

Abstract
Engineered combinatorial libraries derived from small protein scaffolds represent a powerful tool for generating novel binders with high affinity, required specificity and designed inhibitory function. This work was aimed to generate a collection of recombinant binders of human interleukin-23 receptor (IL-23R), which is a key element of proinflammatory IL-23-mediated signaling. A library of variants derived from the three-helix bundle scaffold of the albumin-binding domain (ABD) of streptococcal protein G and ribosome display were used to select for high-affinity binders of recombinant extracellular IL-23R. A collection of 34 IL-23R-binding proteins (called REX binders), corresponding to 18 different sequence variants, was used to identify a group of ligands that inhibited binding of the recombinant p19 subunit of IL-23, or the biologically active human IL-23 cytokine, to the recombinant IL-23R or soluble IL-23R-IgG chimera. The strongest competitors for IL-23R binding in ELISA were confirmed to recognize human IL-23R-IgG in surface plasmon resonance experiments, estimating the binding affinity in the sub- to nanomolar range. We further demonstrated that several REX variants bind to human leukemic cell lines K-562, THP-1 and Jurkat, and this binding correlated with IL-23R cell-surface expression. The REX125, REX009 and REX128 variants competed with the p19 protein for binding to THP-1 cells. Moreover, the presence of REX125, REX009 and REX115 variants significantly inhibited the IL-23-driven expansion of IL-17-producing primary human CD4+ T-cells. Thus, we conclude that unique IL-23R antagonists derived from the ABD scaffold were generated that might be useful in designing novel anti-inflammatory biologicals. Proteins 2014; 82:975–989.
Keywords: cytokine, psoriasis, engineered binding protein, protein scaffold, combinatorial library, ribosome display
INTRODUCTION
Autoimmune diseases such as psoriasis, Crohn′s disease, rheumatoid arthritis, or multiple sclerosis have recently been found to be associated with IL-23-mediated signaling promoted by IL-23 receptor-expressing TH-17 and other lymphocyte subsets.1–7 In these cell types, dendritic cell-released IL-23 cytokine, consisting of a unique p19 subunit and a common p40 subunit shared with IL-12,8,9 activates signaling via interaction of the p19 subunit with its cognate cell surface receptor, IL-23R, while the p40 subunit of IL-23 binds to IL-12 receptor β110,11 (Fig. 1). Synergistic tethering of the IL-23 heterodimer to both receptor units leads to receptor heterodimerization followed by quaternary complex formation and triggering of the Jak/Stat signaling cascade, involving Jak2, Tyk2, Stat1, Stat3, Stat4, and Stat5.10 Transduction of signal activates the transcription machinery and results in secretion of a cocktail of inflammatory modulators such as IL-17A, IL-17F, IL-22, and of certain chemokines that stimulate keratinocytes and other cell types, thereby playing a pivotal role in pro-inflammatory processes (reviewed in Ref.12).
Figure 1.
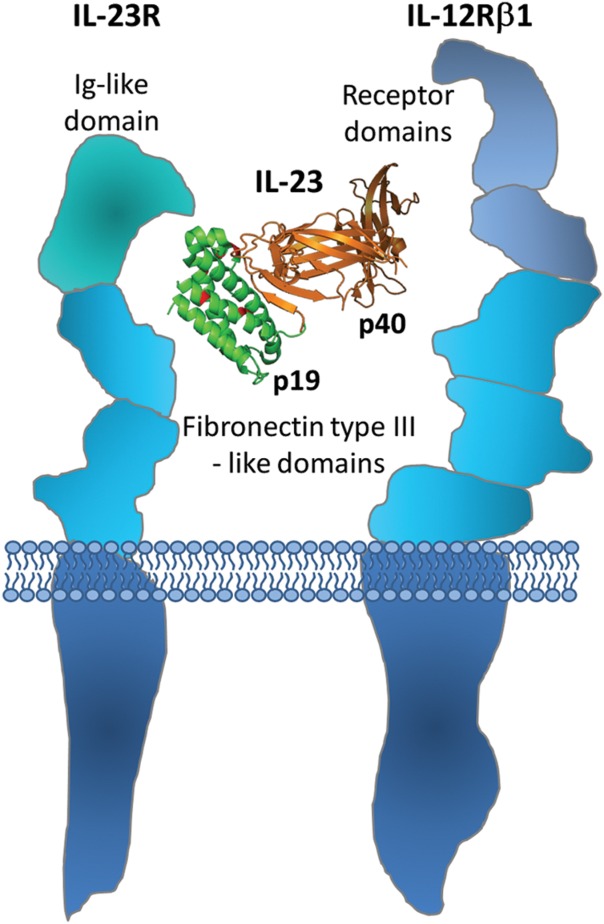
Scheme demonstrating the interaction of IL-23 cytokine with IL-23 receptor complex. The alpha subunit of IL-23, the p19 protein, interacts with IL-23 receptor domains, while the beta subunit, the p40 protein, binds to a putative cytokine homology region of IL-12β1 receptor domains, thus promoting intracellular signaling.
Efficient therapeutic intervention, preventing hyperproliferation of keratinocytes in clinical manifestations of psoriasis, depends on the blockade of interaction between p19/p40 subunits of IL-23 and their cognate cell membrane receptors.13–15 Recently it has been demonstrated that monoclonal antibody-based drug Stelara (ustekinumab, Janssen Biotech), blocking the p40 subunit of IL-23 and preventing it from the interaction with IL-12 receptor β1, reached an excellent efficacy in the treatment of medium to severe form of psoriasis.16–18 However, this drug inhibits the binding of the common p40 subunit, shared both by IL-12 and IL-23, and thus interferes with two different signaling pathways. This often leads to complications, including cardiovascular side effects or higher risk of cancer development. Novel therapeutic strategies, therefore, require development of novel IL-23R antagonists that will separate IL-23-mediated signaling from the IL-12 cascade, thus preserving TH-1 cell differentiation and immunity. Recently, anti-p19 specific antibodies MK-3222 (Merck), CNTO 1959 (Janssen Biotech) and AMG 139 (Amgen/MedImmune)19 have been developed.
As an alternative to conventional monoclonal antibody-based drugs, artificial ligands derived from small protein scaffolds attract attention as robust diagnostic probes and next generation protein therapeutics.20–22 Among different structure-instructed approaches, three-helix bundle scaffolds have recently demonstrated sufficient thermal stability, solubility and mutability for being used as a proof-of-concept domain suitable for generation of highly complex combinatorial libraries.23–26 The well-established Affibody molecules originally selected from Protein A domain-Z-based libraries by phage display selection approaches are currently being used as practical binders for in vitro detection, in vivo diagnostics or high-affinity bioanalytical procedures.27–29 Preservation of folding function together with easy scaffold modifications and low molecular weight, allowing excellent tissue penetration, move the Affibody-derived binders close to the therapeutic use.
The albumin-binding domain of streptococcal protein G30–33 is another example of three-helix bundle scaffold being successfully used for the construction of combinatorial libraries. Recently we have demonstrated that randomization of 11 residues of a flat helical surface, formed by two helices with an inter-link loop (Fig. 2), was sufficient to yield a combinatorial library of a theoretical complexity of 1016 codon variants that was then successfully used for the selection of high-affinity binders of human IFN-γ.34 In this type of library, natural HSA-binding affinity of the ABD domain was compromised in favor of newly engineered affinity for the chosen target. Alternatively, another group randomized 11 residues of a different ABD scaffold surface to generate a combinatorial library that yielded new affinity yet preserved the original HSA binding. This type of “dual-affinity library” was used to select binders of human TNF-α35 and ErbB3.36
Figure 2.
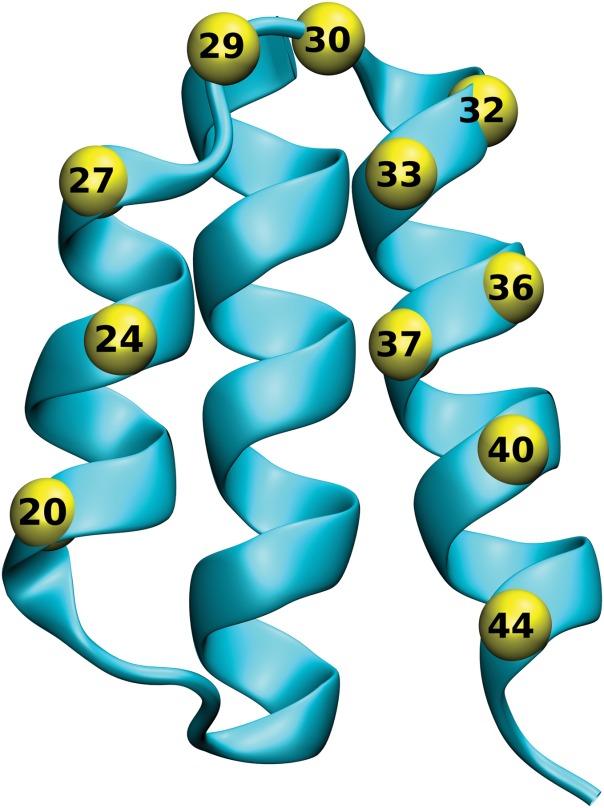
Location of randomized positions in the ABD scaffold. The protein structure of the ABD domain of streptrococcal protein G (PDBID 1GJT) is shown in ribbon representation, with the Cα positions of the 11 residues selected for randomization shown as yellow spheres.
IL-23 receptor belongs to the class-I cytokine receptor family and shares typical features with tandem fibronectin-type III (FnIII) domains containing a hallmark pattern of disulfide bonds and WQPWS sequence tag similar to a conserved WSXWS cytokine receptor consensus located in the transmembrane-proximal FnIII domain.10 Both domains form a cytokine-binding homology region (CHR) which, in concert with a terminal Ig-like domain, is believed to play a substantial role in IL-23 binding.
