
Fig. 4.
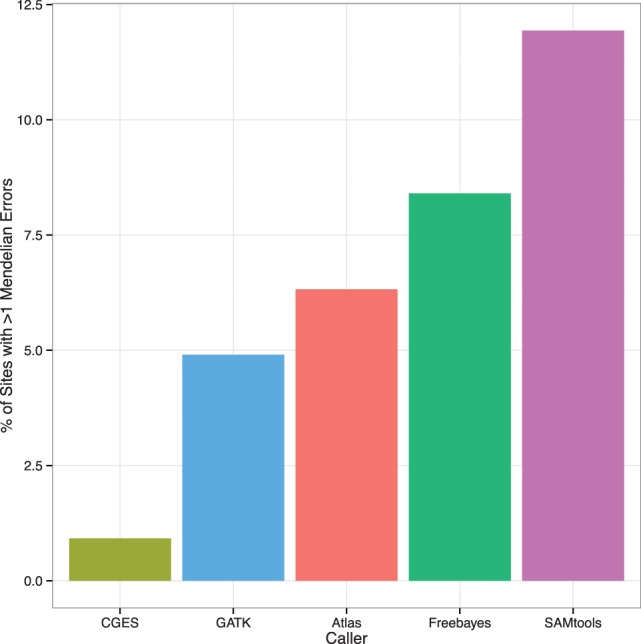
The variant site Mendelian error rate (vMER). The vMER is calculated as the total number of MEs in a VCF file divided by the total number of genotypes with the potential for Mendelian inconsistency (i.e. offspring genotypes with parental genotypes known) in a VCF file. This measure describes the proportion of all offspring genotypes that are inconsistent with parental genotypes present in the VCF