

Abstract
Stimulation of group I mGluRs elicits several forms of translation-dependent neuronal plasticity including epileptogenesis. The translation process underlying plasticity induction is controlled by repressors including the fragile X mental retardation protein (FMRP). In the absence of FMRP-mediated repression, a condition that occurs in a mouse model (Fmr1−/−) of fragile X syndrome, group I mGluR-activated translation is exaggerated causing enhanced seizure propensity. We now show that glutamate exposure (10 μm for 30 min) reduced FMRP levels in wild-type mouse hippocampal slices. Downregulation of FMRP was dependent on group I mGluR activation and was blocked by a proteasome inhibitor (MG-132). Following glutamate exposure, synaptic stimulation induced prolonged epileptiform discharges with properties similar to those observed in Fmr1−/− preparations. In both cases, prolonged epileptiform discharges were blocked by group I mGluR antagonists (LY367385 + MPEP) and their induction was prevented by protein synthesis inhibitor (anisomycin). The results suggest that stimulation of group I mGluRs during glutamate exposure caused proteolysis of FMRP. Reduction of FMRP led to enhanced synaptic group I mGluR-mediated translation. Elevated translation facilitated the recruitment of group I mGluR-mediated prolonged epileptiform discharges.
Keywords: epilepsy, metabotropic glutamate receptor, plasticity, protein synthesis, proteolysis, seizure
Introduction
While it is generally recognized that excessive exposure of cortical neurons to glutamate under conditions such as traumatic brain injury and stroke can cause cell death and epileptogenesis (glutamate toxicity; Nilsson et al., 1990; Bullock et al., 1995, 1998; Dávalos et al., 1997; Smolders et al., 1997; Sun et al., 2001; Rakhade and Jensen, 2009), the cellular mechanisms underlying such toxicity are not as yet widely understood. We show in this study that glutamate exposure altered synaptically activated group I mGluR responses to facilitate the recruitment of prolonged epileptiform discharges.
Stimulation of group I mGluRs elicits rapid and robust protein synthesis in cortical neurons (Weiler et al., 1997; Osterweil et al., 2010). Newly synthesized proteins produce long-term changes in synaptic (LTD; Huber et al., 2000) and intrinsic (Chuang et al., 2005; Young et al., 2008; Bianchi et al., 2009) properties of hippocampal neurons in the CA1 and CA3 regions, respectively. The latter changes in the CA3 region underlie enhanced recurrent seizure discharges and epileptogenesis (Bianchi et al., 2009). Enhanced seizure propensity is indicated in vitro by the appearance of synaptically induced prolonged epileptiform discharges in the hippocampus (Chuang et al., 2005; Bianchi et al., 2009). Prolonged epileptiform discharges are not normal responses elicited synaptically in wild-type hippocampal preparations but are expressed in fragile X syndrome model mouse (Fmr1−/−) preparations (Chuang et al., 2005; Bianchi et al., 2009). The appearance of prolonged epileptiform discharges requires aberrantly elevated group I mGluR responses secondary to enhanced protein synthesis (Merlin et al., 1998; Zhao et al., 2004, 2011; Young et al., 2013).
A key regulator of group I mGluR-stimulated protein synthesis in cortical neurons is fragile X mental retardation protein (FMRP), a translation repressor (Laggerbauer et al., 2001; Li et al., 2001; Bear et al., 2004). When FMRP is absent, as in the case of fragile X syndrome model mouse (Fmr1−/−), enhanced protein synthesis elicited by synaptic stimulation of group I mGluRs causes the activation of prolonged epileptiform discharges (Chuang et al., 2005). Our recent studies show that the function of FMRP itself is regulated by group I mGluRs (Zhao et al., 2011). Thus, in the wild-type hippocampus, stimulation of group I mGluRs by the agonist DHPG causes proteolysis of FMRP via the ubiquitin-proteasome system (Hou et al., 2006; Zhao et al., 2011; Nalavadi et al., 2012). In this study, we examined if (1) exposure of hippocampal cells caused FMRP proteolysis via activation of group I mGluRs and (2) with reduced translation repression (due to downregulation of FMRP), synaptic activation of group I mGluRs can activate protein synthesis (Zhao et al., 2011) so that prolonged epileptiform discharges are now activatable by synaptic stimulation.
Materials and Methods
Animals.
Fmr1−/− (FVB.129P2-Fmr1tm1Cgr/J) and control (FVB.129P2-Pde6b+Tyrc-ch/AntJ) mice were purchased from The Jackson Laboratory. Three-week-old male mice were used.
Slice preparation.
Transverse hippocampal slices (400 μm thick) were prepared as described previously (Chuang et al., 2005). Briefly, slices were obtained from the dorsal half of each hippocampus. During slicing, the hippocampi were submerged in an ice-cold, low-Ca2+/high-Mg2+ buffer, consisting of the following (in mm): 124 NaCl, 26 NaHCO3, 2.5 KCl, 8 MgCl2, 0.5 CaCl2, and 10 d-glucose, bubbled with 95% O2 and 5% CO2. After slicing, slices were incubated in a slice-holding chamber maintained at 32.5°C for at least 90 min containing normal solution ACSF consisting of the following (in mm): 124 NaCl, 26 NaHCO3, 5 KCl, 1.6 MgCl2, 2 CaCl2, and 10 d-glucose bubbled with 95% O2 and 5% CO2, pH 7.4. For electrophysiological experiments, slices were subjected to glutamate or drug exposure and then transferred to an interface chamber maintained at 33.5°C. Slices were perfused with ACSF for 60–90 min before intracellular recording. For Western blot experiments, slices were collected from the slice-holding chamber immediately following drug application.
Electrophysiology.
Microelectrodes were pulled from glass tubes (1 mm outer diameter with glass filament inside; World Precision Instruments) using a micropipette puller (Sutter Instruments). Microelectrodes were filled with 2 m potassium acetate and the resistances were ∼70 MΩ. Signals were amplified (Axoclamp 2B; Molecular Devices) and recorded simultaneously in a computer running pClamp 6 (Molecular Devices) and on a chart recorder (Gould).
Drugs.