The molecular structure of the IL-23/IL-23R complex is not available yet, therefore, designing efficient inhibitors of IL-23 function with a promising therapeutic potential remains cumbersome. Here we describe generation of a set of novel recombinant antagonists of the human IL-23 receptor. Their inhibitory potency on IL-23 function is demonstrated on several arrangements of in vitro binding assays, cell-surface competition experiments and ex vivo functional assays. Our data further document that the three-helix bundle scaffold of ABD is suitable for development of anti-inflammatory IL-23 receptor-based next generation therapeutics.
MATERIALS AND METHODS
Antibodies and detection agents
Monoclonal antibodies (mAbs) anti-human IL-23R-allophycocyanin (APC) (mouse IgG2b) specific for the human IL-23 receptor and IgG2b isotype control-APC (mouse IgG2b) were obtained from R&D Systems, Minneapolis, MN. Mouse anti-p19 mAb was purchased from Biolegend, San Diego, CA. Cy5-conjugated goat anti-mouse IgG (F(ab′)2 fragment) was obtained from Jackson ImmunoResearch Laboratories, West Grove, PA. Streptavidin-phycoerythrin was purchased from eBioscience, San Diego, CA.
Cell lines and growth conditions
The cell lines used in the experiments were a human acute monocytic leukemia cell line, THP-1 (ATCC number: TIB-202), a human leukemic cell line, K−562 (ATCC number: CCL-243) and a human T-cell lymphoma cell line, Jurkat (ATCC number: TIB-152). The cells were grown in RPMI-1640 medium (Sigma-Aldrich, St. Louis, MO) supplemented with 10% fetal calf serum (FCS) (GIBCO, Grand Island, N.Y.) and antibiotic antimycotic solution (ATB) (Sigma-Aldrich, St. Louis, MO).
Production of recombinant IL-23R
cDNA coding for the extracellular part (fragment Gly24-Asn350) of the human IL-23 receptor (IL-23R, GenBank: AF461422.1) was amplified by PCR using forward primer IL23Rex-F-Nco-his (ATTACCATGGGCAGCAGCCACCATCATCATCATCACAGCAGCGGAATTACAAATATAAACTGCTCTGG), containing the start codon and the His6-tag sequence, and a reverse primer IL23Rex-R-Xho(GGGCACCTTACTTCTGACAACTGAC TCGAGATAT) bearing the TGA stop codon. The resulting PCR product was inserted into the pET-28b vector (Novagen, Germany) using NcoI and XhoI cloning sites and introduced in Escherichia coli TOP10 cells. The obtained plasmid was used for exIL-23R protein production in E. coli SHuffle strain (SHuffle® T7 Express Competent E. coli, New England Biolabs, Ipswich, MA). Bacteria were grown in liquid LB media with kanamycin (60 µg/L) at 30°C, induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG). The exIL-23R protein was extracted from isolated inclusion bodies with 8M urea in TN buffer (50 mM Tris, 150 mM NaCl, pH = 8.0) and purified by Ni-NTA affinity chromatography.
For protein production targeted into bacterial periplasm, the exIL-23R cDNA was inserted downstream of the pelB leader sequence into the pET-26b vector (Novagen, Germany) using the same restriction sites as above and introduced into the E. coli BL21 (λDE3) strain. The LB broth culture with kanamycin (60 µg/L) was grown at 30°C to reach cell density OD600 = 1.0, protein production was induced by 1 mM IPTG, and protein was harvested after 4 h. The resulting expression led to the production of an insoluble protein that was extracted after the sonication by 8M urea in the TN buffer and purified on a Ni-NTA column.
Production of recombinant p19 subunit of IL-23
The DH-p19 recombinant form of the p19 subunit of IL-23 (calculated Mw 23.3 kDa) was produced in fusion to an N-terminal double-His6-TEV purification tag containing the TEV protease consensus cleavage site. Synthetic, codon-optimized p19 cDNA (GENEART, Germany) was inserted into the pET-28b vector as a NcoI-XhoI fragment and the DH-p19 protein was produced in E. coli BL21 (λDE3) host cells. The DH-p19 protein was extracted from inclusion bodies and purified in Ni-NTA agarose.
Alternatively, soluble p19 protein was produced in the form of a protein fusion to maltose-binding protein (MBP) carrying a double-His6-tag at the N-terminus (DH-MBP-p19, calculated Mw 69 kDa). Primers p19-F-NheI (GGGCTAGC TAGCAGAGCTGTGCCTGGGGGC) and p19-R-XhoI (GCGCCTCGAGGGGACTCAGGGTTGC TGCTC) were used to amplify DNA encoding p19 for insertion into a pET28b-derived vector into which sequences coding for double-His6–MBP-TEV-MCS-TEV-His6 were introduced. The double-His6–MBP-TEV-p19-TEV-His6 fusion protein was produced in the cytoplasmic fraction of E. coli BL21 (λDE3) cells and purified on a 5 mL His-Trap column using ÄKTA purifier (GE Healthcare, UK) and stepwise gradient of 250, 500, and 1000 mM imidazole.
Test of the binding activity of exIL-23R and p19 by ELISA
The recombinant extracellular portion of the IL-23 receptor (exIL-23R) or the p19 protein were immobilized directly on the NUNC Polysorp 96-well plate surface in coating buffer (100 mM bicarbonate/carbonate solution, pH = 9.6) at a concentration of 5 to 10 µg/mL at ∼7°C overnight. The plate was washed with PBS buffer containing 0.05% Tween (PBST) and blocked by 1% BSA in the same buffer (PBSTB). Serial dilutions of purified DH-MBP-p19 or DH-p19 recombinant proteins were prepared in PBSTB buffer and p19 binding was detected using mouse anti-human IL-23 (anti-p19) polyclonal antibody followed by goat anti-mouse IgG horseradish peroxidase (HRP) conjugate (BioLegend, San Diego, CA), both diluted in PBSTB 1:1000. exIL-23R binding to immobilized DH-MBP-p19 was detected using goat anti-IL-23R polyclonal antibody (1:250) followed by secondary rabbit anti-goat HRP conjugate (1:1000) (R&D Systems, Minneapolis, MN). OPD substrate (Sigma-Aldrich, St. Luis, MO) was used as HRP substrate in citrate buffer (3.31% sodium citrate tribasic dihydrate, phosphoric acid, pH = 5.0), reactions were stopped with 2M sulfuric acid and absorbance was read at 492 nm.
ABD library construction and ribosome display selection of REX binders
Combinatorial DNA library was generated as described previously.34 HPLC-purified synthetic oligonucleotides were used. The forward primer ABDLIB-setB1c (5′-TTAGCTGAAGCTAAAGTCTTAGCTAACAGAGAACTTGACAAATATGGAGTAAGTGAC-3′) and the reverse primer setB-rev (5′-ACCGCGGATCCAGGTAA-3′) were used for PCR. The latter had distinct codons randomized at defined positions (5′-ACCGCGGATCCAGGTAAMNNAGCTAAAATMNNATCTATMNNMNNTTTTACMNNMNNAACMNNMNNGGCMNNGTTGATMNNGTTCTTGTAMNNGTCACTTACTCCATATTTGTC-3′), in which M represents C/A, and N any nucleotides out of A, G, C, or T. To serve as a protein spacer for ribosome display, the tolA gene (GENE ID: 946625 tolA) was amplified from E. coli K12 strain genomic DNA, using the primer pairs ABDLIB-tolA-link (5′-TTACCTGGATCCGCGGTCGGTTCGAGCTCCAAGCTTGGATCTGGT GGCCAGAAGCAA-3′) and tolArev (5′-TTTCCGCTCGAGCTACGGTTT GAAGTCCAATGGCGC-3′). The obtained products were linked to the randomized ABD sequences using amplification with primer pairs EWT5-ABDfor1 (5′-TTCCTCCATGGGTATGAGAGGATCGCATCACCATCACCATCACTTAGCTGAAGCTAAAGTCTTA-3′) and tolArev. To add the T7 promoter and ribosome binding site sequences, the obtained DNA fragment was subjected to further PCR amplifications with the set of primers T7B (5′-ATACGAAATTAATACGACTCACTATAGGGAGACCACAACGG-3′), SD-EW (5′-GGGAGACCACAACGGTTTCCCTCTAGAAATAATTTTGTTTAACTTTAAGAAGGAGATATACCATGGGTATGAGAGGATCG-3′) and tolAk (5′-CCGCACACCAGTAAGGTGTG CGGTTTCAGTTGCCGCTTTCTTTCT-3′), generating a DNA library of ABD variants lacking the downstream stop codon. The assembled library was in vitro transcribed/translated in a single step reaction using E. coli extract (EasyXpress Protein Synthesis Mini Kit, QIAGEN, Germany) and used for the selection of translated binders in ribosome display screening.
For the selection of binders, wells of Maxisorp plates (NUNC, Denmark) were coated with decreasing concentrations of recombinant H-exIL-23R protein (round 1 and 2: 25 µg/mL, round 3: 10 µg/mL, round 4: 4 µg/mL, round 5: 1 µg/mL) and blocked with 3% BSA. Preselection was performed in wells coated with BSA only. The plate wells were washed three times with TBS (50 mM Tris-HCl pH 7.4, 150 mM NaCl), followed by 10 times washing with cold WBT (50 mM Tris-acetate, pH 7.0, 150 mM NaCl, 50 mM MgAc with increasing concentrations of Tween-20 (round 1 and 2: 0.05%, round 3: 0.25%, round 4: 0.5%, round 5: 1%). To release mRNA from the bound ribosome complex, elution with elution buffer (50 mM Tris-acetate, pH 7.5, 150 mM NaCl, 50 mM EDTA) containing 50 µg/mL of Saccharomyces cerevisiae RNA as a carrier was performed. Purified RNA was transcribed into cDNA using specific reverse transcription with the setB-rev reverse primer, and annealing to the 3′end of the ABD cDNA. Double-strand DNA was next obtained by PCR using EWT5-ABDfor1 and setB-rev primers. The final amplified DNA encoding selected ABD variants contained T7 promoter and RBS sequences and a truncated tolA fragment.
