical and subcortical structures that are involved in visceral pain are the thalamus from which signals further ascend to different parts of the brain i.e., the limbic system (insula, cingulate cortex and prefrontal cortex), the primary (discriminating pain) and secondary (recognizing and remembering pain) somatosensory cortex[30]. In particular the insula has an important function in pain perception from the gut[78]. The functional relationship between these areas was described with DTI for healthy controls who underwent rectal distension[79]. Important areas for pain experience, influenced by cognitive, affective and emotional components, are processed in the limbic system. Other structures involved are: the amygdala, periaquaductal gray matter, reticular formation and hypothalamus. These structures are mostly related to pro- and antinociceptive control such as descending pain control[80].
CP: A MRI study with DTI in CP patients showed increased diffusivity in grey matter regions of the insula and cingulate cortex suggesting microstructural changes of pain associated brain areas. These observations appeared to be directly correlated to the pain experienced by patients. Another MRI volumetry CP study supported these findings and showed cortical thinning in similar brain areas (the limbic system)[81]. Brain areas that are associated with descending pain modulation e.g., the cingulate cortex, hypothalamus and periaqueductal grey matter showed cortical thinning in some studies with CP patients. These results might explain impaired descending inhibition in chronic pancreatitis[28,81]. Overall, in CP patients different brain areas that are involved in visceral pain processing showed a decrease in cortical thickness. Whether these changes are due to chronic pain and how these changes influence pain processing is unknown at the moment.
Visceral pain conditions: Studies in other abdominal visceral pain syndromes are scarce. However, similar results to studies in CP were found in patients with inflammatory bowel disease when they were compared to healthy controls[82].
Clinical application of (f)MRI: At present there are no studies using (f)MRI to observe therapeutic effects or disease progression in CP.
To summarize: Similarly to EEG studies, (f)MRI studies have shown for CP patients and other visceral pain syndromes that changes in brain activity are present particularly in areas that are related to pain processing such as the limbic system, hypothalamus and periaqueductal regions. However the role of pain in these changes and how this influences pain perception is poorly understood at the moment.
Is altered central processing (still) dependent on peripheral nociceptive drive?
Central sensitization manifest as spreading hyperalgesia can ultimately become independent of peripheral nociceptive input and no longer respond to treatments targeting the source of nociception and/or achieving peripheral deafferentation i.e., nerve blocks and opioids. Changes in central pain processing independent of peripheral nociceptive input were supported by a study involving CP patients who had a splanchnic denervation to reduce pain, but where ca. 75% continued to experience painful and exhibit widespread hyperalgesia (4 years) after a technically successful procedure, suggesting real central autonomy[54,83]. Further literature on the reversibility of central sensitisation is scarce. One study described two different groups of patients with osteoarthritis after hip replacement surgery, one that showed reversibility of hyperalgesia and a descending inhibitory modulation deficit and another group that had ongoing pain without changes in hyperalgesia and no changes in central inhibition suggesting the presence of central autonomy[18].
IMPLEMENTING A SYSTEMATIC MECHANISM-ORIENTATED APPROACH TO CHRONIC PAIN IN CLINICAL PRACTICE
Source of nociception
QST performed at the site of the nociceptive focus can help identify the source of nociception and provide insight into the nature and aggressiveness of the nociceptive input involved (e.g., visceral pain). EEG and (f)MRI diagnostics have no role in this context.
Altered nociceptive transmission
QST performed close to the site of nociception can be used to help diagnose peripheral sensitization (local, primary hyperalgesia, usually thermal) and nerve damage (classically thermal hypoalgesia and hypoaesthesia in the territory of the nerve in question)[84-87]. Theoretically, evoked potential EEG studies could be used to quantify alterations in nociceptive transmission. However, most EP studies only involve large fibre non-nociceptive somatosensory processing; there are only a few such studies involving nociception-relevant small fibres (e.g., laser EPs).
Role of QST in describing altered central pain processing
QST measured close and distant to the site of pain allows differentiation between segmental (spinal central sensitization) or generalized (supraspinal central sensitization) hyperalgesia. Stimulation of different tissues (e.g., electrical skin stimulation, mechanical stimulation of muscle by pressure algometry) can further help understand the source of pain and spread of associated altered pain processing. Dynamic QST measurements such as the CPM paradigm are helpful in diagnosing shifts in descending nociceptive modulation.
Is altered central processing (still) dependent on peripheral nociceptive drive?