For electrophysiology, pharmacological agents were delivered to the hippocampal slices through continuous perfusion. Depending on the flow rate, the final concentration was reached within 15 min. For Western blot experiments, agents were directly applied to the holding chamber. Pretreatment of slices with various agents lasted for 45 min. Anisomycin, CNQX disodium, CPP, LY367385, and N-[(phenylmethoxy)carbonyl]-l- leucyl-N-[(1S)-1-formyl-3-methylbutyl]-l- leucinamide (MG-132) were purchased from Tocris Bioscience. Other chemicals were from Sigma. Antibodies for FMRP and α-tubulin were purchased from Millipore. Secondary antibodies were from Cell Signaling Technology. MG-132, MPEP, and anisomycin were prepared in DMSO stock solutions. Stock solutions were aliquoted and stored at −20°C.
Sample preparation.
Hippocampal slices were collected from the slice-holding chamber and snap frozen on dry ice. Each sample containing six slices was stored at −80°C until biochemical analysis. Each sample was sonicated in ice-cold homogenization buffer containing the following (in mm): 320 sucrose, 10 HEPES, pH 7.4, 2 EDTA, 1 EGTA, and protease inhibitors (20 μg/ml aprotinin, 20 μg/ml leupeptin, 20 μg/ml Pepstatin A, and 1 mm PMSF), and phosphatase inhibitors (1 NaF, 1 Na3VO4, 2.5 Na4P2O7, and 1 β-glycerophosphate). The homogenate was then mixed with detergents (final: 0.1% SDS, 0.5% DOC, and 1% NP-40). The suspension was cleared by centrifugation. Lysates were aliquoted and stored at −80°C. One aliquot of lysate was used to determine protein concentration with BCA method (Fisher Scientific). For Western blots, samples were mixed with SDS sample buffer (10% glycerol, 5% BME, and 2% SDS, in 62 mm Tris-HCl, pH 6.8) and boiled for 5 min.
Western blots.
Equal amounts of sample were subjected to electrophoresis in 8% or 10% SDS-PAGE gels (1.0 mm; Novex Tris-Glycine; Invitrogen). Gels were then transferred to PVDF membranes for 1 h at a constant voltage of 100 V (mini Trans-Blot Cell; Bio-Rad). The membrane was stained with Ponceau S briefly to verify the quality of transfer. The membrane was blocked in 5% nonfat milk dissolved in TBST buffer (containing the following: 20 mm Tris-HCl, pH 7.6, 15 mm NaCl, and 0.1% Tween 20) for 1 h at room temperature or overnight at 4°C. After blocking, the membrane was probed with primary antibody for 2 h at room temperature or overnight at 4°C. After incubation, the membrane was washed three times for 5 min each with TBST. The membrane was then incubated with HRP-conjugated secondary antibody in TBST for 1 h. It was washed again three times for 5 min each with TBST, and then the membrane was treated with LumiGLO (Cell Signaling Technology) and developed.
Data analysis.
The duration of an individual synchronized discharge was measured from the beginning of the first action potential of the discharge to the repolarization of the last action potential of the discharge. Discharge durations of all synchronized discharges in a 5 min period for each slice in the various experimental conditions were averaged. Synchronized discharges with durations longer than 1.5 s are referred to as prolonged synchronized discharges. Immunoreactive bands in Western blots were scanned with a desktop scanner (EPSON Expression 1680) and quantified with Scion Image software. The optical densities (ODs) of bands of interest were normalized to the OD value of α-tubulin bands. Summary data are reported as means ± SEM. Student's t test was used for the statistical comparison of two sets of data. For analysis of data among groups, one-way ANOVA followed by the Tukey HSD test for repeated measures was applied. In all cases, the level of significant difference was p < 0.05.
Results
Glutamate exposure downregulates FMRP levels
Previous studies show that stimulation of group I mGluRs elicits FMRP proteolysis (Hou et al., 2006; Zhao et al., 2011; Nalavadi et al., 2012) enabling downstream protein synthesis (Hou et al., 2006; Zhao et al., 2011). To examine the effects of tonic glutamate exposure on FMRP levels, hippocampal slices were exposed to glutamate at a series of concentrations (10, 100, and 1000 μm) for 30 min in a slice-holding chamber. The levels of FMRP in hippocampal slices were evaluated using monoclonal 1C3 antibody. Western blot results (Fig. 1A) revealed that glutamate at the lowest concentration tested was sufficient to induce maximal FMRP reduction during glutamate exposure (at 10 μm: 72 ± 6%; at 1000 μm: 69 ± 9%; n = 4; p = 0.77).
Figure 1.
