


Abstract
The preparation and high-throughput sequencing of cDNA libraries from samples of small RNA is a powerful tool to quantify known small RNAs (such as microRNAs) and to discover novel RNA species. Interest in identifying the small RNA repertoire present in tissues and in biofluids has grown substantially with the findings that small RNAs can serve as indicators of biological conditions and disease states. Here we describe a novel and straightforward method to clone cDNA libraries from small quantities of input RNA. This method permits the generation of cDNA libraries from sub-picogram quantities of RNA robustly, efficiently and reproducibly. We demonstrate that the method provides a significant improvement in sensitivity compared to previous cloning methods while maintaining reproducible identification of diverse small RNA species. This method should have widespread applications in a variety of contexts, including biomarker discovery from scarce samples of human tissue or body fluids.
INTRODUCTION
MicroRNAs (miRNAs) are small regulatory RNAs, present in all animal cells, that post-transcriptionally regulate protein-coding messenger RNAs (mRNAs) (1). In addition to being expressed inside cells, miRNAs can be detected in stable complexes circulating in human body fluids (2–5). Circulating miRNAs have since been identified in blood serum/plasma, cerebrospinal fluid, saliva and urine (6), and the profile of expressed miRNAs has been found to be altered in disease states. As such, miRNAs offer the exciting potential for use as non-invasive biomarkers for the diagnosis of disease and/or the monitoring of disease progression and treatment (2–5).
Current methods for identification and quantification of circulating miRNAs include microarray and quantitative real-time polymerase chain reaction (qRT-PCR) hybridization-based techniques (7–9), as well as the identification and quantification of both known and novel miRNAs through deep sequencing of small RNA-derived cDNA libraries (10,11). Importantly, unlike hybridization-based approaches, deep sequencing is not limited by assay content and enables single-nucleotide resolution of RNA species revealing the presence of unannotated miRNAs including novel miRNAs and isoforms of known miRNAs (isoMirs) (12).
Conventional approaches to preparing cDNA libraries from small RNAs (sRNAs) rely on T4 RNA ligase-dependent ligations of adapter oligonucleotides to the 3′ and 5′ termini of isolated RNAs. These ligation products are reverse transcribed and PCR amplified to generate a cDNA library suitable for high-throughput sequencing (for examples see (10) and (13)). One limitation of existing approaches is the need to begin with input RNA quantities in the hundreds of picogram (pg) to microgram (μg) range to allow for losses during the various enzymatic reactions and at multiple gel purification steps (as many as four) prior to PCR amplification (14). Recent innovations include employing a single RNA-adapter ligation followed by reverse transcription (RT) and circularization of cDNA. However, these approaches still require micrograms of starting material (11,15). Commercially available cDNA library preparation kits are optimized for input RNA in the microgram range, and moreover can become prohibitively expensive for multiple sample applications, and offer little latitude for customization.
Recent studies identifying small RNAs as biomarkers for disease (reviewed in (16) and (17)) highlight the potential of applying cDNA cloning and deep sequencing in clinical settings. However, low quantities (LQ) of material found in biofluids and other clinically relevant samples pose significant challenges for cDNA library preparation. Recent advances in LQ cDNA cloning include the preparation of miRNA cDNA libraries from 5 ng of total RNA extracted from human blood plasma (14). Single cell RNA-seq methods have been described for surveying messenger RNAs, but not for small noncoding RNAs (18–22). Therefore, for such applications where sequences are unknown, sample is scarce and especially where quantities of input RNA are in the sub-ng and pg range, novel methods are required for the efficient cloning of cDNA libraries from miRNAs and other small noncoding RNAs.
In this paper, we present the development of a highly sensitive LQ cloning method for the generation of cDNA libraries from very small quantities of RNA (pg and sub-pg range) isolated from clinical samples of human blood plasma. The method incorporates several novel components including (i) the reduction of gel purification steps, (ii) seamless transition between ligation and RT using sequential reactions in a single tube and (iii) incorporation of biotinylated nucleotides in the RT reaction to permit efficient purification of cDNA prior to PCR.
MATERIALS AND METHODS
Blood draw and plasma isolation
3 × 10 ml blood was collected in ethylenediaminetetraacetic acid (EDTA) blood collection tubes and spun at 1100 × g for 20 min at 4°C. Plasma from all three tubes was combined into one large cryovial, aliquoted into 1 ml cryovials, and stored at −80°C.