In this case, central sensitization is present but no longer dependent on ongoing nociceptive input. Thus (trial) treatments aiming to deafferent the nociceptive source (e.g., nerve block or nerve transection) will not be accompanied by changes in central pain processing (e.g., spreading hyperalgesia) as measured by QST. As flanking - mainly experimental - procedures, EEG and (f)MRI have made it possible to directly demonstrate cortical reorganization, altered connectivity and modulation in chronic pain conditions.
Clinical use
Diagnostics: At our institution, QST has proven useful to diagnose and monitor changes in pain processing accompanying chronic pain. Our research and clinical experience suggest that implementation of a systematic mechanism-orientated approach to pain based on a simple diagnostic QST is both feasible and desirable in clinical pain practice. To this end we have instituted a simple QST screening paradigm, which all difficult chronic pain patients undergo [the Nijmegen-Aalborg screening QST (NASQ)][18]. The NASQ paradigm includes four measurement points measured bilaterally (close and distant to the site of pain, thus providing topographical information), two stimulation modalities (electric and pressure stimulation) and a CPM paradigm (cold pressor task). Details are provided in Table 1[18].
Table 1.
Nijmegen-Aalborg screening quantitative sensory testing paradigm[18]
| NASQ paradigm | |
| Standard QST | |
| Sites (bilateral) | Trapezius muscle, thenar eminence, rectus femoris, abductor hallucis, site of pain |
| Thresholds | Pressure pain, electric detection, electric pain detection, electric pain tolerance |
| Conditioned pain modulation | |
| Sites | 1 Ice-water bucket (non-dominant hand) |
| 2 Thresholds on rectus femoris | |
| Thresholds (before ice-water/180 s after) | Pressure pain, electric pain tolerance |
Quantitative sensory testing (QST) measurements to detect central sensitization and pro- or anti-nociceptive shifts in descending pain modulation. NASQ: Nijmegen-Aalborg screening QST.
The NASQ paradigm is well accepted by patients, easy to perform and learn, and can be completed within 30 min. Thermal QST testing can be added to test specifically for peripheral nerve damage[18,88].
Regarding clinical use of EEG and (f)MRI in chronic pain, the literature remains scarce. Furthermore both investigations are onerous, time consuming and expensive. Therefore we do not at present recommend their use in daily clinical practice for chronic pain patients, reserving these techniques for research.
Therapeutics: The new approach to pain in CP presented here allows for holistic and systematic management of CP pain. Such a systematic mechanism-orientated approach not only facilitates the diagnosis and prognosis of chronic pain, it also provides the possibility of monitoring signs of chronic pain progression. As such, it forms the basis for more rational choice of treatment options to maximize treatment response, together with subsequent ongoing monitoring of effectiveness of chronic pain treatment.
Table 2 provides a summary of our systematic mechanism-orientated approach to chronic pain, such as pancreatitis pain, as implemented at our institution. The scheme is based on the literature discussed in this review and our own clinical experience and practice.
Table 2.
Schematic for systematic mechanism-orientated approach to chronic pancreatitis pain
| Questions | Issue | QST | Therapy |
| Nociceptive source? | Site/agressiveness | Local hyperalgesia | Treat or deafferent |
| Nociceptive transmission? | Nerve damage | Territorial thermal hyperalgesia | Treat (cave CS!) |
| Central pain processing? | Central sensitisation | Spreading hyperalgesia | Antihyperalgesia (ketamine, gabapentinoids) |
| Pronociceptive modulation | Sensitisation to CPM paradigm | Activate DI (TCA, NRI) | |
| Autonomy of central pain processing? | Autonomy | No changes in thresholds after therapy | Traget altered central processing |
Autonomy means that alterations in central pain processing have become independent of peripheral nociceptive drive. CPM: Conditioned pain modulation; DI: Descending inhibition; TCA: Tricyclic antidepressant; NRI: Noradrenaline reuptake inhibitor; QST: Quantitative sensory testing. This figure is based on the original figure of ref [18].
CONCLUSION
Intense abdominal pain is the dominant feature of CP. In this review we propose a new systematic mechanism-orientated approach to the chronic pain of CP. Multiple studies support that pain in CP is similar to other visceral pain syndromes such as inflammatory bowel disease. Increasing evidence has shown that changes in central pain processing are present and comparable in CP and other abdominal visceral pain syndromes. The data suggest that changes in pain processing due to chronic visceral pain are common and necessitate a targeted and mechanism-orientated diagnostic and therapeutic approach. This management approach needs to be holistic, including not only traditional treatments addressing the pancreas as a nociceptive source, but also specifically searching for - and therapeutically targeting - alterations in CNS processing of pain.