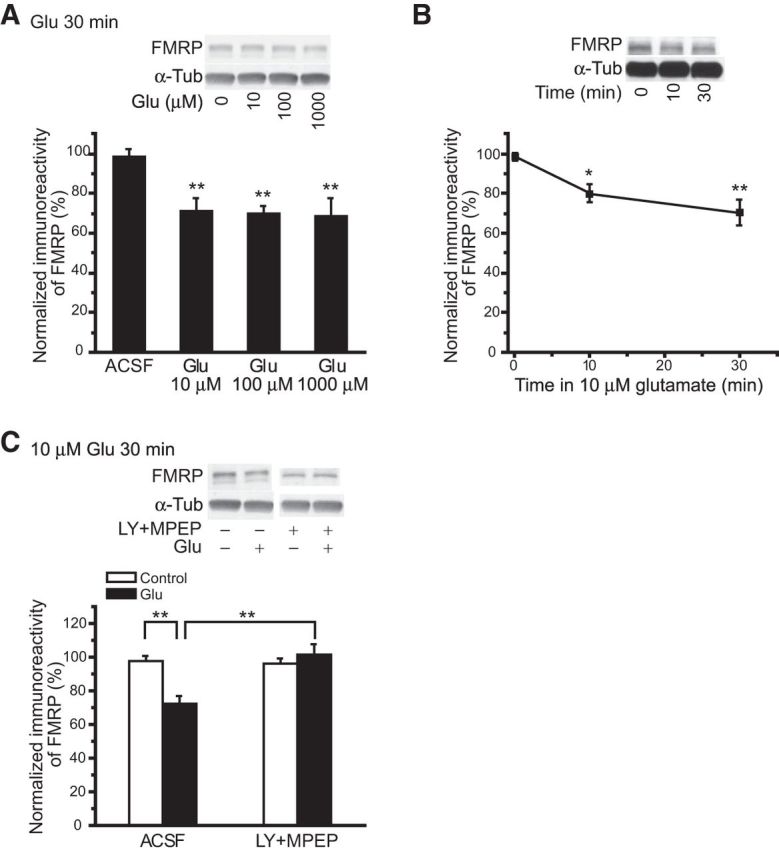
Glutamate exposure caused group I mGluR-dependent decreases in FMRP levels. A, Dose responses of FMRP levels to glutamate exposure. Wild-type hippocampal slices were maintained in a slice-holding chamber and exposed to glutamate of increasing concentrations for 30 min. Glutamate effectively decreased FMRP levels at all concentrations tested, and increasing glutamate concentration did not affect either the direction or the amplitude of glutamate-induced changes of FMRP levels. B, Time course of the changes of FMRP levels by glutamate exposure. Slices were collected at 0, 10, and 30 min following the addition of 10 μm glutamate. FMRP was quickly decreased by glutamate exposure and reached a plateau level as early as 10 min. C, Effects of mGluR antagonists (100 μm LY367385 and 50 μm MPEP) on glutamate-induced downregulation of FMRP. In control experiments (ACSF), 10 μm glutamate exposure for 30 min significantly decreased FMRP levels (filled column). However, in the presence of mGluR antagonists (LY +MPEP), glutamate exposure was no longer effective in eliciting FMRP downregulation. Representative Western blots are displayed above summary data plots. α-Tubulin (α-Tub) levels, used as loading controls, are also displayed. For summary data, the immunoreactivity of FMRP bands was normalized to the immunoreactivity of their corresponding α-tubulin bands. For this and the following figures, ***p < 0.001, **p < 0.01, and *p < 0.05.
In parallel experiments, the time course of FMRP level changes by glutamate exposure was examined. Slices were collected at time points of 0, 10, and 30 min after glutamate exposure (10 μm). Glutamate induced a rapid decrease of FMRP level (Fig. 1B; at 0 min: 98 ± 2%; at 10 min: 80 ± 4%; at 30 min: 70 ± 6%; one-way ANOVA, p < 0.01; n = 4), and there was no significant difference in FMRP levels between 10 and 30 min (n = 4; p = 0.26).
The receptor involvement in FMRP downregulation by glutamate exposure was examined using group I mGluR antagonists. Slices were treated with LY367385 and MPEP (100 and 50 μm, respectively) before (45 min) and during glutamate exposure (10 μm for 30 min). FMRP level remained stable when slices were exposed to antagonists alone or to antagonists plus glutamate (Fig. 1C; in LY + MPEP alone: 97 ± 3%; in LY + MPEP + Glu: 102 ± 6%; n = 6; p = 0.41). On the other hand, antagonists of AMPA and NMDA receptors (CNQX and CPP, 20 μm each) did not affect FMRP downregulation during glutamate exposure (in CNQX + CPP alone: 98 ± 4%; in CNQX + CPP + Glu: 96 ± 3%; n = 4; p = 0.75).
Glutamate exposure alters population responses in the hippocampus
Short interictal-like epileptiform discharges (<1 s) are elicited by application of bicuculline (Bic), a GABAA antagonist, in the hippocampus (Traub and Wong, 1982). These population responses were examined in slices subjected to vehicle (control) or glutamate exposure (10 μm for 30 min). In control experiments, addition of bicuculline consistently elicited short interictal-like epileptiform discharges and the duration of epileptiform discharges remained stable with extended bicuculline perfusion (up to 3 h; Fig. 2Aa,B, empty columns; in control slices; at Bic 10 min: 398 ± 49 ms; at Bic 40 min: 405 ± 52 ms; n = 8; p = 0.92).
Figure 2.
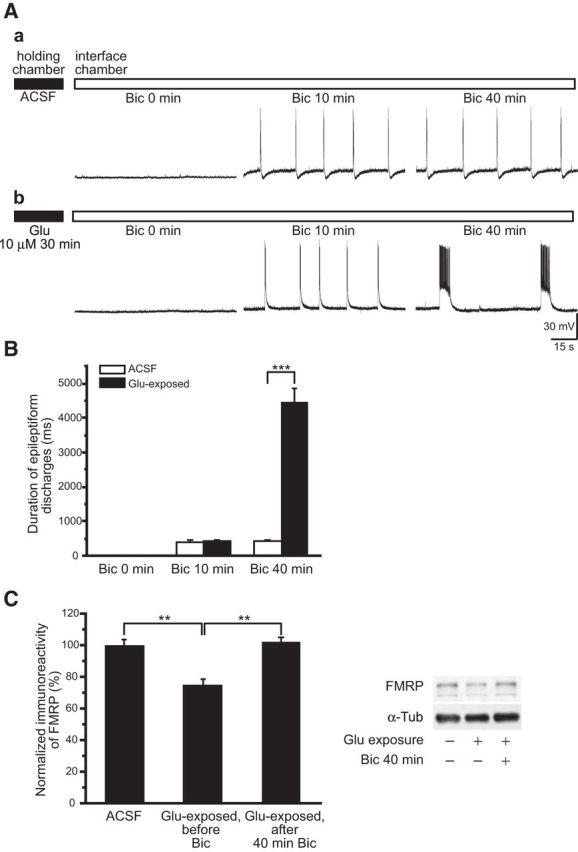
Distinct prolonged epileptiform discharges were elicited in glutamate-exposed slices. Slices were maintained in a slice-holding chamber (filled horizontal bar) for glutamate exposure, and then transferred to an interface chamber for continuous bicuculline perfusion (empty horizontal bar). A, Representative intracellular recordings obtained at 0, 10, and 40 min following the addition of 50 μm Bic to the perfusion solution. Aa, In control slices, bicuculline elicited stable, short epileptiform discharges (Bic 10 min), and the short epileptiform discharges remained stable with continuous bicuculline perfusion (Bic 40 min). Ab, In slices exposed to glutamate (Glu 10 μm 30 min), short epileptiform discharges elicited by bicuculline (Bic 10 min) were followed by an abrupt appearance of prolonged epileptiform discharges that gradually dominated population responses (Bic 40 min). B, Average durations of epileptiform discharges following continuous bicuculline perfusion in control (ACSF, empty columns) and glutamate-exposed slices (filled columns). The average durations of initial epileptiform discharges at Bic 10 min in control and in glutamate-exposed slices were not significantly different. In contrast, at Bic 40 min the average duration of the epileptiform discharges in glutamate-exposed slices was significantly longer than that of control slices due to the occurrence of prolonged epileptiform discharges. C, FMRP levels in slices in ACSF, following glutamate exposure (Glu-exposed, before Bic), and after 40 min bicuculline in the recording chamber (Glu-exposed, after 40 min Bic). FMRP in ACSF was 100 ± 3.5%, n = 6; in Glu-exposed, before Bic: 75 ± 3.6%, n = 6; and in Glu-exposed, after 40 min Bic: 102 ± 2.5%; n = 6; one-way ANOVA; **p < 0.01).