Two groups of ABD binder cDNA libraries, obtained by reverse transcription after the third or the fifth round of a ribosome display selection campaign, were obtained and cloned as NcoI and XhoI fragments34 in the pET-28b vector containing an in-frame inserted full length tolA DNA sequence. Later, the AviTag in vivo biotinylation sequence (GLNDIFEAQKIEWHE) was added to the C-terminus of the TolA spacer to allow biotinylated protein detection by streptavidin. The AviTag sequence was introduced using PCR with forward primer EWT5-ABDfor1 and reverse primer tolA-AVIrev1 (TTTCCGCTCGAGCTATTCGTGCCATTCGATTTTCTGAGCCTCGAAGATGTCGTTCAGGCCCGGTTTGAAGTCCAATGGCGC). The final His6-REX-TolA-AVI fusion proteins were produced as biotinylated proteins in the E. coli BL21 (λDE3) BirA strain expressing biotin ligase (BirA) in the presence of 50 µM d-biotin in the LB medium and following induction with 2 mM IPTG. The soluble proteins were purified from cell extracts on Ni-NTA agarose columns.
For all binding assays, a control TolA fusion protein with the original albumin binding domain (ABDwt) was used as a negative control. To construct ABDwt with TolA and AviTag sequences (His6-ABDwt-TolA-AVI), PCR amplification with ABDwt-tolA plasmid DNA as a template and forward primer EWT5-ABDfor1 (as mentioned above) and reverse primer ABDrev (TTACTAGGATCCAGGTAATGCAGCTAAAATTTC) was used. The amplified PCR product was digested by NcoI and BamHI enzymes and ligated into the pET-28b vector carrying the tolA-AVI sequence downstream of the BamHI site. The resulting expressed His6-ABDwt-TolA-AVI protein was produced and purified in the same way as REX variants described above.
Screening of IL-23R-binding REX variants by ELISA
For binding assays, selected clones were picked, the inserted sequence was verified by DNA sequencing and proteins were produced in the E. coli BL21 (λDE3) BirA strain as previously described,34 where appropriate REX proteins purified on Ni-NTA columns were used. Two different sandwich layouts were used for the binding assays. In the first case, NUNC Polysorp plates were coated directly with exIL-23R protein (5 µg/mL, recombinant variant produced in E. coli SHuffle strain) in coating buffer at low temperature (∼7°C) overnight and the washed plates were blocked by PBSTB. The serially diluted cell lysates or purified REX proteins were applied in PBSTB and the amount of bound biotinylated REX proteins was detected using streptavidin Poly-HRP conjugate (1:5000). In the second setup, the plates were coated with streptavidin (1 µg/mL) in coating buffer, and biotinylated REX proteins (5 µg/mL) were immobilized through binding to streptavidin. Serially diluted exIL-23R in PBSTB was added into the wells and receptor binding to immobilized REX variants was detected by goat anti-IL-23Rex polyclonal antibody (1:250) sandwich with secondary rabbit anti-goat HRP conjugate (1:1000, R&D Systems, Minneapolis, MN).
Sequence analysis and clustering of selected REX variants
DNA constructs of selected clones expressing full-length REX variants were sequenced. Amino acid multiple sequence alignment of all selected clones and construction of the similarity tree were performed using the ClustalW program. The tree is presented as a phenogram rendered by the Phylodendron online service (http://iubio.bio.indiana.edu/treeapp).
Competition ELISA assay
Maxisorp or Polysorp plates (NUNC, Denmark) were coated with recombinant H-exIL-23R or pelB-exIL-23R and binding of DH-MBP-p19 or DH-p19 as analytes was detected using antibodies as above. Alternatively, plates were coated with DH-MBP-p19 or DH-p19 and binding of IL-23R variants was detected using the corresponding antibodies; 20 nM DH-MBP-p19, 50 nM DH-p19, and 60 nM exIL-23R or pelB-exIL-23R in PBSTB were used as constant concentrations of analytes in the experiments where the concentration of the competitor REX binders varied.
Three of the best inhibitory variants, REX125, REX115, and REX009, were examined for binding to immobilized recombinant human IL-23R-IgG chimera (R&D Systems, Minneapolis, MN), produced as soluble and glycosylated protein secreted by a mouse myeloma cell line. A Polysorp plate was coated with 1 to 2 µg/mL IL-23R-IgG chimera diluted in coating buffer, and 20 nM DH-MBP-p19 protein or 23 nM human IL-23 (R&D Systems, Minneapolis, MN) were used to compete with REX ligands.
Fluorescence-based thermal-shift assay
Protein samples (0.1 mg/mL) in HEPES, and 5× Sypro Orange dye (Sigma-Aldrich, St. Luis, MO) were added into 25 µL total volume. Using the real-time PCR Detection System CFX96 Touch (Bio-Rad Laboratories), the proteins were incubated in a thermal gradient from 20°C to 80°C at increments of 0.5°C and with 30 s-hold intervals. The degree of protein unfolding was monitored by the FRET (fluorescence resonance energy transfer) channel that captured the spectral properties of Sypro Orange unfolded protein complexes (excitation wavelength ≈470 nm and emission wavelength ≈570 nm). The data were analyzed by CFX Manager software and the melting temperatures were determined using the first derivative spectra.
Surface plasmon resonance measurement
Surface plasmon resonance measurements were carried out using custom SPR biosensors (Institute of Photonics and Electronics, Prague, Czech Republic) with four independent sensing spots.37 The REX proteins were immobilized to the SPR chip using the protocol described in Ref.38. Briefly, the SPR sensor chip was coated with self-assembled monolayer of HSC11(EG)2-OH and HSC11(EG)3OCH2COOH alkenthiols. The functionalized SPR chip was mounted to the SPR sensor and all the subsequent molecular interactions were monitored in real-time at temperature 25°C and flow rate 30 μL/min. The REX009, REX125 and ABDwt proteins were diluted in SA buffer (10 mM sodium acetate, pH 5.0 at 25°C) at 5 µg/mL concentration. Each protein was immobilized to a separate sensing spot via amide-bond-forming chemistry. The noncovalently bound proteins were then washed away with a buffer of high ionic strength (PBSNa; 1.4 mM KH2PO4, 8 mM Na2HPO4, 2.7 mM KCl, 0.75M NaCl, pH 7.4 at 25°C). The remaining carboxylic groups were deactivated with 5-min injection of 1M ethanolamine-hydrochloride (pH 8.5). After a baseline was established in the SA buffer, the solution of IL23-R was pumped in the sensor for 10 min. The measurement was repeated on three different chips. The interaction kinetics was compensated for reference sensor response (measured in channel with ABDWT) and analyzed with BiaEvaluation software (GE Healthcare, Uppsala, Sweden).
Detection of cell surface IL-23R and binding of REX variants, p19 or IL-23 to the surface of cultured human cells
All binding assays were performed in HBSS buffer (10 mM HEPES, pH 7.4, 140 mM NaCl, 5 mM KCl) complemented with 2 mM CaCl2, 2 mM MgCl2 and 1% (v/v) FCS (cHBSS) in 96-well culture plates (Nunc, Roskilde, Denmark).
For staining of the IL-23R molecules on the cell surface, 5 × 105 cells were incubated for 30 min at 4°C in 50 µL of cHBSS buffer containing anti-human IL-23R-APC (1:5 dilution) or isotype control-APC (1:5 dilution) mAbs. For REX binding assay, 5 × 105 cells were incubated in 100 µL of cHBSS with biotinylated REX binders or ABDwt controls (10 µg/mL) for 30 min at 4°C, washed with cHBSS, and the cell-bound biotinylated REX-TolA-AVI or ABDwt-TolA-AVI was stained with streptavidin-phycoerythrin (dilution 1:400) for 30 min at 4°C. For DH-p19 binding assay, 5 × 105 cells were incubated in 100 µL of cHBSS with or without DH-p19 (10 µg/mL) for 30 min at 4°C. The cells were washed with cHBSS and the cell-bound DH-p19 was stained with mouse anti-p19 mAb (1:50 dilution) for 30 min at 4°C and after washing with goat anti-mouse IgG antibody labeled with Cy5 (1:50 dilution) for 30 min at 4°C.
Cells were washed, resuspended in 100 µL of HBSS and analyzed by flow cytometry in a FACS LSR II instrument (BD Biosciences, San Jose, CA) in the presence of 5 µg/mL of propidium iodide. Appropriate gatings were used to exclude cell aggregates and dead cells and binding data were deduced from the mean fluorescence intensities (MFI).
Competition between DH-p19 and REX ligands for binding to THP-1 cells
For blocking of REX binding to the IL-23 receptor molecule by DH-p19, THP-1 cells (2 × 105) were preincubated for 15 min at 4°C in the presence of serially-diluted DH-p19 in 50 µL of cHBSS buffer. Biotinylated REX-TolA-AVI clones (or ABDwt-TolA-AVI negative control) were added to the cells in the continuous presence of DH-p19 in 50 µL of cHBSS buffer to a final concentration of 13 nM and incubated at 4°C for 30 min. The cells were washed with cHBSS and the bound biotinylated REX ligands or ABDwt were detected by streptavidin-phycoerythrin conjugate (dilution 1:400) for 30 min at 4°C. Cells were washed, resuspended in 100 µL of HBSS and analyzed by flow cytometry as described above.