As shown in this review, QST, EEG and (f)MRI can be useful diagnostic instruments to analyze central pain processing and help us in finding optimal mechanism-orientated treatments for pain in CP and other chronic visceral pain syndromes. Future research should define the presence and pattern of altered pain processing for specific chronic pain disorders and compare this with a healthy population using diagnostic tools such as QST, EEG and fMRI. Apart from characterization of hyperalgesia and descending pain modulation further questions need to be addressed. How does hyperalgesia develop over time? How is this influenced by disease progression and our treatments? What is the impact of gender and psychological state? Can we predict patients who are prone to chronic pain and altered central pain processing? The only way to increase our knowledge in this respect is to measure the effect of pain and nociception on central pain processing in large-scale clinical studies using QST, EEG or fMRI before and after interventions and during disease progression[77]. This will help us evaluate therapies and guide us to the proper treatment for a specific patient at a specific disease stages. Such personalized medicine is the key to improved pain treatment and may pave the way to new and more effective therapeutic approaches.
ACKNOWLEDGMENTS
We like to thank Dr. Jan-Maarten Luursema for providing us with the design of the pictures in this manuscript.
Footnotes
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: July 3, 2014
First decision: July 21, 2014
Article in press: November 19, 2014
P- Reviewer: Dimcevski G, Demir IE, Ruckert F S- Editor: Gou SX L- Editor: A E- Editor: Wang CH
References
- 1.Steer ML, Waxman I, Freedman S. Chronic pancreatitis. N Engl J Med. 1995;332:1482–1490. doi: 10.1056/NEJM199506013322206. [DOI] [PubMed] [Google Scholar]
- 2.Amann ST, Yadav D, Barmada MM, O’Connell M, Kennard ED, Anderson M, Baillie J, Sherman S, Romagnuolo J, Hawes RH, et al. Physical and mental quality of life in chronic pancreatitis: a case-control study from the North American Pancreatitis Study 2 cohort. Pancreas. 2013;42:293–300. doi: 10.1097/MPA.0b013e31826532e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bang UC, Benfield T, Hyldstrup L, Bendtsen F, Beck Jensen JE. Mortality, cancer, and comorbidities associated with chronic pancreatitis: a Danish nationwide matched-cohort study. Gastroenterology. 2014;146:989–994. doi: 10.1053/j.gastro.2013.12.033. [DOI] [PubMed] [Google Scholar]
- 4.Cervero F, Laird JM. Visceral pain. Lancet. 1999;353:2145–2148. doi: 10.1016/S0140-6736(99)01306-9. [DOI] [PubMed] [Google Scholar]
- 5.Jadad AR, Browman GP. The WHO analgesic ladder for cancer pain management. Stepping up the quality of its evaluation. JAMA. 1995;274:1870–1873. [PubMed] [Google Scholar]
- 6.Issa Y, van Santvoort HC, van Goor H, Cahen DL, Bruno MJ, Boermeester MA. Surgical and endoscopic treatment of pain in chronic pancreatitis: a multidisciplinary update. Dig Surg. 2013;30:35–50. doi: 10.1159/000350153. [DOI] [PubMed] [Google Scholar]
- 7.Buscher HC, van Goor H, Wilder-Smith OH. Effect of thoracoscopic splanchnic denervation on pain processing in chronic pancreatitis patients. Eur J Pain. 2007;11:437–443. doi: 10.1016/j.ejpain.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 8.Andrén-Sandberg A, Hoem D, Gislason H. Pain management in chronic pancreatitis. Eur J Gastroenterol Hepatol. 2002;14:957–970. doi: 10.1097/00042737-200209000-00006. [DOI] [PubMed] [Google Scholar]
- 9.Drewes AM, Krarup AL, Detlefsen S, Malmstrøm ML, Dimcevski G, Funch-Jensen P. Pain in chronic pancreatitis: the role of neuropathic pain mechanisms. Gut. 2008;57:1616–1627. doi: 10.1136/gut.2007.146621. [DOI] [PubMed] [Google Scholar]