In slices exposed to glutamate, addition of bicuculline first induced short interictal-like epileptiform discharges (Fig. 2Ab, middle panel) as observed in control slices, but with time prolonged epileptiform discharges of 2–7 s in duration appeared and they ultimately became the dominant population responses (Fig. 2Ab,2B, filled columns; in glutamate-exposed slices; at Bic 10 min: 427 ± 40 ms; at Bic 40 min: 4474 ± 394 ms; n = 11; p < 0.001).
FMRP level was assessed in slices 40 min after bicuculline in the recording chamber. A rebound of FMRP by 25–30% at the 40 min time point was noted (Fig. 2C). Stimulation of group I mGluRs elicits rapid translation of its target mRNAs including FMRP mRNA (Weiler et al., 1997). The rebound in FMRP most probably reflects enhanced translation by sustained synaptic activation of group I mGluRs during bicuculline exposure. These results are consistent with those shown in slices treated with DHPG (Zhao et al., 2011) where an initial transient decrease in FMRP, peaking at ∼10 min, is followed by an increase at 30 min of DHPG exposure.
Group I mGluRs are involved in the activation of prolonged epileptiform discharges
The receptors engaged in the synaptic activation of prolonged epileptiform discharges were examined. In this series of experiments, all slices examined were pre-exposed to glutamate (10 μm for 30 min). Two sets of experiments were performed. First, group I mGluR antagonists (100 μm LY367385 and 50 μm MPEP) were applied to the slices during glutamate exposure (Fig. 3Aa). Subsequent application of bicuculline in ACSF no longer caused the appearance of prolonged epileptiform discharges (Fig. 3Aa, middle and right). Bicuculline induced only short epileptiform discharges that remained stable for the duration of the recordings (up to 3 h; Figs. 3Aa, middle and right, 4C; LY + MPEP; at Bic 10 min: 396 ± 34 ms; at Bic 40 min: 427 ± 33 ms; n = 8; p = 0.51). In a second set of experiments, once prolonged epileptiform discharges were established in slices exposed to glutamate, addition of LY367385 and MPEP suppressed the occurrence of prolonged epileptiform discharges unmasking typical short epileptiform discharges (Figs. 3Ab, middle, 4D; LY + MPEP; control: 4681 ± 440 ms; LY + MPEP: 452 ± 51 ms; n = 6; p < 0.001). Subsequent washout of the group I mGluR antagonists caused reemergence of prolonged epileptiform discharges (Fig. 3Ab, right).
Figure 3.
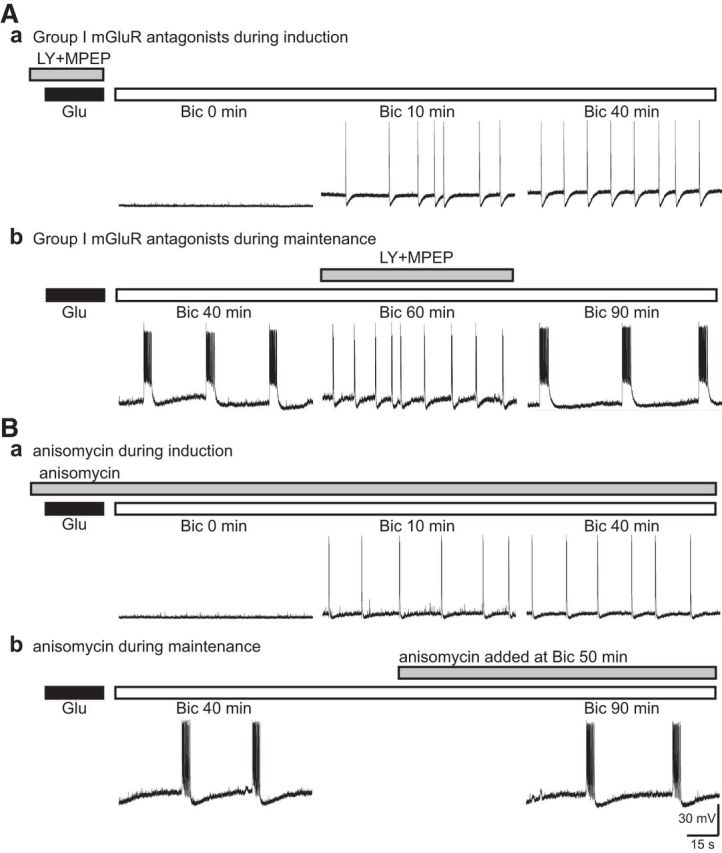
Group I mGluR signaling was required for the induction of prolonged epileptiform discharges. Representative records from glutamate-exposed slices obtained in the presence of group I mGluR antagonists or anisomycin (gray bar). Aa, In the presence of group I mGluR antagonists, 100 μm LY367385 and 50 μm MPEP (LY + MPEP), bicuculline induced short epileptiform discharges (Bic 10 min) that remained stable (Bic 40 min) for the duration of the recording. Ab, Group I mGluR antagonists (LY + MPEP) were added after perfusion with bicuculline for 40 min. Bicuculline elicited prolonged epileptiform discharges (Bic 40 min). Addition of the Group I mGluR antagonists blocked the prolonged epileptiform discharges unmasking short epileptiform discharges (Bic 60 min). Upon group I mGluR antagonist washout, prolonged epileptiform discharges reappeared and dominated population responses (Bic 90 min). Ba, In the presence of anisomycin (20 μm), bicuculline induced short epileptiform discharges (Bic 10 min) but failed to elicit prolonged epileptiform discharges with further perfusion (Bic 40 min). Bb, Once prolonged epileptiform discharges were established following bicuculline application (Bic 40 min), addition of anisomycin did not affect the occurrence of prolonged epileptiform discharges (Bic 90 min).