IL-23-dependent ex vivo expansion of human IL-17-producing T-cells in PBMC suspensions
Peripheral blood drawn into EDTA-containing tubes was used to obtain purified mononuclear cells (PBMCs) on Ficoll-Paque gradients (Pharmacia, Uppsala, Sweden). PBMCs were washed once with PBS and resuspended in complete RPMI 1640 media (RPMI 1640 supplemented with 10% heat-inactivated FCS, 100 U/mL penicillin, 100 μg/mL streptomycin sulfate and 1.7 mM sodium glutamate). PBMCs were adjusted to 2 × 106 cells/mL and activated on a 96-well plate pre-coated with anti-CD3 (MEM-57, 10 μg/mL, Exbio Praha a.s., Praha, Czech Republic) in the presence of co-stimulatory antibodies against CD28 and CD49d (1 μg/mL, BD Biosciences, San Jose, CA), in the presence of IL-23 (10 ng/mL) and IL-2 (100 U/mL). REX009, REX115, REX125, REX128 binders (7 μg/mL) or control ABDwt were added and cells were incubated for three days at 37°C. After overnight rest at 37°C, cells were re-stimulated again (as above) for 6 h at 37°C (last 4 h exocytosis was blocked with Brefeldin A (10 μg/mL, Sigma-Aldrich). Next, cells were stained with antibodies to CD8 Horizon V-500 (BDB) for 15 min in the dark, washed with PBS containing 0.1% sodium azide and 2% gelatin from cold fish (Sigma-Aldrich) and fixed using FACS lysing solution/FACS Perm 2 (BDB) according to the manufacturer′s instructions. Upon further washing, cells were next stained for CD3 (PerCP-Cy5.5, eBioscience, San Diego, CA), for CD4 (ECD, Immunotech, Marseille, France) and with antibodies for IFN-γ (PE Cy7), IL-2 (APC), IL-17 (PB) (eBioscience) and CD154 (PE, Immunotech). Cells were washed again and measured in flow cytometr BD FACS Aria III (BDB). Absolute cell counts were obtained using BD Truecount (BDB). Viable, nucleated cells were counterstained with Syto-16 and DAPI (Invitrogen).
RESULTS
Production of recombinant human IL-23R and p19 proteins
The human IL-23 receptor gene consists of 10 exons that code for a 629 amino acid-long transmembrane receptor molecule. For the purpose of being used as a target in ribosome display selection of ABD scaffold-derived ligands, we produced a recombinant form of the extracellular domain of IL-23R (exIL-23R) comprising the residues 24 to 350. This part of IL-23R contains five cysteine pairs encoded within sequences forming both fibronectin type III and terminal Ig-like domains. Therefore, the E. coli SHuffle strain supporting formation of disulphide bridges in bacterial cytoplasm was used to express the N-terminally His6-tagged IL-23R (H-exIL-23R). The protein was extracted with 8 M urea from inclusion bodies formed in bacterial cells. Alternatively, we cloned the same receptor cDNA fragment into the pET-26b vector containing a periplasm-targeting pelB leader sequence (pelB-exIL-23R) and expressed the receptor in E. coli BL21 (λDE3) strain. However, the protein was also produced as insoluble fraction and was, therefore, refolded from urea-containing extracts. Both proteins were affinity-purified by Ni-NTA chromatography and refolded by dilution from urea-solutions prior to use in ELISA binding assays and ribosome display selection. The identity of the proteins was confirmed by Western blots using anti-His and anti-IL-23R antibodies (data not shown).
For studies of the interactions between IL-23R and IL-23 cytokine, we also produced a recombinant form of the 170 amino acid residue-long p19 subunit of human IL-23. A 23 kDa fusion p19 protein, consisting of a mature portion of p19 with an N-terminal double-polyhistidinyl tag (DH-p19), was produced into inclusion bodies in E. coli cells, extracted with 8M urea, and DH-p19 was affinity purified by Ni-NTA chromatography. Its identity was confirmed by detection with anti-IL-23 antibody on Western blots (data not shown).
In search for solubility-mediating modifications of the p19 protein using several different solubility-supporting fusion tags, the maltose-binding protein (MBP) in combination with a C-terminal modification were found to support the solubility of the produced p19-MBP fusion protein as the only found variant. In this case, the 69 kDa N-terminally double-His6-MBP-p19 fusion protein contained a C-terminal prolongation, installing an additional 40 aa solubility-supporting sequence (LEKKTCTSRASSTTTTTTEIRLLTKPERKLSWLLPPLSNN). Yet this modification was found unintentionally, all other tested C-terminal modifications, including removal of this sequence by a stop codon termination and replacement by a single-his tag, Streptag or FLAG tag sequence consensus, converted the DH-MBP-p19 protein into the insoluble form. Therefore, this soluble version of p19 was used for further studies as an affinity purified product.
To verify that the recombinant refolded exIL-23R protein still bound the recombinant form of the p19 subunit, ELISA experiments with immobilized exIL-23R were performed. As shown in Figure 3, both the refolded DH-p19 protein and the soluble purified DH-MBP-p19 specifically bound to immobilized H-exIL-23R in a saturable manner. This was further confirmed in a reversed setup where the coated DH-MBP-p19 or refolded DH-p19 protein bound the refolded soluble H-exIL-23R protein (data not shown). These data suggested that the refolded recombinant exIL-23R protein maintained the capacity to specifically bind the p19 subunit of IL-23. Therefore, this protein could be used as a target in ribosome display for selection of IL-23R-specific binders derived from the ABD scaffold.
Figure 3.
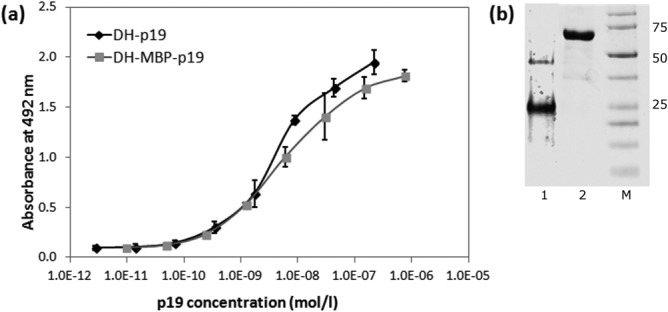
Recombinant extracellular IL-23 receptor binds the recombinant p19 subunit of IL-23. (a) The extracellular portion (residues 24 to 350) of human IL-23 receptor was expressed in E. coli SHuffle cells, purified and refolded from 8M urea extracts of inclusion bodies and coated on ELISA plates. Binding of soluble DH-MBP-p19 fusion protein or of refolded DH-p19 protein to immobilized receptor was detected by anti-human IL-23 (p19) polyclonal antibody sandwich with secondary anti-IgG-HRP. Error bars represent standard deviations. (b) SDS-PAGE of DH-p19 (lane 1) and DH-MBP-p19 (lane 2). The DH-p19 and DH-MBP-p19 proteins were purified from E. coli cell lysates on Ni-NTA and separated on 12,5% polyacrylamide gel stained by Coomassie blue. M indicates the protein marker.
Ribosome display selection of human IL-23R binders
Recently we have demonstrated that randomization of 11 residues in the ABD domain scaffold was sufficient for identification of binders of human interferon-γ with nanomolar range of affinity.34 We therefore used the same approach for the generation of a combinatorial ABD library of a theoretical complexity of 1014 protein variants (Fig. 2). In combination with ribosome display screening, we identified a collection of binders raised against recombinant H-exIL23R. To be able to produce in vivo biotinylated ABD variants required for verification of their binding affinity to H-exIL-23R by ELISA, ABD variants found after three- or five-round selection campaigns (called REX binders) were modified by installing the AviTag sequence downstream of TolA C-terminus. ELISA-positive cell lysates of REX-TolA-AVI clones were selected for further analysis using Western blot. DNA sequences of 34 REX binders were sequentially analyzed and their amino acid sequence similarity compared. As shown in Figure 4, we identified 18 different REX variants, but displaying significant sequence redundancy. All 18 biotinylated REX-TolA-AVI proteins were purified in Ni-NTA agarose and the binding affinity to both immobilized H-exIL-23R and pelB-H-exIL-23R receptor proteins was analyzed using ELISA (data not shown). Analysis of several ELISA-negative REX-TolA-AVI variants by restriction digestion and DNA sequencing revealed that these clones lack the ABD cDNA sequence, thus forming the spliced REX version His6-TolA-AVI. One of such clones, called ΔABD, was purified and used for further experiments as another important negative control over the original His6-ABDwt-TolA-AVI.
Figure 4.

Similarity tree of polypeptide sequences of the obtained REX variants binding exIL-23R. Sequence analysis of 34 cloned REX binders of exIL-23R obtained by ribosome display selection revealed 18 unique sequence variants. For similarity analysis, only the sequences between residues 20 and 46 were compared, as the N-terminal amino acid positions 1 to 19 were nonrandomized.
Identification of inhibitory REX variants
Based on the results of binding assays performed for all 18 different REX-TolA-AVI variants, 15 clones of the complete REX collection were investigated for their ability to inhibit p19/IL-23 binding. To this goal, we used competition ELISA with the immobilized H-exIL-23R and a constant amount of DH-MBP-p19 as an analyte, spiked with an increasing level of the REX-TolA-AVI variants. We found that 11 of the 15 tested REX variants inhibited p19 binding in the micromolar to nanomolar concentration range. The results of the competition ELISA for the four best REX variants are presented in Figure 5. For these inhibitory variants, the sequence similarity comparison is documented in Table 1. Sequence analysis of all tested variants indicated that two of the inhibitory variants, REX001 and REX009, contain one cysteine residue in the 11 randomized positions.