- 10.Drewes AM, Gratkowski M, Sami SA, Dimcevski G, Funch-Jensen P, Arendt-Nielsen L. Is the pain in chronic pancreatitis of neuropathic origin? Support from EEG studies during experimental pain. World J Gastroenterol. 2008;14:4020–4027. doi: 10.3748/wjg.14.4020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bouwense SA, Buscher HC, van Goor H, Wilder-Smith OH. S-ketamine modulates hyperalgesia in patients with chronic pancreatitis pain. Reg Anesth Pain Med. 2011;36:303–307. doi: 10.1097/AAP.0b013e3182177022. [DOI] [PubMed] [Google Scholar]
- 12.Bouwense SA, Olesen SS, Drewes AM, Poley JW, van Goor H, Wilder-Smith OH. Effects of pregabalin on central sensitization in patients with chronic pancreatitis in a randomized, controlled trial. PLoS One. 2012;7:e42096. doi: 10.1371/journal.pone.0042096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buscher HC, Wilder-Smith OH, van Goor H. Chronic pancreatitis patients show hyperalgesia of central origin: a pilot study. Eur J Pain. 2006;10:363–370. doi: 10.1016/j.ejpain.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 14.Moore RA, Straube S, Wiffen PJ, Derry S, McQuay HJ. Pregabalin for acute and chronic pain in adults. Cochrane Database Syst Rev. 2009;(3):CD007076. doi: 10.1002/14651858.CD007076.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Olesen SS, Bouwense SA, Wilder-Smith OH, van Goor H, Drewes AM. Pregabalin reduces pain in patients with chronic pancreatitis in a randomized, controlled trial. Gastroenterology. 2011;141:536–543. doi: 10.1053/j.gastro.2011.04.003. [DOI] [PubMed] [Google Scholar]
- 16.Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288:1765–1769. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- 17.Coderre TJ, Katz J, Vaccarino AL, Melzack R. Contribution of central neuroplasticity to pathological pain: review of clinical and experimental evidence. Pain. 1993;52:259–285. doi: 10.1016/0304-3959(93)90161-H. [DOI] [PubMed] [Google Scholar]
- 18.Wilder-Smith OH. A Paradigm-Shift in Pain Medicine. USA: River Publishers; 2013. [Google Scholar]
- 19.Schaible HG, Vanegas H. How do we manage chronic pain? Baillieres Best Pract Res Clin Rheumatol. 2000;14:797–811. doi: 10.1053/berh.2000.0114. [DOI] [PubMed] [Google Scholar]
- 20.Neziri AY, Bersinger NA, Andersen OK, Arendt-Nielsen L, Mueller MD, Curatolo M. Correlation between altered central pain processing and concentration of peritoneal fluid inflammatory cytokines in endometriosis patients with chronic pelvic pain. Reg Anesth Pain Med. 2014;39:181–184. doi: 10.1097/AAP.0000000000000068. [DOI] [PubMed] [Google Scholar]
- 21.Yarnitsky D, Granot M, Nahman-Averbuch H, Khamaisi M, Granovsky Y. Conditioned pain modulation predicts duloxetine efficacy in painful diabetic neuropathy. Pain. 2012;153:1193–1198. doi: 10.1016/j.pain.2012.02.021. [DOI] [PubMed] [Google Scholar]
- 22.Lindblom K. Technique and results in myelography and disc puncture. Acta radiol. 1950;34:321–330. doi: 10.3109/00016925009135278. [DOI] [PubMed] [Google Scholar]
- 23.Lindblom U, Tapper DN. Terminal properties of a vibro-tactile sensor. Exp Neurol. 1967;17:1–15. doi: 10.1016/0014-4886(67)90117-3. [DOI] [PubMed] [Google Scholar]
- 24.Geber C, Klein T, Azad S, Birklein F, Gierthmühlen J, Huge V, Lauchart M, Nitzsche D, Stengel M, Valet M, et al. Test-retest and interobserver reliability of quantitative sensory testing according to the protocol of the German Research Network on Neuropathic Pain (DFNS): a multi-centre study. Pain. 2011;152:548–556. doi: 10.1016/j.pain.2010.11.013. [DOI] [PubMed] [Google Scholar]
- 25.Olesen SS, van Goor H, Bouwense SA, Wilder-Smith OH, Drewes AM. Reliability of static and dynamic quantitative sensory testing in patients with painful chronic pancreatitis. Reg Anesth Pain Med. 2012;37:530–536. doi: 10.1097/AAP.0b013e3182632c40. [DOI] [PubMed] [Google Scholar]