Figure 4.
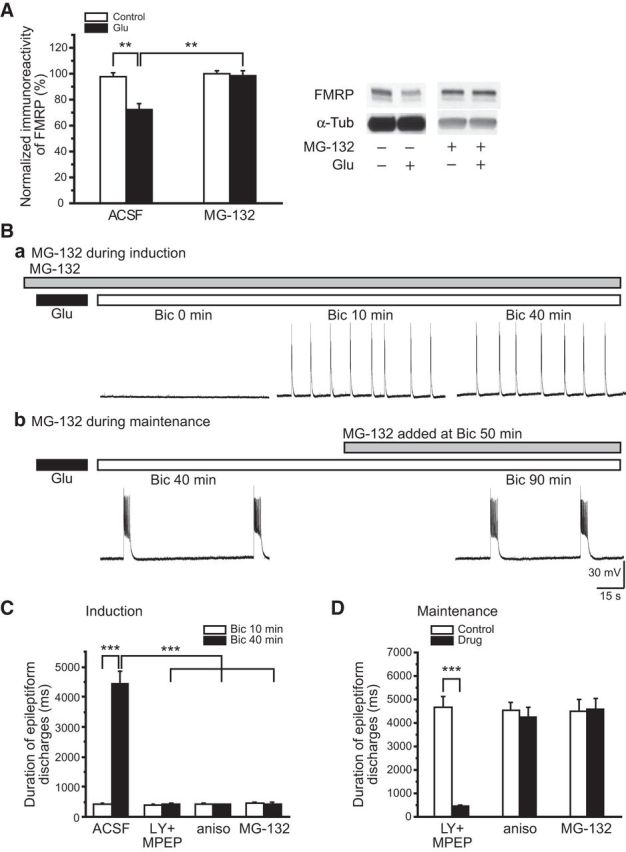
Proteasome activity was required for FMRP downregulation and the induction of prolonged epileptiform discharges in glutamate-exposed slices. A, Summary data of the effect of glutamate exposure on FMRP levels in the absence (ACSF) and presence (MG-132) of a proteasome inhibitor, MG-132 (1 μm). In the presence of MG-132, FMRP levels remained stable (MG-132, empty column) and were not altered by glutamate exposure (MG-132, filled column). Representative Western Blots are shown on the right. Ba, In the presence of MG-132 (gray bar), bicuculline perfusion after glutamate exposure elicited short epileptiform discharges (Bic 10 min) with durations that remained stable with extended perfusion (Bic 40 min). Bb, MG-132 was applied after prolonged epileptiform discharges were induced in glutamate-exposed slices by bicuculline (Bic 40 min). Addition of MG-132 had little effect on ongoing prolonged epileptiform discharges (Bic 90 min). C, Summary data of the effects of mGluR antagonists (LY + MPEP), anisomycin (aniso), and MG-132 (MG-132) on the induction of prolonged epileptiform discharges in glutamate-exposed slices. There were no significant differences in initial short epileptiform discharges among mGluR antagonists, anisomycin, or MG-132-treated slices (at Bic 10 min: in control, 427 ± 40 ms, n = 11; in LY + MPEP, 396 ± 34 ms, n = 8; in anisomycin, 418 ± 32 ms, n = 8; in MG-132, 452 ± 44 ms, n = 8; one-way ANOVA, p = 0.80). With further bicuculline perfusion, prolonged epileptiform discharges were elicited only in the control group but not in the mGluR antagonist or MG-132 groups (at Bic 40 min: in control, 4474 ± 394 ms, n = 11; in LY + MPEP, 427 ± 33 ms, n = 8; in anisomycin, 411 ± 23 ms, n = 8; in MG-132, 422 ± 49 ms, n = 8; one-way ANOVA, p < 0.001). D, Summary data showing the effects of mGluR antagonists, anisomycin, and MG-132 on ongoing prolonged epileptiform discharges. Addition of LY367385 and MPEP effectively suppressed the occurrence of prolonged epileptiform discharges (LY + MPEP: control, 4681 ± 440 ms; after drug, 452 ± 51 ms; n = 6; p < 0.001). However, addition of anisomycin or MG-132 afterward had little effects on prolonged epileptiform discharges (anisomycin: control, 4553 ± 301 ms; after drug, 4240 ± 429 ms, n = 6; p = 0.56; MG-132: control, 4485 ± 495 ms; after drug, 4587 ± 435 ms, n = 6; p = 0.89).
Group I mGluRs elicit a number of long-term cellular changes via protein synthesis-dependent processes in the hippocampus (Chuang et al., 2005; Young et al., 2008; Bianchi et al., 2009). The engagement of protein synthesis in the induction of prolonged epileptiform discharges was examined by testing whether application of a protein synthesis inhibitor (anisomycin, 20 μm) affected the occurrence of prolonged epileptiform discharges in glutamate-exposed slices. Slices were treated with anisomycin during glutamate exposure and bicuculline application. The duration of short epileptiform discharges recorded under this condition remained stable even with extended perfusion (up to 3 h; Figs. 3Ba, middle and right, 4C, aniso; at Bic 10 min: 418 ± 32 ms; at Bic 40 min: 411 ± 23 ms; n = 8; p = 0.86). In parallel experiments, anisomycin was introduced after prolonged epileptiform discharges were established. In this case, anisomycin had no effect on the duration and frequency of ongoing prolonged epileptiform discharges (Figs. 3Bb, left and right, 4D, aniso; control: 4553 ± 301 ms; anisomycin: 4240 ± 429 ms; n = 6; p = 0.56).