Nucleic acid manipulations
1.7 ml siliconized (low retention) microcentrifuge tubes were used whenever possible to facilitate maximum recovery of material. For all reactions done in a PCR machine, either 0.2 ml strip tubes or 0.2 ml 96-well plates were used depending on sample size and number.
Synthetic miRNA mixtures
Obtained from Life Technologies, referred to as ‘LT-miRmix’ (personal communication and Supplementary Table S2), and Rui Yi lab, referred to as ‘29-miRmix’ (Supplementary Table S3) (23).
Total RNA isolation
For total RNA extraction from samples of human blood plasma, 250 μl plasma aliquots (stored at −80°C) were thawed on ice and cleared by centrifugation at 16,000 rpm for 15 min at 4°C. 200 μl of the supernatant was removed and total RNA was extracted using Trizol, followed by extraction with phenol/chloroform, and ethanol precipitation at −80°C overnight using polyacryl carrier (Molecular Research Center) and 3M KAc. Precipitate was recovered by centrifugation, washed with 200 μl 70% ethanol, resuspended in 10.0 μl RNase free water and stored at −80°C.
Determination of plasma equivalents
‘Plasma equivalents’ refer to the relative amount of plasma used to generate cDNA libraries from RNA isolated from human blood plasma. Using the protocol described above, 10 μl of RNA corresponds to approximately 200 μl of plasma.
Quantification of miRNA in total RNA from human blood plasma
Approximate quantity of miRNA in samples of total RNA isolated from human blood plasma was determined using miR TaqMan real-time qPCR assays (Life Technologies), with reference to a standard curve generated using a known quantity of LT-miRmix. RT was performed using the miR-223 specific stem-loop primer (ID 002295) and the MicroRNA RT Kit (both Life Technologies). Reaction conditions followed manufacturer's instructions: 16°C 30 min, 42°C 30 min, 85°C 5 min. One microliter of the RT reaction was used as template in reactions containing 1× miRNA-specific TaqMan primers/probes in combination with 1× TaqMan GeneExpression Master Mix (Life Technologies) according to the manufacturer's instructions in a total reaction volume of 10.0 μl. Samples were split and run as 3 × 3.0 μl reactions along with a no template sample as a negative control. PCR reaction conditions followed manufacturer's instructions: 50°C 2 min, 95°C 10 min, 40× (95°C 15 s, 60°C 1 min). Assays were run on a 7900HT Fast Real-Time instrument (Life Technologies).