- 26.Pavlaković G, Petzke F. The role of quantitative sensory testing in the evaluation of musculoskeletal pain conditions. Curr Rheumatol Rep. 2010;12:455–461. doi: 10.1007/s11926-010-0131-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bouwense SA, Olesen SS, Drewes AM, Frøkjær JB, van Goor H, Wilder-Smith OH. Is altered central pain processing related to disease stage in chronic pancreatitis patients with pain? An exploratory study. PLoS One. 2013;8:e55460. doi: 10.1371/journal.pone.0055460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Olesen SS, Brock C, Krarup AL, Funch-Jensen P, Arendt-Nielsen L, Wilder-Smith OH, Drewes AM. Descending inhibitory pain modulation is impaired in patients with chronic pancreatitis. Clin Gastroenterol Hepatol. 2010;8:724–730. doi: 10.1016/j.cgh.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 29.Kirschbaum WR, Stehle HC. Electroencephalographic studies of patients with peptic ulcer and functional gastric disorders. Electroencephalogr Clin Neurophysiol. 1953;5:513–520. doi: 10.1016/0013-4694(53)90027-3. [DOI] [PubMed] [Google Scholar]
- 30.Frokjaer JB, Olesen SS, Graversen C, Andresen T, Lelic D, Drewes AM. Neuroimaging of the human visceral pain system-A methodological review. Scand J Pain. 2011;2:95–104. doi: 10.1016/j.sjpain.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 31.Niedermeyer E, Krauss GL, Peyser CE. The electroencephalogram and mental activation. Clin Electroencephalogr. 1989;20:215–227. doi: 10.1177/155005948902000409. [DOI] [PubMed] [Google Scholar]
- 32.Jensen O, Bonnefond M, VanRullen R. An oscillatory mechanism for prioritizing salient unattended stimuli. Trends Cogn Sci. 2012;16:200–206. doi: 10.1016/j.tics.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 33.de Vries M, Wilder-Smith OH, Jongsma ML, van den Broeke EN, Arns M, van Goor H, van Rijn CM. Altered resting state EEG in chronic pancreatitis patients: toward a marker for chronic pain. J Pain Res. 2013;6:815–824. doi: 10.2147/JPR.S50919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Olesen SS, Hansen TM, Graversen C, Steimle K, Wilder-Smith OH, Drewes AM. Slowed EEG rhythmicity in patients with chronic pancreatitis: evidence of abnormal cerebral pain processing? Eur J Gastroenterol Hepatol. 2011;23:418–424. doi: 10.1097/MEG.0b013e3283457b09. [DOI] [PubMed] [Google Scholar]
- 35.van den Broeke EN, Wilder-Smith OH, van Goor H, Vissers KC, van Rijn CM. Patients with persistent pain after breast cancer treatment show enhanced alpha activity in spontaneous EEG. Pain Med. 2013;14:1893–1899. doi: 10.1111/pme.12216. [DOI] [PubMed] [Google Scholar]
- 36.Peterson NN, Schroeder CE, Arezzo JC. Neural generators of early cortical somatosensory evoked potentials in the awake monkey. Electroencephalogr Clin Neurophysiol. 1995;96:248–260. doi: 10.1016/0168-5597(95)00006-e. [DOI] [PubMed] [Google Scholar]
- 37.Sur S, Sinha VK. Event-related potential: An overview. Ind Psychiatry J. 2009;18:70–73. doi: 10.4103/0972-6748.57865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nissen TD, Brock C, Graversen C, Coen SJ, Hultin L, Aziz Q, Lykkesfeldt J, Drewes AM. Translational aspects of rectal evoked potentials: a comparative study in rats and humans. Am J Physiol Gastrointest Liver Physiol. 2013;305:G119–G128. doi: 10.1152/ajpgi.00403.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garcia-Larrea L. Objective pain diagnostics: clinical neurophysiology. Neurophysiol Clin. 2012;42:187–197. doi: 10.1016/j.neucli.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 40.Kramer JL, Haefeli J, Jutzeler CR. An objective measure of stimulus-evoked pain. J Neurosci. 2012;32:12981–12982. doi: 10.1523/JNEUROSCI.3175-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.De Broucker T, Willer JC. [Comparative study of the nociceptive reflex and late components of the evoked somatosensory potential during stimulation of the sural nerve in healthy subjects] Rev Electroencephalogr Neurophysiol Clin. 1985;15:149–153. doi: 10.1016/s0370-4475(85)80019-8. [DOI] [PubMed] [Google Scholar]
- 42.Baumgärtner U, Treede RD. Are there nociceptive-specific brain potentials? J Neurophysiol. 2009;102:3073–3074; author reply 3075-3076. doi: 10.1152/jn.00588.2009. [DOI] [PubMed] [Google Scholar]