Glutamate exposure proteolyzes FMRP
Previous studies suggest that downregulation of FMRP is an obligatory step for group I mGluR-mediated protein synthesis and plasticity in wild-type preparations (Hou et al., 2006; Zhao et al., 2011). FMRP downregulation is mediated by the ubiquitin-proteasome system (UPS). The involvement of UPS in FMRP downregulation elicited by glutamate exposure was examined using MG-132, a potent proteasome inhibitor (Lee and Goldberg, 1996). Slices were treated with MG-132 before (1 μm for 45 min) and during glutamate exposure (10 μm for 30 min). Western blot results showed that glutamate no longer affected FMRP levels in the presence of MG-132 (glutamate exposure in the presence of MG-132; Fig. 4A; in MG-312 alone: 100 ± 2%; in MG-312 + Glu: 98 ± 4%; n = 6; p = 0.75).
MG-132 applied during glutamate exposure prevented synaptic activation of prolonged epileptiform discharges. In slices with glutamate exposure in the presence of MG-132, addition of bicuculline elicited stable short epileptiform discharges for the duration of the recordings (up to 3 h; Fig. 4Ba,C, MG-132; at Bic 10 min: 452 ± 44 ms; at Bic 40 min: 422 ± 49 ms; n = 8; p = 0.65).
Additional data showed that after glutamate exposure and associated FMRP proteolysis, perfusion of MG-132 did not prevent the activation of prolonged epileptiform discharges (Fig. 4Bb,D, MG-132; control: 4485 ± 495 ms; MG-132: 4587 ± 435 ms; n = 6; p = 0.89).
FMRP is the target of the anti-epileptogenic action of MG-132
MG-132 affects all cellular proteins subjected to UPS action. Thus, the prevention of prolonged epileptiform discharges could result from changes in any one or more of these proteins. We evaluated the specific role of FMRP downregulation using Fmr1−/− mice. Here, proteasome activity affects all proteins except FMRP, which is absent. Application of MG-132 to Fmr1−/− preparations did not affect the activation of prolonged epileptiform discharges (Fig. 5Ab, right, B, Fmr1−/−; at Bic 40 min: control, 4424 ± 558 ms; MG-132: 4609 ± 804 ms; n = 7; p = 0.84) suggesting that the MG-132 inhibition of prolonged epileptiform discharges is not due to nonspecific actions on cellular proteins other than FMRP.
Figure 5.

MG-132 failed to block the induction of prolonged epileptiform discharges in glutamate-exposed Fmr1−/− slices. Slices were prepared from Fmr1−/− mice and subjected to glutamate exposure and bicuculline perfusion. Aa, In control experiments, bicuculline elicited prolonged epileptiform discharges (Bic 40 min, average duration: 4424 ± 558 ms) following a brief appearance of short epileptiform discharges (Bic 10 min, average duration: 392 ± 53 ms; n = 7; p < 0.001). Ab, Slices were treated with MG-132 (gray bar) before and during glutamate exposure and bicuculline perfusion. Unlike that observed in wild-type glutamate-exposed slices (Fig. 4Ba), MG-132 was no longer effective in blocking the induction of prolonged epileptiform discharges and bicuculline induced prolonged epileptiform discharges (Bic 40 min) after the initial short epileptiform discharges (Bic 10 min). B, Summary data of the average duration of the epileptiform discharges induced by bicuculline in wild-type and Fmr1−/− glutamate-exposed slices in the absence (empty columns) or presence (filled columns) of MG-132. MG-132 blocked the induction of prolonged epileptiform discharges in wild-type, but not in Fmr1−/−, preparations.
Discussion
The results of this study suggest that exposure of hippocampal neurons to glutamate altered synaptic responses in the hippocampus to facilitate the activation of group I mGluR-mediated epileptogenesis. Data show that glutamate exposure caused a group I mGluR-mediated proteolysis of FMRP via the ubiquitin-proteasome system (Figs. 1, 4A). Downregulation of FMRP enabled the synaptic induction of prolonged epileptiform discharges via group I mGluR stimulation (Figs. 2, 3Aa, 4C). The data further indicate that the induction of prolonged epileptiform discharges is a protein synthesis-dependent process (Figs. 3Ba, 4C) subjected to FMRP regulation and that prolonged epileptiform discharges, once induced, are maintained by endogenous group I mGluR activities (Fig. 3Bb).
FMRP regulation of prolonged epileptiform discharges
In the hippocampus, the constitutive level of neuronal FMRP is sufficient to arrest group I mGluR-activated translation (Zhao et al., 2011). Because of the regulatory action of FMRP, synaptic stimulation in hippocampal circuits does not normally elicit prolonged epileptiform discharges that require de novo protein synthesis for their production. Such discharges are only elicited in fragile X syndrome model mouse preparations where FMRP is absent (Chuang et al., 2005).
Activation of group I mGluRs elicits parallel responses: signaling cascades for protein synthesis and, in tandem, proteolysis of FMRP in wild-type neurons (Hou et al., 2006; Zhao et al., 2011; Nalavadi et al., 2012). Group I mGluRs are stimulated by recurrent synapses between CA3 pyramidal cells (Lee et al., 2002). Our data suggest that stimulation of group I mGluRs through synchronized activation of these recurrent synapses in the presence of bicuculline is not sufficient to elicit FMRP downregulation and thus allow for the group I mGluR-mediated protein synthesis necessary for prolonged epileptiform discharge activation. In contrast, during glutamate exposure, more persistent and tonic stimulation of group I mGluRs causes sufficient FMRP proteolysis to enable induction of prolonged epileptiform discharges during synaptic stimulation (Figs. 1, 2).