Figure 5.
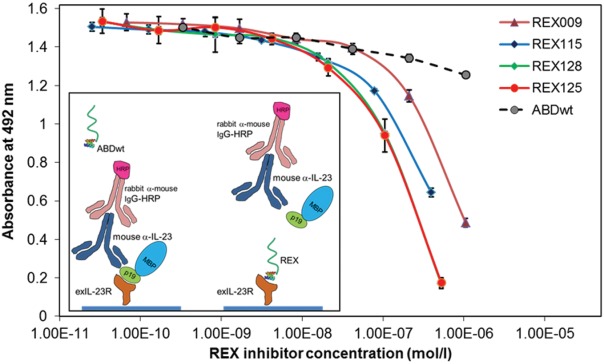
REX binders compete with the soluble recombinant p19 subunit of IL-23 for binding to immobilized exIL-23R. REX-TolA-AVI variants were serially diluted in PBST solution containing 20 nM DH-MBP-p19 as ligand of immobilized exIL-23R. Bound p19 was detected with anti-IL-23 (p19) polyclonal antibody sandwich with secondary anti-IgG-HRP. ABDwt-TolA-AVI, recognizing human serum albumin, served as a negative control. Error bars are shown as standard deviations.
Table 1.
Sequence Similarity Comparison of REX Binders
| 20 | 21 | 22 | 23 | 24 | 25 | 26 | 27 | 28 | 29 | 30 | 31 | 32 | 33 | 34 | 35 | 36 | 37 | 38 | 39 | 40 | 41 | 42 | 43 | 44 | 45 | 46 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ABDwt | Y | Y | K | N | L | I | N | N | A | K | T | V | E | G | V | K | A | L | I | D | E | I | L | A | A | L | P |
| REX009 | Y | Y | K | N | R | I | N | P | A | C | H | V | L | S | V | K | S | N | I | D | W | I | L | A | S | L | P |
| REX115 | V | Y | K | N | T | I | N | I | A | I | P | V | R | V | V | K | R | V | I | D | W | I | L | A | V | L | P |
| REX125 | H | Y | K | N | W | I | N | P | A | R | R | V | R | P | V | K | W | L | I | D | A | I | L | A | A | L | P |
| REX128 | R | Y | K | N | S | I | N | R | A | L | P | V | A | A | V | K | W | A | L | D | L | I | L | A | W | L | P |
Parental nonmutated ABD (ABDwt) of streptococcal protein G (G148_GA3) was aligned with the randomized portions of sequenced REX clones that were selected in ribosome display for IL-23R binding and belong to the best binders. Grey boxes indicate the 11 positions at which the residues of ABD (aa 20 to 46) were randomized. The dark box marks an unintended mutation I38L found in REX128. In the nonrandomized N-terminal part of ABD (residues 1–19), the LAEAKVLANRELDKYGVSD amino acid sequence was present. Multiple alignment was performed in ClustalW.
To further confirm the inhibitory potency of the REX binders, we performed competition ELISA with a different receptor-cytokine protein pair. REX inhibitors found in the previous setup of the competition ELISA also inhibited binding of the DH-p19 protein to the coated pelB-H-exIL-23R (not shown). In correlation with the result shown in Figure 5, all tested REX binders were found to suppress the p19 binding. To verify that the found competition potency of the tested REX clones can be attributed to the specific binding of the REX proteins to the refolded receptor molecule rather than to non-specific or misfolded protein binding, we performed additional competition assays with the same protein pairs but in the opposite ELISA layouts in which DH-MBP-p19 or DH-p19 were immobilized on the plastic plate. These results did confirm previous data (not shown).
To investigate whether inhibitory REX binders recognize the soluble and glycosylated form of IL-23R, we performed ELISA experiments with a commercial product of recombinant human IL-23R-IgG chimera (R&D Systems), secreted by an NS0-derived murine myeloma cell line. As shown in Figure 6, increasing concentration of REX125, REX115, and REX009 binders decreased the binding of DH-MBP-p19 to the immobilized IL-23R-IgG. A similar inhibitory effect of the REX binders was also found for the recombinant Sf 21 (baculovirus)-derived human single-chain IL-23 (R&D Systems) (not shown). Thus, we conclude that we identified REX variants with the ability to suppress p19/IL-23 binding to IL-23R, as demonstrated in several layouts of direct ELISA.
Figure 6.
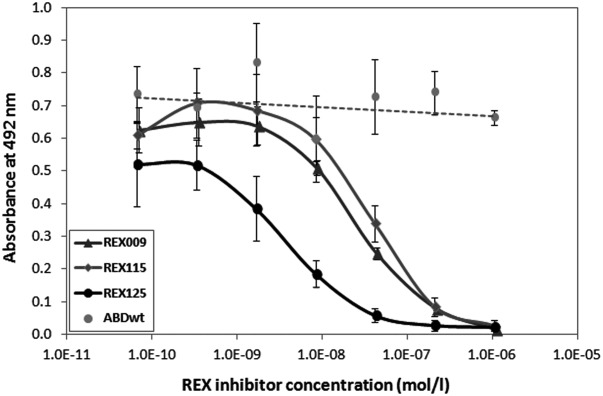
REX binders compete with p19 subunit-mediated binding of IL-23 to the IL-23R-IgG receptor chimera. The IL-23R-IgG receptor chimera was immobilized on an ELISA plate and serially diluted inhibitory REX-TolA-AVI ligands were used to compete for binding with 20 nM of DH-MBP-p19. Bound p19 was detected with anti-IL-23 (p19) polyclonal antibody sandwich with secondary anti-IgG-HRP. ABDwt-TolA-AVI served as a negative control. Error bars represent standard deviations.
Biophysical characterization of REX inhibitors
SPR biosensor binding analysis was used to further characterize the binding interactions between IL-23R and the REX ligands. The chip of a 4-channel SPR biosensor was functionalized with self-assembled monolayer of alkanethiols, to which His6-REX-TolA-AVI and His6-ABDwt-TolA-AVI proteins were attached. The response to IL-23R-IgG chimera interaction of such SPR biosensor is shown in Figure 7 for sensor surfaces functionalized with REX125, REX009 and control ABDwt proteins, respectively. It can be seen that the sensor response to IL-23R-IgG was much higher in the channels coated with the REX proteins than in the reference channel functionalized with ABDwt. Interestingly, the interaction of IL-23R-IgG with the REX variants was very sensitive to pH and salt composition of the running buffer. The complex was stable at pH 5.0 of the sodium acetate (SA) buffer, while it was very rapidly dissociated at pH 7.4 in PBS buffer. Similar results were obtained when IL-23R-IgG was immobilized to the sensor surface and REX proteins were flowed over the sensor (data not shown). The reference-compensated sensor responses to binding of IL-23R-IgG chimera to immobilized REX009, or REX125, respectively, at pH 5 were analyzed using an 1:1 interaction model in the kinetic BiaEvaluation software. Global fit of four concentrations in the range of 20–200nM indicated a dissociation constant Kd = 1.3 ± 0.7 × 10−9M for REX009. The binding affinity for REX125 could not be precisely determined due to a more complex interaction mode, but it was estimated to be in the order of 10−9M.
Figure 7.
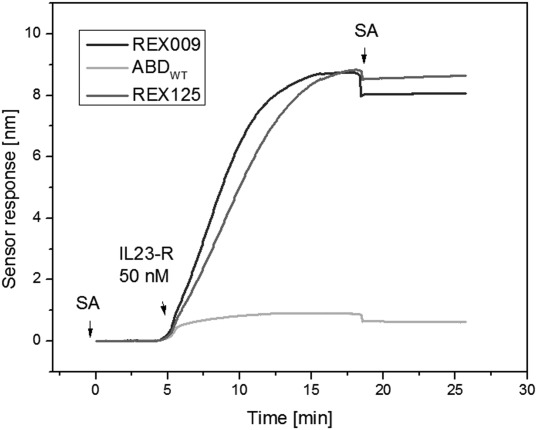
Immobilized REX ligands bind the soluble IL-23R-IgG receptor chimera. The REX-TolA-AVI variants REX009 and REX125, or ABDwt-TolA-AVI (ABDWT), were attached to the surface of SPR sensor over which the IL-23R-IgG receptor chimera was passed at 50 nM concentration. SA indicates sodium acetate buffer.
To investigate the thermal stability of the strongest inhibitory binders, we performed the fluorescence-based thermal shift assay (data not shown). Melting temperatures (Tm) for REX125 and REX128 were found to be 56 and 56.5°C, respectively, and for REX009 between 50 and 53°C, while the Tm value for WT control was found to be 58°C. This suggested that the randomization of mutable residues of the ABD domain did not significantly affect the basic stability of the scaffold structure.
REX ligands competitively inhibit p19 subunit binding to the native IL-23 receptor on human cells
To explore whether the REX ligands bind to the native IL-23 receptor molecules on the cell surface, we first investigated cultured human K-562 and Jurkat leukemic cells for the expression of IL-23R. We found that both K-562 and Jurkat cells express IL-23R [Fig. 8(a,b)] and this expression is further increased upon T-cell activation, as documented 24 h after the induction [Fig. 8(c)]. We also found that THP-1 cells express more IL-23R than K-562, as documented in Figure 9(a) by IL-23R-specific polyclonal antibody and, therefore, both these cell lines were further used as targets for REX ligand-binding assay. As shown in Figure 9(b), all of the tested REX variants bound to K-562 as well as THP-1 cells, and the extent of REX ligand binding correlated well with the observed level of IL-23R expression on the surface of both cell types [Fig. 9(a)]. To further demonstrate that binding of REX ligands correlates with IL-23R expression, we performed a cell-surface binding test with Jurkat cells that express lower amounts of IL-23 receptor per cell than the K-562 cells [Fig. 9(c)]. As shown in Figure 9(d), all tested REX binders exhibited reduced binding to Jurkat cells in comparison with the K562 cells, in agreement with the reduced IL-23R expression on Jurkat cells. These results strongly suggested that the generated REX ligands can bind to native IL-23R molecules exposed on the human cell surface.