- 43.Mouraux A, Iannetti GD. Nociceptive laser-evoked brain potentials do not reflect nociceptive-specific neural activity. J Neurophysiol. 2009;101:3258–3269. doi: 10.1152/jn.91181.2008. [DOI] [PubMed] [Google Scholar]
- 44.Turner R. Magnetic resonance imaging of brain function. Am J Physiol Imaging. 1992;7:136–145. [PubMed] [Google Scholar]
- 45.Nucifora PG, Verma R, Lee SK, Melhem ER. Diffusion-tensor MR imaging and tractography: exploring brain microstructure and connectivity. Radiology. 2007;245:367–384. doi: 10.1148/radiol.2452060445. [DOI] [PubMed] [Google Scholar]
- 46.Detre JA, Leigh JS, Williams DS, Koretsky AP. Perfusion imaging. Magn Reson Med. 1992;23:37–45. doi: 10.1002/mrm.1910230106. [DOI] [PubMed] [Google Scholar]
- 47.Tracey I, Johns E. The pain matrix: reloaded or reborn as we image tonic pain using arterial spin labelling. Pain. 2010;148:359–360. doi: 10.1016/j.pain.2009.11.009. [DOI] [PubMed] [Google Scholar]
- 48.Napadow V, LaCount L, Park K, As-Sanie S, Clauw DJ, Harris RE. Intrinsic brain connectivity in fibromyalgia is associated with chronic pain intensity. Arthritis Rheum. 2010;62:2545–2555. doi: 10.1002/art.27497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abdallah AA, Krige JE, Bornman PC. Biliary tract obstruction in chronic pancreatitis. HPB (Oxford) 2007;9:421–428. doi: 10.1080/13651820701774883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Andrén-Sandberg A, Dervenis C. Pancreatic pseudocysts in the 21st century. Part I: classification, pathophysiology, anatomic considerations and treatment. JOP. 2004;5:8–24. [PubMed] [Google Scholar]
- 51.Barreto SG, Saccone GT. Pancreatic nociception--revisiting the physiology and pathophysiology. Pancreatology. 2012;12:104–112. doi: 10.1016/j.pan.2012.02.010. [DOI] [PubMed] [Google Scholar]
- 52.Demir IE, Tieftrunk E, Maak M, Friess H, Ceyhan GO. Pain mechanisms in chronic pancreatitis: of a master and his fire. Langenbecks Arch Surg. 2011;396:151–160. doi: 10.1007/s00423-010-0731-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vijungco JD, Prinz RA. Management of biliary and duodenal complications of chronic pancreatitis. World J Surg. 2003;27:1258–1270. doi: 10.1007/s00268-003-7246-7. [DOI] [PubMed] [Google Scholar]
- 54.Bouwense SA, Buscher HC, van Goor H, Wilder-Smith OH. Has central sensitization become independent of nociceptive input in chronic pancreatitis patients who fail thoracoscopic splanchnicectomy? Reg Anesth Pain Med. 2011;36:531–536. doi: 10.1097/AAP.0b013e31822e0d4a. [DOI] [PubMed] [Google Scholar]
- 55.Ceyhan GO, Bergmann F, Kadihasanoglu M, Altintas B, Demir IE, Hinz U, Müller MW, Giese T, Büchler MW, Giese NA, et al. Pancreatic neuropathy and neuropathic pain--a comprehensive pathomorphological study of 546 cases. Gastroenterology. 2009;136:177–186.e1. doi: 10.1053/j.gastro.2008.09.029. [DOI] [PubMed] [Google Scholar]
- 56.Demir IE, Schorn S, Schremmer-Danninger E, Wang K, Kehl T, Giese NA, Algül H, Friess H, Ceyhan GO. Perineural mast cells are specifically enriched in pancreatic neuritis and neuropathic pain in pancreatic cancer and chronic pancreatitis. PLoS One. 2013;8:e60529. doi: 10.1371/journal.pone.0060529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ceyhan GO, Demir IE, Maak M, Friess H. Fate of nerves in chronic pancreatitis: Neural remodeling and pancreatic neuropathy. Best Pract Res Clin Gastroenterol. 2010;24:311–322. doi: 10.1016/j.bpg.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 58.Bockman DE, Buchler M, Malfertheiner P, Beger HG. Analysis of nerves in chronic pancreatitis. Gastroenterology. 1988;94:1459–1469. doi: 10.1016/0016-5085(88)90687-7. [DOI] [PubMed] [Google Scholar]