Downregulation of FMRP during glutamate exposure is dependent on group I mGluR activation and subsequent UPS-mediated proteolysis. Inhibition either of group I mGluRs or of proteasome activity blocked FMRP downregulation and prevented the induction of prolonged epileptiform discharges (Figs. 3A, 4). The ineffectiveness of proteasome inhibitor on the induction of prolonged epileptiform discharges in Fmr1−/− slices (Fig. 5) points to the importance of FMRP preservation from proteolysis by proteasome inhibitor as the response guarding against prolonged epileptiform discharge activation. It remains possible that glutamate exposure also affects the stability of some proteins associated with FMRP signaling, such as AGO1-miR-125a inhibitory complex (Muddashetty et al., 2011), which also regulates translation. Accordingly, an alternative explanation for the ineffectiveness of proteasome inhibitor in Fmr1−/− preparation is that these other protein(s) are constitutively downregulated in Fmr1−/− neurons so that they are not at a sufficient level to effectively regulate translation in the Fmr1−/− preparation.
Upon glutamate exposure, FMRP is quickly degraded and then becomes stable at a reduced level of ∼70% of control even with extended exposure time or increased glutamate concentration. These results suggest that FMRP proteolysis by glutamate is a saturatable process. This finding is consistent with that observed in a previous study in slices treated with group I mGluR agonist DHPG (Zhao et al., 2011) where a stable level of FMRP is retained at 72%. Other studies have shown that group I mGluR stimulation elicits a rapid FMRP dephosphorylation, a step required for subsequent UPS-mediated proteolysis (Nalavadi et al., 2012). Thus, it is possible that the upper limit of glutamate-induced proteolysis in FMRP level may be set by the amount of FMRP available for the dephosphorylation process elicited by group I mGluR activation.
Induction and maintenance of prolonged epileptiform discharges
Induction of group I mGluR-mediated prolonged epileptiform discharges involves the long-term activation of several excitatory cellular responses in CA3 pyramidal cells (Chuang et al., 2005; Young et al., 2008; Bianchi et al., 2009). These include suppression of the spike afterhyperpolarization, activation of a voltage-dependent inward current (ImGluR(V)), and increase in cellular input resistance. Long-term activation of these cellular responses by group I mGluR stimulation is critically dependent on de novo protein synthesis (Bianchi et al., 2009). When protein synthesis is blocked pharmacologically by anisomycin, excitatory responses are elicited transiently upon receptor stimulation but are not maintained (Young et al., 2008, 2013; Bianchi et al., 2009). Similarly, in the presence of protein synthesis inhibitors, synaptic stimulation no longer elicited prolonged epileptiform discharges in glutamate-exposed slices (Figs. 3B, 4C). A question that can be asked is how does group I mGluR-induced protein synthesis cause the excitatory responses to become persistently active (long-term activation)? Data reveal that the protein synthesis-dependent process changes the properties of group I mGluRs such that the receptors themselves become constitutively active (Lodge et al., 2013; Young et al., 2013). Thus, following induction, constitutively active group I mGluRs continually elicit long-term activation of excitatory cellular responses without requiring ligand-mediated activation (Lodge et al., 2013; Young et al., 2013).
Because of the involvement of constitutively active receptors, maintenance of all group I mGluR excitatory cellular responses are reversibly blocked by group I mGluR antagonists. Previous data show that prolonged epileptiform discharges directly activated by group I mGluR agonists are sustained by the concerted action of persistent excitatory cellular responses (Bianchi et al., 2009; Young et al., 2013). The finding that synaptically induced prolonged epileptiform discharges in glutamate-exposed slices were also reversibly blocked by group I mGluR antagonists (Fig. 3Ab) is consistent with the hypothesis that group I mGluRs play a role both in the induction and maintenance of these discharges.
In sum, the results suggest the following scheme for the effects of glutamate exposure: downregulation of FMRP in glutamate enables group I mGluR-mediated protein synthesis to facilitate the generation of prolonged epileptiform discharges that are sustained by excitatory cellular responses activated by endogenous group I mGluR activity.
Excessive glutamate exposure is an initial brain insult commonly seen in stroke, status epilepticus (SE), and traumatic brain injury (TBI). Data suggested that overstimulation of glutamate receptors is a contributor to the development of recurrent epileptic seizures (for review, see McNamara et al., 2006). The in vitro results here suggest that glutamate exposure facilitates the emergence of group I mGluR-mediated epileptiform discharges. Our results do not provide any indication of whether such responses are elicited in vivo upon glutamate exposure. The data do suggest that the integrity of FMRP is a point of vulnerability that is compromised upon glutamate exposure and is thus a candidate site to be evaluated for long-term effects of glutamate exposure including epileptogenesis.
Footnotes
This work has been supported by the National Institutes of Health Grant NS35481 and the FRAXA Research Foundation.
The authors declare no competing financial interests.