Figure 8.
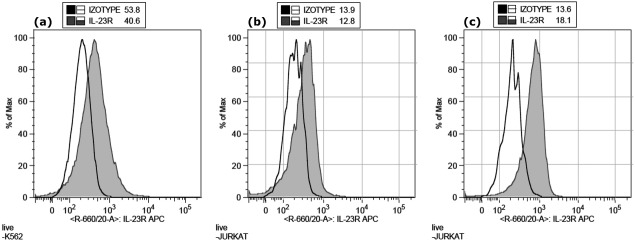
Expression of IL-23R on K562 cells (a), before stimulation on Jurkat cells (b) and 24 h after the induction of Jurkat cells with anti-CD3 (c). The cells were stained with anti-human IL-23R-APC or isotype-APC control mAbs and analyzed by flow cytometry.
Figure 9.
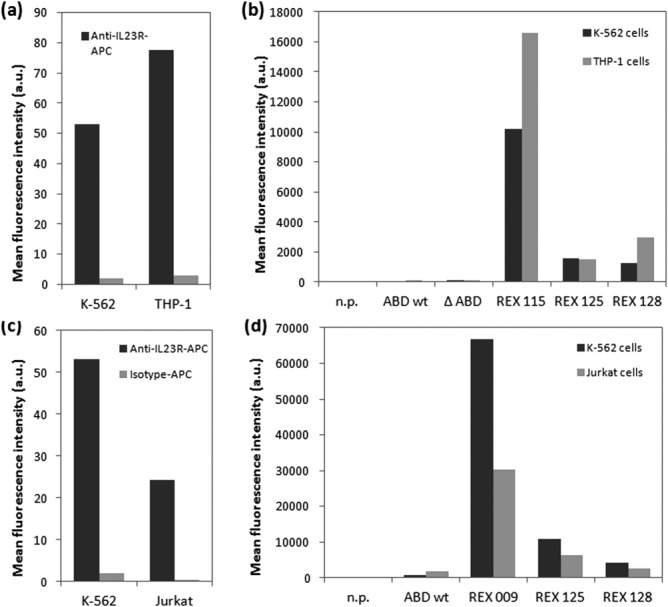
Binding of REX variants to human cell lines correlates with IL-23R expression levels. (a) Expression of IL-23 receptor molecules on the surface of cultured human K-562 and THP-1 cells was detected by flow cytometry using anti-human IL-23R-APC or isotype control-APC mAbs. (b) Binding of REX-TolA-AVI variants to K-562 and THP-1 cells. The cells were incubated with in vivo biotinylated REX-TolA-AVI proteins or negative ABD-TolA-AVI controls (ABDwt, ΔABD) and the cell-bound proteins were stained with streptavidin-phycoerythrin and analyzed by flow cytometry. In the n.p. control, the REX binders and ABD controls were omitted. (c) Comparison of IL-23 receptor expression on K-562 and Jurkat cell surface. The cells were stained with anti-human IL-23R-APC or isotype-APC control mAbs and analyzed by flow cytometry. (d) Comparison of REX ligand binding to K-562 and Jurkat cells. The cells were incubated with biotinylated REX variants or ABDwt control and cell-bound proteins were detected with streptavidin-phycoerythrin by flow cytometry.
To corroborate this observation, we performed assays in which the p19 protein was allowed to compete with REX ligands for the binding to IL-23R on the cell surface. For this purpose, we used THP-1 cells expressing higher levels of IL-23R [Fig. 9(a)], to which also higher amounts of DH-p19 protein were bound than to K-562 cells [Fig. 9(a)]. As shown in Figure 10(b), at increasing concentrations of the DH-p19 protein, the binding of all three tested REX ligands to THP-1 cells was significantly inhibited.
Figure 10.
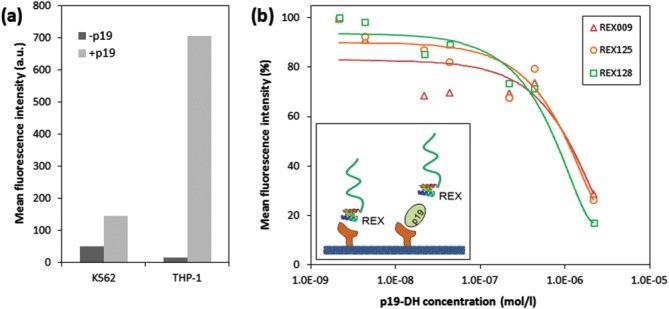
REX inhibitory variants compete with the p19 subunit of IL-23 for binding to THP-1 cells. (a) Comparison of DH-p19 protein binding to THP-1 and K-562 cells. Cell-bound DH-p19 molecules were detected by flow cytometry using a mouse anti-IL-23 mAb goat anti-mouse IgG antibody sandwich labeled with Cy5. (b) Micromolar concentrations of DH-p19 block binding of REX variants to THP-1 cells. The cells (2 × 105) were preincubated with indicated concentrations of DH-p19 before biotinylated REX-TolA-AVI or ABDwt-TolA-AVI control were added to a final concentration of 13 nM and incubation was continued for 30 min at 4°C. After washing of cells, the bound ligands were detected by flow cytometry using streptavidin-phycoerythrin. The results are representative of two independent but confirming experiments. Measured values for each of REX variants were supplemented by the particular polynomial trendline.
REX binders inhibit IL-23-dependent ex vivo expansion of IL-17-positive CD4+ T-cells
The sum of the above-outlined results indicated that at least several of the generated REX ligands could bind to native IL-23R molecules on human cells and block the p19-subunit-mediated binding of the IL-23 cytokine to its receptor. Therefore, we assessed ex vivo whether the REX ligands could inhibit the IL-23 signaling function on primary human T-cells. We determined whether blocking of IL-23R by excess of REX ligands would inhibit IL-23-mediated expansion of primary human Th-17 lymphocytes. Mononuclear cells were isolated from the blood of healthy donors and stimulated with anti-CD3 monoclonal antibody and co-stimulated by activating antibodies binding CD28 and CD49d in the continued presence of the Th-17 conditioning cytokines IL-23 and IL-2. As shown in Figure 11, over the 3 days of ex vivo cultivation of PBMC suspensions, a marked decrease of Th-17 cell expansion (decrease of counts of IL-17-secreting, IFN-γ non-secreting T-cells) was observed when excess of the REX ligands was present, as compared with mock treatment or presence of the ABD-WT control. In PBMC suspensions, the enhancement of Th17+ cells after IL-23-mediated induction is documented as a difference between non-induced cells (sample NO IL-23, 0.52 ± 0.11, n = 6, P = 0.0001) and normalized counts of IL-23 stimulation (IL-23 only, =1), shown as a boxplot in Figure 11(b). The addition of ABD-WT as a control into the PBMC suspensions had little effect on Th-17+ cell expansion (0.88 ± 0.26, n = 7, P = 0.2892). In contrast, all tested REX variants significantly inhibited Th-17+ cell expansion: REX009 (0.62 ± 0.32, n = 7, P = 0.0559), REX115 (0.55 ± 0.20, n = 5, P = 0.0074) and REX125 (0.42 ± 0.17, n = 5, P = 0.0004), as documented in Figure 11(b).
Figure 11.

REX binders inhibit IL-23-dependent ex vivo expansion of IL-17-producing CD4+ T-cells. PBMCs from seven healthy volunteers were activated using anti-CD3 and co-stimulation in the presence of IL-23 and IL-2 and in the presence or absence of indicated REX ligands for 3 days. (a) Detection of IL-17-producing cells that were gated using FSC, SSC, CD3+, and CD4+. The number in the right-hand bottom corner indicates the percentage of the gated cell population producing solely IL-17 (IFN-γ secretion marks TH−1 type response). (b, c) The amount of IL-17-producing cells recovered from PBMC (b) or separated T-cell (c) samples of healthy donors after 3 days of activation in the presence or absence of the REX ligands. Total cell numbers were normalized to cell counts in IL-23/IL-2-treated samples. Boxes range from 25th to 75th percentiles with a line at the median. Whiskers reach 10th and 90th percentiles. Outlying values are indicated as individual circles.
To verify whether the observed inhibitory effect of REX binders in PBMC suspensions can be attributed to the T-cell-dependent cell expansion, we repeated the same experiment in samples with separated T cells isolated from the same PBMC donors. As shown in Figure 11(c), the IL-23-dependent Th-17+ cell count enrichment was also detected (NO IL-23, 0.77 ± 0.08, n = 4, P = 0.0116). T-cell suspensions with added ABD-WT control exhibited no inhibitory effect on T-cell expansion (0.99 ± 0.26, n = 4, P = 0.9901). In striking contrast to this observation, REX variants demonstrated an immunosuppressive effect on T-cell expansion as follows: REX115 (0.69 ± 0.17, n = 3, P = 0.0883), REX125 (0.65 ± 0.16, n = 3, P = 0.0671) and REX009 (0.80 ± 0.01, n = 2, P = 0.0139).
In summary, the collected data indicate that the tested REX variants inhibited IL-23-dependent Th-17+ cell expansion to the level of no-IL-23-induced samples (REX009) or even below this level (REX115 and REX125) and this finding is valid for both PBMC and separated T-cell samples. While IL-23 was shown to promote development of Th-17+ T-cells in man,39 the mode of its action might be fixing the Th-17 commitment rather than “de novo” Th-17 development, survival or proliferation enhancement.40 As shown here, REX ligands might block the IL-23 signaling function on human T-cells through selective binding and occupation of the IL-23 receptor.