- 59.Poulsen JL, Olesen SS, Malver LP, Frøkjær JB, Drewes AM. Pain and chronic pancreatitis: a complex interplay of multiple mechanisms. World J Gastroenterol. 2013;19:7282–7291. doi: 10.3748/wjg.v19.i42.7282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Friess H, Zhu ZW, di Mola FF, Kulli C, Graber HU, Andren-Sandberg A, Zimmermann A, Korc M, Reinshagen M, Büchler MW. Nerve growth factor and its high-affinity receptor in chronic pancreatitis. Ann Surg. 1999;230:615–624. doi: 10.1097/00000658-199911000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhu ZW, Friess H, Wang L, Zimmermann A, Büchler MW. Brain-derived neurotrophic factor (BDNF) is upregulated and associated with pain in chronic pancreatitis. Dig Dis Sci. 2001;46:1633–1639. doi: 10.1023/a:1010684916863. [DOI] [PubMed] [Google Scholar]
- 62.Anand P, Aziz Q, Willert R, van Oudenhove L. Peripheral and central mechanisms of visceral sensitization in man. Neurogastroenterol Motil. 2007;19:29–46. doi: 10.1111/j.1365-2982.2006.00873.x. [DOI] [PubMed] [Google Scholar]
- 63.Dimcevski G, Sami SA, Funch-Jensen P, Le Pera D, Valeriani M, Arendt-Nielsen L, Drewes AM. Pain in chronic pancreatitis: the role of reorganization in the central nervous system. Gastroenterology. 2007;132:1546–1556. doi: 10.1053/j.gastro.2007.01.037. [DOI] [PubMed] [Google Scholar]
- 64.Arendt-Nielsen L, Laursen RJ, Drewes AM. Referred pain as an indicator for neural plasticity. Prog Brain Res. 2000;129:343–356. doi: 10.1016/s0079-6123(00)29026-2. [DOI] [PubMed] [Google Scholar]
- 65.Drewes AM, Pedersen J, Liu W, Arendt-Nielsen L, Gregersen H. Controlled mechanical distension of the human oesophagus: sensory and biomechanical findings. Scand J Gastroenterol. 2003;38:27–35. [PubMed] [Google Scholar]
- 66.Drewes AM, Reddy H, Pedersen J, Funch-Jensen P, Gregersen H, Arendt-Nielsen L. Multimodal pain stimulations in patients with grade B oesophagitis. Gut. 2006;55:926–932. doi: 10.1136/gut.2005.067769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Drewes AM, Frøkjaer JB, Larsen E, Reddy H, Arendt-Nielsen L, Gregersen H. Pain and mechanical properties of the rectum in patients with active ulcerative colitis. Inflamm Bowel Dis. 2006;12:294–303. doi: 10.1097/01.MIB.0000209365.09189.04. [DOI] [PubMed] [Google Scholar]
- 68.Faure C, Giguère L. Functional gastrointestinal disorders and visceral hypersensitivity in children and adolescents suffering from Crohn’s disease. Inflamm Bowel Dis. 2008;14:1569–1574. doi: 10.1002/ibd.20506. [DOI] [PubMed] [Google Scholar]
- 69.Galeazzi F, Lucà MG, Lanaro D, D’Incà R, D’Odorico A, Sturniolo GC, Mastropaolo G. Esophageal hyperalgesia in patients with ulcerative colitis: role of experimental stress. Am J Gastroenterol. 2001;96:2590–2595. doi: 10.1111/j.1572-0241.2001.04102.x. [DOI] [PubMed] [Google Scholar]
- 70.Bernstein CN, Niazi N, Robert M, Mertz H, Kodner A, Munakata J, Naliboff B, Mayer EA. Rectal afferent function in patients with inflammatory and functional intestinal disorders. Pain. 1996;66:151–161. doi: 10.1016/0304-3959(96)03062-x. [DOI] [PubMed] [Google Scholar]
- 71.Fass R, Naliboff B, Higa L, Johnson C, Kodner A, Munakata J, Ngo J, Mayer EA. Differential effect of long-term esophageal acid exposure on mechanosensitivity and chemosensitivity in humans. Gastroenterology. 1998;115:1363–1373. doi: 10.1016/s0016-5085(98)70014-9. [DOI] [PubMed] [Google Scholar]
- 72.Mertz H, Fullerton S, Naliboff B, Mayer EA. Symptoms and visceral perception in severe functional and organic dyspepsia. Gut. 1998;42:814–822. doi: 10.1136/gut.42.6.814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bouwense SA, Ahmed Ali U, ten Broek RP, Issa Y, van Eijck CH, Wilder-Smith OH, van Goor H. Altered central pain processing after pancreatic surgery for chronic pancreatitis. Br J Surg. 2013;100:1797–1804. doi: 10.1002/bjs.9322. [DOI] [PubMed] [Google Scholar]
- 74.Stawowy M, Drewes AM, Arendt-Nielsen L, Funch-Jensen P. Somatosensory changes in the referred pain area before and after cholecystectomy in patients with uncomplicated gallstone disease. Scand J Gastroenterol. 2006;41:833–837. doi: 10.1080/00365520500463332. [DOI] [PubMed] [Google Scholar]
- 75.Wilder-Smith CH, Hill L, Wilkins J, Denny L. Effects of morphine and tramadol on somatic and visceral sensory function and gastrointestinal motility after abdominal surgery. Anesthesiology. 1999;91:639–647. doi: 10.1097/00000542-199909000-00013. [DOI] [PubMed] [Google Scholar]