Oligonucleotide substrates, adapters and primers
3′ adapter, 5′ adapters, RT oligonucleotides and PCR primers (Tables 1 and 2) were obtained from Integrated DNA Technologies (IDT). RT oligonucleotides were HPLC or PAGE purified by IDT.
Table 1. 3′ adapter and reverse transcription oligonucleotide sequences.
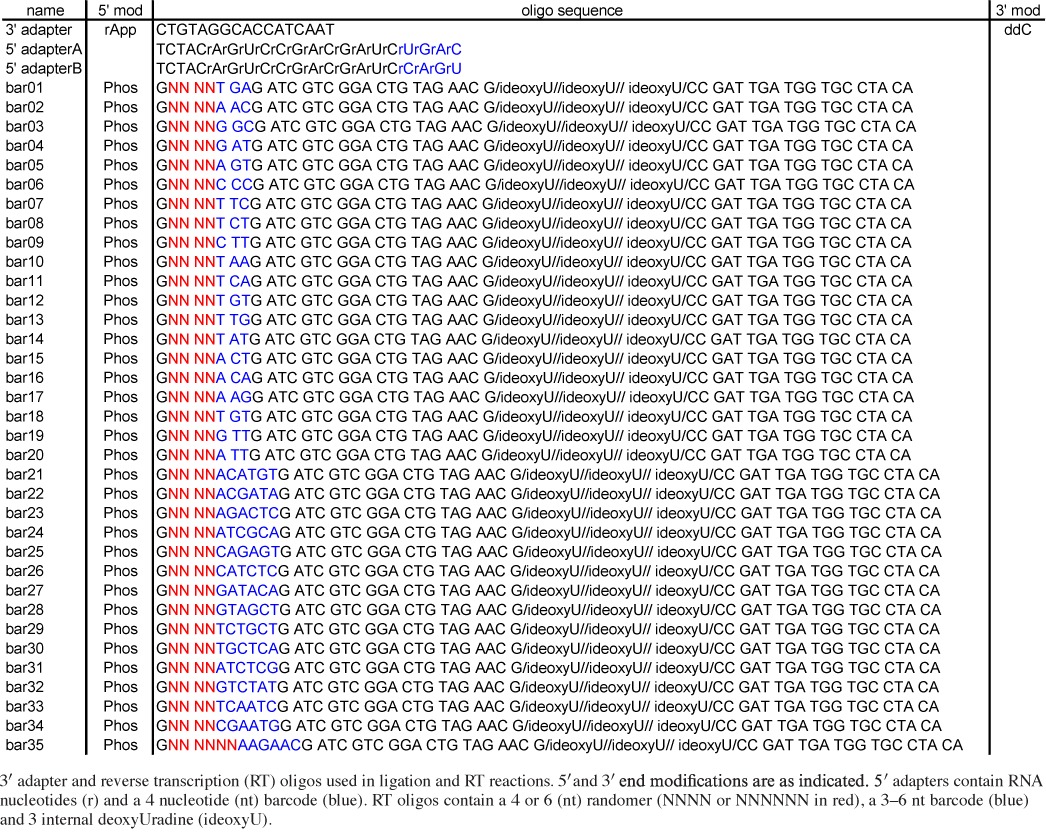
Table 2. PCR oligonucleotide sequences.
| Name | PCR round | End | Sequence | End |
|---|---|---|---|---|
| Ion A short | 1 | 5′ | ATTGATGGTGCCTACAG | 3′ |
| Ion P1 short | 1 | 5′ | GATCTACAGTCCGACGATC | 3′ |
| Ion P1 long | 2 | 5′ | CCATCTCATCCCTGCGTGTCTCCGACTCAGATTGATGGTGCCTACAG | 3′ |
| Ion A long | 2 | 5′ | CCACTACGCCTCCGCTTTCCTCTCTATGGGCAGTCGGTGATCTACAGTCCGACGATC | 3′ |
| DP3 | 1 | 5′ | ATTGATGGTGCCTACAG | 3′ |
| DP5 | 1 | 5′ | GTTCTACAGTCCGACGATC | 3′ |
| Solexa 3 | 2 | 5′ | ACAGCAGAAGACGGCATACGAATTGATGGTGCCTACAG | 3′ |
| Solexa 5 | 2 | 5′ | AATGATACGGCGACCACCGACAGGTTCAGAGTTCTACAGTCCGACGAT | 3′ |
DNA oligonucleotides used in first (1) and second (2) rounds of PCR amplification for sequencing on the Ion Torrent (indicated by prefix ‘Ion’) or Illumina (DP3, DP5, Solexa 3 and Solexa 5) platforms.
Pre-annealing of reverse transcription primer and 3′ linker oligonucleotide
5.0 μl of 20 µM modban 3′ adapter (IDT miRNA cloning linker 1) (1) and 5.0 μl of 20 µM RT oligonucleotide (Table 1) were incubated in 20.0 μl total volume with annealing buffer (10 mM Tris-HCl pH 7.5, 50 mM NaCl, 1.0 mM EDTA). Annealing reactions were performed in a thermocycler with a 105°C heated lid as follows: 95°C 1 min, cool 1°C every minute for 85 min.
1.0 pmol of annealed product (3′ adapter::RT oligo) was analyzed on an 8% non-denaturing polyacrylamide gel (29:1) to confirm annealing.
3′ ligation reactions
5.0 pmol of annealed 3′ adapter::RT oligo product was incubated with various quantities of either a synthetic miRNA mixture or total RNA from human plasma (Table 3). 10.0 μl reactions contained 50 mM Tris-HCl pH 7.5, 10 mM MgCl2, 10 mM DTT, 0.1 µg/μl BSA, 15% DMSO and 1.0 μl T4 Rnl2 truncated K227Q (24). The T4 Rnl2tr K227Q was prepared using Addgene plasmid 14072 as described in (25). Samples were incubated for 6 h at 30°C in a PCR machine with a 105°C heated lid.