DISCUSSION
In this work, we aimed to generate novel immunomodulatory binders suppressing the function of human IL-23 receptor, a crucial molecule of the IL-23-mediated signaling pathway. As a valuable alternative to conventionally developed neutralizing antibodies, we thought to use the three-helix bundle scaffold-derived combinatorial library of albumin-binding domain variants as a primary source for selection of IL-23 receptor binders. To this goal, we used our recently constructed high complex library that has been successfully used for selection of high-affinity binders of human IFN-γ.34 Using campaigns of ribosome display selection, we generated 18 sequence variants of high-affinity IL-23R binders, much less in comparison to the selection of IFN-γ binders described before,34 suggesting that a limited number of high-affinity epitopes were available as targets. This is further supported by a high redundancy of the found REX variants demonstrating that the selected high-stringency conditions used for high-affinity binder screening in ribosome display were properly adjusted. Among the 18 found sequence variants, 11 were observed to inhibit binding of p19 to the bacterial product of human IL-23R, suggesting that several surface-exposed epitopes are critical for the ligand-receptor interaction. Among seven best inhibiting clones, two (REX001 and REX009) contain a randomized cysteine residue located in the inter-loop between helices 2 and 3 (residues 29, 30). Another of the best inhibitory clones, REX125, includes an interesting randomized sequence pattern 24W-27P-29R-30R-32R-33P-36W with an intrinsic symmetry 24W-27P-30R-33P-36W between helices 2 and 3 that could be beneficial for the ligand binding. In addition, proline residues are known to break α-helical structures, so this may lead to a changed ABD scaffold geometry, affecting C- and N-terminal ends of the helices 2 and 3, respectively, and thereby to an increased flexibility of the inter-loop, supporting the binder′s affinity. Yet the detailed molecular structure of REX-IL-23R complexes needs to be further investigated, and installation of proline residues in amino acid positions 27–29-30–32-33 in the case of all the best inhibitory binders (REX009, REX115, REX125, REX128) attracts attention as cysteine and proline residues are being typically designed to be eliminated from the randomization during the combinatorial library construction.
Interestingly, the REX005 binder was found to inhibit the binding of the p19 protein to the used bacterial receptor products. This inhibitory effect was then confirmed in competition ELISA using a 46 amino acid synthetic form of ABD (not shown), excluding the possibility that the TolA spacer protein affects the binding or inhibition. However, this clone does not bind to cell-surface IL-23R expressed in THP-1 and K-562 cells. It is possible that glycosylation of the Ig-like domain in the N-glycosylation consensus (residues 29, 47, and 81) or in the distal FnIII domain (residues 141 and 181) prevents the binding to the cell-surface receptor. However, we cannot exclude that REX005 recognizes an unnatural epitope found in the bacterial receptor protein, yet this binder interacts with the cytoplasm-produced E. coli Shuffle strain as well as periplasm-targeted BL21 (λDE3) cell products.
To demonstrate that REX binders recognize cell-surface IL-23R, we thought to prepare cell transfectants expressing human IL-23R on the cell surface. To this goal we tested several different cell lines and found that all of them, including K-562, Jurkat, THP-1, HEK 293, COS-7, or CHO cells, to some extent bind anti-IL23R antibodies. In addition, several other human cell lines such as HeLa, A431 and NKT, or mouse NIH 3T3 cells, have been described to express IL-23R. Therefore, we decided to demonstrate specific binding of the generated REX variants using the correlation of cell-surface REX binding with IL-23R expression, in combination with a p19-specific competition binding assay. We clearly demonstrated that REX009, REX125 and REX128 compete with the p19 protein for binding to IL-23R-expressing and p19-protein-binding THP-1 cells. This level of evidence in combination with the ex vivo immunosuppressing effect on the PBMC-derived Th-17+ cell expansion brought us to the conclusion that we generated a collection of novel antagonists of the IL-23 receptor. This is documented by a marked decrease of REX125-affected (decrease 52%, P = 0.0029) and REX115-affected (38%, P = 0.0317) Th-17+ cell counts compared with the value of the control ABD-WT in PBMC cell suspensions, reaching the values below the counts of IL-23-non-stimulated samples. As ex vivo experiments were done with bacterial LPS-free REX proteins, we attribute the inhibitory effect solely to the function of the tested REX variants. The expansion of the Th-17+ population in the presence of IL-23 is manifested by a comparison between IL-23-positive and IL-23-negative control samples, further suggesting that a significant amount of IL-23+ cells have already been present in the no-IL-23 PBMC as well as separated T-cell samples. This is in correlation with the known fact41 that also other cells, including γδ T-cells, or certain natural-killer cell populations, express the IL-23 receptor. It is important to mention that REX binders tend to decrease the Th-17+ cell counts even under the level of IL-23-negative control in the case of separated T-cell samples [Fig. 11(c)]. However, we also observed a slight decrease (12%) in the Th-17+ cell counts when we used the ABD-WT control in comparison to the untreated samples of PBMC [see sample IL-23 only, Fig. 11(b)], yet this decrease was absent in the samples of separated T-cells (0.01% decrease, P = 0.9901) [Fig. 11(c)]. We do not know the principal reason for this phenomenon found in the PBMC samples, but it could be caused by non-specific binding of ABD-WT to IL-23R or other cell receptors, especially under the conditions of more than 100-fold molar excess of the used ABD/REX molecules. This would correspond to the observed nonspecific binding of the ABD-WT control in ELISA experiments under the conditions of high concentrations. Another possibility is that the TolA spacer protein still maintains some effect on cell stimulation, although we show in Figure 9(b) that neither ABD-WT-TolA-AVI nor ΔABD-TolA-AVI control proteins substantially bind to the cell surface of human cells. This possibility was, however, excluded in further experiments in which we compared the inhibitory effect of a long version of REX009-TolA-AVI with a constructed short REX009-AVI variant (lacking the helical TolA spacer protein), demonstrating that the same ex vivo inhibitory effect on T-cell expansion was reached with non-TolA-containing REX009 protein (data not shown).
Collectively, we demonstrated that the three-helix bundle scaffold of the ABD domain can be modified to generate high-affinity binders with immunomodulatory function. To our knowledge, this is the first example of described ABD-derived receptor antagonists with ex vivo immunosuppressive function. The unique REX inhibitory binders might be a useful clue for designing novel anti-IL-23R-based therapeutics, especially when the precise structural mode-of-function of the IL-23/IL-23R complex is not available. In addition, the small 5 kD size of the ABD domain brings new alternatives for skin-penetration-based drug delivery systems that might be essential for psoriasis treatment.
Acknowledgments
The authors thank Petra Kadlčáková for excellent technical assistance.
REFERENCES
- 1.Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, Vega F, Yu N, Wang J, Singh K, Zonin F, Vaisberg E, Churakova T, Liu M, Gorman D, Wagner J, Zurawski S, Liu Y, Abrams JS, Moore KW, Rennick D, de Waal-Malefyt R, Hannum C, Bazan JF, Kastelein RA. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715–725. doi: 10.1016/s1074-7613(00)00070-4. [DOI] [PubMed] [Google Scholar]
- 2.Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, Zurawski S, Wiekowski M, Lira SA, Gorman D, Kastelein RA, Sedgwick JD. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 3.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chan JR, Blumenschein W, Murphy E, Diveu C, Wiekowski M, Abbondanzo S, Lucian L, Geissler R, Brodie S, Kimball AB, Gorman DM, Smith K, de Waal Malefyt R, Kastelein RA, McClanahan TK, Bowman EP. IL-23 stimulates epidermal hyperplasia via TNF and IL-20R2-dependent mechanisms with implications for psoriasis pathogenesis. J Exp Med. 2006;203:2577–2587. doi: 10.1084/jem.20060244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Capon F, Di Meglio P, Szaub J, Prescott NJ, Dunster C, Baumber L, Timms K, Gutin A, Abkevic V, Burden AD, Lanchbury J, Barker JN, Trembath RC, Nestle FO. Sequence variants in the genes for the interleukin-23 receptor (IL23R) and its ligand (IL12B) confer protection against psoriasis. Hum Genet. 2007;122:201–206. doi: 10.1007/s00439-007-0397-0. [DOI] [PubMed] [Google Scholar]