- 76.Olesen SS, Frøkjær JB, Lelic D, Valeriani M, Drewes AM. Pain-associated adaptive cortical reorganisation in chronic pancreatitis. Pancreatology. 2010;10:742–751. doi: 10.1159/000321644. [DOI] [PubMed] [Google Scholar]
- 77.Graversen C, Olesen SS, Olesen AE, Steimle K, Farina D, Wilder-Smith OH, Bouwense SA, van Goor H, Drewes AM. The analgesic effect of pregabalin in patients with chronic pain is reflected by changes in pharmaco-EEG spectral indices. Br J Clin Pharmacol. 2012;73:363–372. doi: 10.1111/j.1365-2125.2011.04104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mayer EA, Aziz Q, Coen S, Kern M, Labus JS, Lane R, Kuo B, Naliboff B, Tracey I. Brain imaging approaches to the study of functional GI disorders: a Rome working team report. Neurogastroenterol Motil. 2009;21:579–596. doi: 10.1111/j.1365-2982.2009.01304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Moisset X, Bouhassira D, Denis D, Dominique G, Benoit C, Sabaté JM. Anatomical connections between brain areas activated during rectal distension in healthy volunteers: a visceral pain network. Eur J Pain. 2010;14:142–148. doi: 10.1016/j.ejpain.2009.04.011. [DOI] [PubMed] [Google Scholar]
- 80.Willis WD, Westlund KN. Neuroanatomy of the pain system and of the pathways that modulate pain. J Clin Neurophysiol. 1997;14:2–31. doi: 10.1097/00004691-199701000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Frøkjær JB, Bouwense SA, Olesen SS, Lundager FH, Eskildsen SF, van Goor H, Wilder-Smith OH, Drewes AM. Reduced cortical thickness of brain areas involved in pain processing in patients with chronic pancreatitis. Clin Gastroenterol Hepatol. 2012;10:434–438.e1. doi: 10.1016/j.cgh.2011.11.024. [DOI] [PubMed] [Google Scholar]
- 82.Bernstein CN, Frankenstein UN, Rawsthorne P, Pitz M, Summers R, McIntyre MC. Cortical mapping of visceral pain in patients with GI disorders using functional magnetic resonance imaging. Am J Gastroenterol. 2002;97:319–327. doi: 10.1111/j.1572-0241.2002.05464.x. [DOI] [PubMed] [Google Scholar]
- 83.Buscher HC, Schipper EE, Wilder-Smith OH, Jansen JB, van Goor H. Limited effect of thoracoscopic splanchnicectomy in the treatment of severe chronic pancreatitis pain: a prospective long-term analysis of 75 cases. Surgery. 2008;143:715–722. doi: 10.1016/j.surg.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 84.Kosek E, Ordeberg G. Lack of pressure pain modulation by heterotopic noxious conditioning stimulation in patients with painful osteoarthritis before, but not following, surgical pain relief. Pain. 2000;88:69–78. doi: 10.1016/S0304-3959(00)00310-9. [DOI] [PubMed] [Google Scholar]
- 85.Cruccu G, Anand P, Attal N, Garcia-Larrea L, Haanpää M, Jørum E, Serra J, Jensen TS. EFNS guidelines on neuropathic pain assessment. Eur J Neurol. 2004;11:153–162. doi: 10.1111/j.1468-1331.2004.00791.x. [DOI] [PubMed] [Google Scholar]
- 86.Maier C, Baron R, Tölle TR, Binder A, Birbaumer N, Birklein F, Gierthmühlen J, Flor H, Geber C, Huge V, et al. Quantitative sensory testing in the German Research Network on Neuropathic Pain (DFNS): somatosensory abnormalities in 1236 patients with different neuropathic pain syndromes. Pain. 2010;150:439–450. doi: 10.1016/j.pain.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 87.Rolke R, Baron R, Maier C, Tölle TR, Treede RD, Beyer A, Binder A, Birbaumer N, Birklein F, Bötefür IC, et al. Quantitative sensory testing in the German Research Network on Neuropathic Pain (DFNS): standardized protocol and reference values. Pain. 2006;123:231–243. doi: 10.1016/j.pain.2006.01.041. [DOI] [PubMed] [Google Scholar]
- 88.Timmerman H, Wilder-Smith O, van Weel C, Wolff A, Vissers K. Detecting the neuropathic pain component in the clinical setting: a study protocol for validation of screening instruments for the presence of a neuropathic pain component. BMC Neurol. 2014;14:94. doi: 10.1186/1471-2377-14-94. [DOI] [PMC free article] [PubMed] [Google Scholar]