Table 3. cDNA libraries.
| Library ID | Synthetic mixture | Concentration of RNA input | Quantity RNA input (pg) | Sequencing platform | Cloning method | R1 PCR cycle no. | R2 PCR cycle no. | Total read number | % Filtered reads | % Reads mapped | % Reference covered | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | LT-miRmix | 1 fmol | 6.89 | Ion Torrent PGM | LQ | 20 | 8 | 363,895 | 3.8 | 82.1 | 99.7 | |
| 2A | LT-miRmix | 50 fmol | 344.48 | Ion Torrent PGM | LQ | 14 | 12 | 438,625 | 0.6 | 85.9 | 98.3 | |
| 2B | LT-miRmix | 500 amol | 3.44 | Ion Torrent PGM | LQ | 16 | 12 | 350,126 | 2.4 | 81.1 | 99.8 | |
| 2C | LT-miRmix | 100 amol | 0.69 | Ion Torrent PGM | LQ | 18 | 12 | 348,732 | 7.3 | 58.8 | 99.1 | |
| 2D | LT-miRmix | 50 amol | 0.34 | Ion Torrent PGM | LQ | 18 | 12 | 274,635 | 7.5 | 61.6 | 98.9 | |
| 3 | LT-miRmix | 50 fmol | 344.48 | Illumina HiSeq2000 | LQ | 18 | 10 | 2,479,439 | 6.7 | 95.7 | 97.9 | |
| 4A | LT-miRmix | 50 fmol | 344.48 | Illumina HiSeq2000 | LQ | 14 | 12 | 2,290,950 | 0.7 | 95.0 | 98.4 | |
| 4B | LT-miRmix | 500 amol | 3.44 | Illumina HiSeq2000 | LQ | 16 | 12 | 1,946,371 | 3.9 | 86.9 | 99.9 | |
| 4C | LT-miRmix | 100 amol | 0.69 | Illumina HiSeq2000 | LQ | 18 | 12 | 1,505,284 | 9.9 | 53.1 | 99.5 | |
| A: synthetic miRNA mixture | 4D | LT-miRmix | 50 amol | 0.34 | Illumina HiSeq2000 | LQ | 18 | 12 | 608,838 | 7.7 | 51.6 | 99.2 |
| 5A | LT-miRmix | 9,100 fmol | 62,699.00 | Illumina HiSeq2000 | 2-Linker | 14 | 8 | 1,718,684 | 95.2 | 90.9 | ||
| 5B | LT-miRmix | 900 fmol | 6,201.00 | Illumina HiSeq2000 | 2-Linker | 14 | 8 | 2,411,058 | 94.3 | 92.3 | ||
| 9A | 29-miRmix | 2,500 fmol | 19,725.00 | Ion Torrent proton | LQ | 12 | 10 | 10,644,245 | 6.9 | 92.6 | 100.0 | |
| 9B | 29-miRmix | 1,250 fmol | 9,862.50 | Ion Torrent proton | LQ | 12 | 10 | 9,693,704 | 5.8 | 93.7 | 100.0 | |
| 9D | 29-miRmix | 50 fmol | 344.50 | Ion Torrent proton | LQ | 14 | 10 | 6,241,100 | 6.9 | 92.5 | 100.0 | |
| 9E | 29-miRmix | 1 fmol | 7.89 | Ion Torrent proton | LQ | 22 | 10 | 6,548,076 | 8.1 | 84.4 | 100.0 | |
| 9F | 29-miRmix | 500 amol | 3.44 | Ion Torrent proton | LQ | 22 | 10 | 1,678,663 | 14.7 | 72.9 | 100.0 | |
| 10A | LT-miRmix | 500 amol | 3.44 | Ion Torrent proton | LQ | 24 | 10 | 5,347,655 | 7.9 | 89.2 | 95.4 | |
| 10B | LT-miRmix | 500 amol | 3.44 | Ion Torrent proton | LQ + heat | 24 | 10 | 2,346,229 | 9.3 | 87.3 | 93.4 | |
| 10C | LT-miRmix | 50 fmol | 344.48 | Ion Torrent proton | LQ | 15 | 12 | 5,787,993 | 3.9 | 89.1 | 98.4 | |
| 10D | LT-miRmix | 50 fmol | 344.48 | Ion Torrent proton | LQ − biotin | 15 | 12 | 3,302,266 | 4.9 | 89.0 | 98.0 | |
| Estimated miRNA concentration | Plasma equivalents | Total no. of miRNAs represented | ||||||||||
| 6A | 100 amol | 10 µl | Ion Torrent PGM | LQ | 20 | 12 | 338,867 | 5.4 | 77.1 | 184 | ||
| 6B | 100 amol | 10 µl | Ion Torrent PGM | LQ | 22 | 12 | 337,375 | 13.0 | 79.5 | 136 | ||
| 6C | 100 amol | 10 µl | Ion Torrent PGM | LQ | 20 | 12 | 313,864 | 8.3 | 76.8 | 196 | ||
| 6D | 500 amol | 51 µl | Ion Torrent PGM | LQ | 20 | 12 | 296,922 | 3.7 | 86.9 | 223 | ||
| B: total RNA from human blood plasma | 7A | 90 amol | 110 µl | Ion Torrent proton | LQ | 20 | 10 | 4,754,997 | 2.2 | 96.4 | 127 | |
| 7B | 125 amol | 110 µl | Ion Torrent proton | LQ | 20 | 10 | 3,562,371 | 2.7 | 95.6 | 137 | ||