- 6.Vaknin-Dembinsky A, Balashov K, Weiner HL. IL-23 is increased in dendritic cells in multiple sclerosis and down-regulation of IL-23 by antisense oligos increases dendritic cell IL-10 production. J Immunol. 2006;176:7768–7774. doi: 10.4049/jimmunol.176.12.7768. [DOI] [PubMed] [Google Scholar]
- 7.Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, Steinhart AH, Abraham C, Regueiro M, Griffiths A, Dassopoulos T, Bitton A, Yang H, Targan S, Datta LW, Kistner EO, Schumm LP, Lee AT, Gregersen PK, Barmada MM, Rotter JI, Nicolae DL, Cho JH. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beyer BM, Ingram R, Ramanathan L, Reichert P, Le HV, Orth V, P Crystal structures of the pro-inflammatory cytokine interleukin-23 and its complex with a high-affinity neutralizing antibody. J Mol Biol. 2008;382:942–955. doi: 10.1016/j.jmb.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 9.Lupardus PJ, Garcia KC. The structure of interleukin-23 reveals the molecular basis of p40 subunit sharing with interleukin-12. J Mol Biol. 2008;382:931–941. doi: 10.1016/j.jmb.2008.07.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parham C, Chirica M, Timans J, Vaisberg E, Travis M, Cheung J, Pflanz S, Zhang R, Singh KP, Vega F, To W, Wagner J, O′Farrell AM, McClanahan T, Zurawski S, Hannum C, Gorman D, Rennick DM, Kastelein RA, de Waal Malefyt R, Moore KW. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R. J Immunol. 2002;168:5699–5708. doi: 10.4049/jimmunol.168.11.5699. [DOI] [PubMed] [Google Scholar]
- 11.Kastelein RA, Hunter CA, Cua DJ. Discovery and biology of IL-23 and IL-27: related but functionally distinct regulators of inflammation. Annu Rev Immunol. 2007;25:221–242. doi: 10.1146/annurev.immunol.22.012703.104758. [DOI] [PubMed] [Google Scholar]
- 12.Boniface K, Blom B, Liu YJ, de Waal Malefyt R. From interleukin-23 to T-helper 17 cells: human T-helper cell differentiation revisited. Immunol Rev. 2008;226:132–146. doi: 10.1111/j.1600-065X.2008.00714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Toichi E, Torres G, McCormick TS, Chang T, Mascelli MA, Kauffman CL, Aria N, Gottlieb AB, Everitt DE, Frederick B, Pendley CE, Cooper KD. An anti-IL-12p40 antibody down-regulates type 1 cytokines, chemokines, and IL-12/IL-23 in psoriasis. J Immunol. 2006;177:4917–4926. doi: 10.4049/jimmunol.177.7.4917. [DOI] [PubMed] [Google Scholar]
- 14.Krueger GG, Langley RG, Leonardi C, Yeilding N, Guzzo C, Wang Y, Dooley LT, Lebwohl M. A human interleukin-12/23 monoclonal antibody for the treatment of psoriasis. New Engl J Med. 2007;356:580–592. doi: 10.1056/NEJMoa062382. [DOI] [PubMed] [Google Scholar]
- 15.Gottlieb AB, Cooper KD, McCormick TS, Toichi E, Everitt DE, Frederick B, Zhu Y, Pendley CE, Graham MA, Mascelli MA. A phase 1, double-blind, placebo-controlled study evaluating single subcutaneous administrations of a human interleukin-12/23 monoclonal antibody in subjects with plaque psoriasis. Curr Med Res Opin. 2007;23:1081–1092. doi: 10.1185/030079907x182112. [DOI] [PubMed] [Google Scholar]
- 16.Leonardi CL, Kimball AB, Papp KA, Yeilding N, Guzzo C, Wang Y, Li S, Dooley LT, Gordon KB. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1) Lancet. 2008;371:1665–1674. doi: 10.1016/S0140-6736(08)60725-4. [DOI] [PubMed] [Google Scholar]
- 17.Papp KA, Langley RG, Lebwohl M, Krueger GG, Szapary P, Yeilding N, Guzzo C, Hsu MC, Wang Y, Li S, Dooley LT, Reich K. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 2) Lancet. 2008;371:1675–1684. doi: 10.1016/S0140-6736(08)60726-6. [DOI] [PubMed] [Google Scholar]
- 18.Schmidt C. Ustekinumab poised to enter the psoriasis market. Nat Biotechnol. 2008;26:1317–1318. doi: 10.1038/nbt1208-1317a. [DOI] [PubMed] [Google Scholar]
- 19.Garber K. Anti-IL-17 mAbs herald new options in psoriasis. Nat Biotechnol. 2012;30:475–477. doi: 10.1038/nbt0612-475. [DOI] [PubMed] [Google Scholar]
- 20.Binz HK, Pluckthun A. Engineered proteins as specific binding reagents. Curr Opin Biotechnol. 2005;16:459–469. doi: 10.1016/j.copbio.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 21.Nygren PA, Skerra A. Binding proteins from alternative scaffolds. J Immunol Methods. 2004;290:3–28. doi: 10.1016/j.jim.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 22.Gronwall C, Stahl S. Engineered affinity proteins—generation and applications. J Biotechnol. 2009;140:254–269. doi: 10.1016/j.jbiotec.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 23.Nord K, Nilsson J, Nilsson B, Uhlen M, Nygren PA. A combinatorial library of an alpha-helical bacterial receptor domain. Protein Eng. 1995;8:601–608. doi: 10.1093/protein/8.6.601. [DOI] [PubMed] [Google Scholar]
- 24.Nord K, Gunneriusson E, Ringdahl J, Stahl S, Uhlen M, Nygren PA. Binding proteins selected from combinatorial libraries of an alpha-helical bacterial receptor domain. Nat Biotechnol. 1997;15:772–777. doi: 10.1038/nbt0897-772. [DOI] [PubMed] [Google Scholar]
- 25.Nygren PA, Uhlen M. Scaffolds for engineering novel binding sites in proteins. Curr Opin Struct Biol. 1997;7:463–469. doi: 10.1016/s0959-440x(97)80108-x. [DOI] [PubMed] [Google Scholar]
- 26.Nord K, Nord O, Uhlen M, Kelley B, Ljungqvist C, Nygren PA. Recombinant human factor VIII-specific affinity ligands selected from phage-displayed combinatorial libraries of protein A. Eur J Biochem. 2001;268:4269–4277. doi: 10.1046/j.1432-1327.2001.02344.x. [DOI] [PubMed] [Google Scholar]
- 27.Nilsson FY, Tolmachev V. Affibody molecules: new protein domains for molecular imaging and targeted tumor therapy. Curr Opin Drug Discov Dev. 2007;10:167–175. [PubMed] [Google Scholar]
- 28.Gronwall C, Sjoberg A, Ramstrom M, Hoiden-Guthenberg I, Hober S, Jonasson P, Stahl S. Affibody-mediated transferrin depletion for proteomics applications. Biotechnol J. 2007;2:1389–1398. doi: 10.1002/biot.200700053. [DOI] [PubMed] [Google Scholar]
- 29.Lofblom J, Feldwisch J, Tolmachev V, Carlsson J, Stahl S, Frejd FY. Affibody molecules: engineered proteins for therapeutic, diagnostic and biotechnological applications. FEBS Lett. 2010;584:2670–2680. doi: 10.1016/j.febslet.2010.04.014. [DOI] [PubMed] [Google Scholar]
- 30.Johansson MU, Frick IM, Nilsson H, Kraulis PJ, Hober S, Jonasson P, Linhult M, Nygren PA, Uhlen M, Bjorck L, Drakenberg T, Forsen S, Wikstrom M. Structure, specificity, and mode of interaction for bacterial albumin-binding modules. J Biol Chem. 2002;277:8114–8120. doi: 10.1074/jbc.M109943200. [DOI] [PubMed] [Google Scholar]
- 31.Kraulis PJ, Jonasson P, Nygren PA, Uhlen M, Jendeberg L, Nilsson B, Kordel J. The serum albumin-binding domain of streptococcal protein G is a three-helical bundle: A heteronuclear NMR study. FEBS Lett. 1996;378:190–194. doi: 10.1016/0014-5793(95)01452-7. [DOI] [PubMed] [Google Scholar]
- 32.Linhult M, Binz HK, Uhlen M, Hober S. Mutational analysis of the interaction between albumin-binding domain from streptococcal protein G and human serum albumin. Protein Sci. 2002;11:206–213. doi: 10.1110/ps.02802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lejon S, Frick IM, Bjorck L, Wikstrom M, Svensson S. Crystal structure and biological implications of a bacterial albumin binding module in complex with human serum albumin. J Biol Chem. 2004;279:42924–42928. doi: 10.1074/jbc.M406957200. [DOI] [PubMed] [Google Scholar]
- 34.Ahmad JN, Li J, Biedermannova L, Kuchar M, Sipova H, Semeradtova A, Cerny J, Petrokova H, Mikulecky P, Polinek J, Stanek O, Vondrasek J, Homola J, Maly J, Osicka R, Sebo P, Maly P. Novel high-affinity binders of human interferon gamma derived from albumin-binding domain of protein G. Proteins: Struct Funct Bioinform. 2012;80:774–789. doi: 10.1002/prot.23234. [DOI] [PubMed] [Google Scholar]
- 35.Nilvebrant J, Alm T, Hober S, Lofblom J. Engineering bispecificity into a single albumin-binding domain. PLoS One. 2011;6:e25791. doi: 10.1371/journal.pone.0025791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nilvebrant J, Astrand M, Lofblom J, Hober S. Development and characterization of small bispecific albumin-binding domains with high affinity for ErbB3. Cell Mol Life Sci. 2013;70:3973–3985. doi: 10.1007/s00018-013-1370-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vaisocherova H, Zitova A, Lachmanova M, Stepanek J, Kralikova S, Liboska R, Rejman D, Rosenberg I, Homola J. Investigating oligonucleotide hybridization at subnanomolar level by surface plasmon resonance biosensor method. Biopolymers. 2006;82:394–398. doi: 10.1002/bip.20433. [DOI] [PubMed] [Google Scholar]
- 38.Sipova H, Sevcu V, Kuchar M, Ahmad JN, Mikulecky P, Osicka R, Maly P, Homola J. Surface plasmon resonance biosensor based on engineered proteins for direct detection of interferon-gamma in diluted blood plasma. Sens Actuators B Chem. 2012;174:306–311. [Google Scholar]
- 39.Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. 2007;8:942–949. doi: 10.1038/ni1496. [DOI] [PubMed] [Google Scholar]
- 40.Stritesky GL, Yeh N, Kaplan MH. IL-23 promotes maintenance but not commitment to the Th17 lineage. J Immunol. 2008;181:5948–5955. doi: 10.4049/jimmunol.181.9.5948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Iwakura Y, Ishigame H, Saijo S, Nakae S. Functional Specialization of Interleukin-17 Family Members. Immunity. 2011;34:149–162. doi: 10.1016/j.immuni.2011.02.012. [DOI] [PubMed] [Google Scholar]