References
- Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27:370–377. doi: 10.1016/j.tins.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Bianchi R, Chuang SC, Zhao W, Young SR, Wong RK. Cellular plasticity for group I mGluR-mediated epileptogenesis. J Neurosci. 2009;29:3497–3507. doi: 10.1523/JNEUROSCI.5447-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullock R, Zauner A, Woodward J, Young HF. Massive persistent release of excitatory amino acids following human occlusive stroke. Stroke. 1995;26:2187–2189. doi: 10.1161/01.STR.26.11.2187. [DOI] [PubMed] [Google Scholar]
- Bullock R, Zauner A, Woodward JJ, Myseros J, Choi SC, Ward JD, Marmarou A, Young HF. Factors affecting excitatory amino acid release following severe human head injury. J Neurosurg. 1998;89:507–518. doi: 10.3171/jns.1998.89.4.0507. [DOI] [PubMed] [Google Scholar]
- Chuang SC, Zhao W, Bauchwitz R, Yan Q, Bianchi R, Wong RK. Prolonged epileptiform discharges induced by altered group I metabotropic glutamate receptor-mediated synaptic responses in hippocampal slices of a fragile X mouse model. J Neurosci. 2005;25:8048–8055. doi: 10.1523/JNEUROSCI.1777-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dávalos A, Castillo J, Serena J, Noya M. Duration of glutamate release after acute ischemic stroke. Stroke. 1997;28:708–710. doi: 10.1161/01.STR.28.4.708. [DOI] [PubMed] [Google Scholar]
- Hou L, Antion MD, Hu D, Spencer CM, Paylor R, Klann E. Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron. 2006;51:441–454. doi: 10.1016/j.neuron.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Huber KM, Kayser MS, Bear MF. Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science. 2000;288:1254–1257. doi: 10.1126/science.288.5469.1254. [DOI] [PubMed] [Google Scholar]
- Laggerbauer B, Ostareck D, Keidel EM, Ostareck-Lederer A, Fischer U. Evidence that fragile X mental retardation protein is a negative regulator of translation. Hum Mol Genet. 2001;10:329–338. doi: 10.1093/hmg/10.4.329. [DOI] [PubMed] [Google Scholar]
- Lee AC, Wong RK, Chuang SC, Shin HS, Bianchi R. Role of synaptic metabotropic glutamate receptors in epileptiform discharges in hippocampal slices. J Neurophysiol. 2002;88:1625–1633. doi: 10.1152/jn.2002.88.4.1625. [DOI] [PubMed] [Google Scholar]
- Lee DH, Goldberg AL. Selective inhibitors of the proteasome-dependent and vacuolar pathways of protein degradation in Saccharomyces cerevisiae. J Biol Chem. 1996;271:27280–27284. doi: 10.1074/jbc.271.44.27280. [DOI] [PubMed] [Google Scholar]
- Li Z, Zhang Y, Ku L, Wilkinson KD, Warren ST, Feng Y. The fragile X mental retardation protein inhibits translation via interacting with mRNA. Nucleic Acids Res. 2001;29:2276–2283. doi: 10.1093/nar/29.11.2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge D, Tidball P, Mercier MS, Lucas SJ, Hanna L, Ceolin L, Kritikos M, Fitzjohn SM, Sherwood JL, Bannister N, Volianskis A, Jane DE, Bortolotto ZA, Collingridge GL. Antagonists reversibly reverse chemical LTD induced by group I, group II and group III metabotropic glutamate receptors. Neuropharmacology. 2013;74:135–146. doi: 10.1016/j.neuropharm.2013.03.011. [DOI] [PubMed] [Google Scholar]
- McNamara JO, Huang YZ, Leonard AS. Molecular signaling mechanisms underlying epileptogenesis. Sci STKE. 2006;2006:re12. doi: 10.1126/stke.3562006re12. [DOI] [PubMed] [Google Scholar]
- Merlin LR, Bergold PJ, Wong RK. Requirement of protein synthesis for group I mGluR-mediated induction of epileptiform discharges. J Neurophysiol. 1998;80:989–993. doi: 10.1152/jn.1998.80.2.989. [DOI] [PubMed] [Google Scholar]
- Muddashetty RS, Nalavadi VC, Gross C, Yao X, Xing L, Laur O, Warren ST, Bassell GJ. Reversible inhibition of PSD-95 mRNA translation by miR-125a, FMRP phosphorylation, and mGluR signaling. Mol Cell. 2011;42:673–688. doi: 10.1016/j.molcel.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalavadi VC, Muddashetty RS, Gross C, Bassell GJ. Dephosphorylation-induced ubiquitination and degradation of FMRP in dendrites: a role in immediate early mGluR-stimulated translation. J Neurosci. 2012;32:2582–2587. doi: 10.1523/JNEUROSCI.5057-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson P, Hillered L, Pontén U, Ungerstedt U. Changes in cortical extracellular levels of energy-related metabolites and amino acids following concussive brain injury in rats. J Cereb Blood Flow Metab. 1990;10:631–637. doi: 10.1038/jcbfm.1990.115. [DOI] [PubMed] [Google Scholar]
- Osterweil EK, Krueger DD, Reinhold K, Bear MF. Hypersensitivity to mGluR5 and ERK1/2 leads to excessive protein synthesis in the hippocampus of a mouse model of fragile X syndrome. J Neurosci. 2010;30:15616–15627. doi: 10.1523/JNEUROSCI.3888-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakhade SN, Jensen FE. Epileptogenesis in the immature brain: emerging mechanisms. Nat Rev Neurol. 2009;5:380–391. doi: 10.1038/nrneurol.2009.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolders I, Khan GM, Manil J, Ebinger G, Michotte Y. NMDA receptor-mediated pilocarpine-induced seizures: characterization in freely moving rats by microdialysis. Br J Pharmacol. 1997;121:1171–1179. doi: 10.1038/sj.bjp.0701231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun DA, Sombati S, DeLorenzo RJ. Glutamate injury-induced epileptogenesis in hippocampal neurons: an in vitro model of stroke-induced “epilepsy.”. Stroke. 2001;32:2344–2350. doi: 10.1161/hs1001.097242. [DOI] [PubMed] [Google Scholar]
- Traub RD, Wong RK. Cellular mechanism of neuronal synchronization in epilepsy. Science. 1982;216:745–747. doi: 10.1126/science.7079735. [DOI] [PubMed] [Google Scholar]
- Weiler IJ, Irwin SA, Klintsova AY, Spencer CM, Brazelton AD, Miyashiro K, Comery TA, Patel B, Eberwine J, Greenough WT. Fragile X mental retardation protein is translated near synapses in response to neurotransmitter activation. Proc Natl Acad Sci U S A. 1997;94:5395–5400. doi: 10.1073/pnas.94.10.5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young SR, Bianchi R, Wong RK. Signaling mechanisms underlying group I mGluR-induced persistent AHP suppression in CA3 hippocampal neurons. J Neurophysiol. 2008;99:1105–1118. doi: 10.1152/jn.00435.2007. [DOI] [PubMed] [Google Scholar]
- Young SR, Chuang SC, Zhao W, Wong RK, Bianchi R. Persistent receptor activity underlies group I mGluR-mediated cellular plasticity in CA3 neuron. J Neurosci. 2013;33:2526–2540. doi: 10.1523/JNEUROSCI.3338-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Bianchi R, Wang M, Wong RK. Extracellular signal-regulated kinase 1/2 is required for the induction of group I metabotropic glutamate receptor-mediated epileptiform discharges. J Neurosci. 2004;24:76–84. doi: 10.1523/JNEUROSCI.4515-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Chuang SC, Bianchi R, Wong RK. Dual regulation of fragile X mental retardation protein by group I metabotropic glutamate receptors controls translation-dependent epileptogenesis in the hippocampus. J Neurosci. 2011;31:725–734. doi: 10.1523/JNEUROSCI.2915-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]