| 8A | 100 amol | 10 µl | Ion Torrent PGM | LQ | 20 | 12 | 197,598 | 3.4 | 68.9 | 211 | ||
| 8B | 250 amol | 25 µl | Ion Torrent PGM | LQ | 20 | 12 | 191,701 | 2.7 | 63.3 | 223 | ||
| 8C | 500 amol | 51 µl | Ion Torrent PGM | LQ | 20 | 12 | 533,535 | 2.1 | 85.5 | 221 | ||
| 8D | 750 amol | 77 µl | Ion Torrent PGM | LQ | 20 | 12 | 635,374 | 1.9 | 88.5 | 209 | ||
| 8E | 1000 amol | 102 µl | Ion Torrent PGM | LQ | 20 | 12 | 316,475 | 2.8 | 78.2 | 202 |
Libraries generated from the synthetic miRNA mixtures or total RNA isolated from human blood plasma. Each library is assigned a number designation associated with individual cloning experiments and, where applicable, a letter when samples were multiplexed. These designations are used throughout the text and figure legends to refer to specific libraries analyzed. The LQ and 2-linker methods are distinguished by black and red text, respectively, in subsequent figures.
Synthetic mixture, RNA concentration, RNA quantity, sequencing platform, cloning method used and total read number obtained are as indicated. LQ + heat refers to the library generated with a 65°C heat step prior to the RT reaction. LQ − biotin refers to the library generated using the LQ method in the absence of biotin. RNA quantity for the miRNA mixture was calculated based on molecular weight of 21 nt ssRNA and by using a standard curve and single Taqman assay for miRNA from human blood plasma (see Supplementary Materials and Methods). Total read number includes all reads ≥17 nt and that contained adapter and randomer (NXG) sequences. ‘% filtered reads’ is the percent of total reads removed by hotspot read filtering. The reference used for mapping the synthetic miRNA mixture was the appropriate reference set generated as explained in the Materials and Methods section and was the human genome for libraries made from total RNA from human blood plasma. ‘% reads mapped’ is the percentage of remaining reads mapped to the appropriate reference set. Reads with more than 10 reads per million were used to determine the ‘% reference coverage’ for the miRNA mixture libraries and to determine the ‘total no. of miRNAs represented’ for plasma RNA.
First-strand cDNA synthesis
RT of ligation products was performed directly in the ligation reaction mixture after completion of ligation by addition of the following: 5.0 μl of 4× RT reaction buffer (100 mM Tris-HCl pH 8.0, 300 mM KCl), 5.0 μl of 4× RT master mix (0.25 mM dGTP, 0.25 mM dTTP, 0.1625 mM dCTP, 0.175 mM dATP, 0.0875 mM Biotin-dCTP (Trilink Biotin-16-AA-2′dCTP), 0.075 mM Biotin-dATP (Metkinen Biotin-11-dATP), 20 units RNase inhibitor, 0.5 units Invitrogen Superscript III Reverse Transcriptase, a thermally stable RT enzyme, lacking terminal transferase activity (Life Technologies product literature). Samples were incubated for 5 min at 45°C followed by 5 min at 85°C in a thermocycler with a 105°C heated lid.
For the LQ − biotin Library (10D), cDNA was generated as described above except with 10 mM dNTP containing equal molar concentrations of dGTP, dTTP, dCTP, dATP, and no biotinylated nucleotides.
Removal of template RNA and ethanol precipitation of cDNA reaction products
To hydrolyze RNA in the sample after RT, 1.8 μl 1M NaOH was added to each RT reaction, samples were incubated for 20 min at 98°C in a PCR machine with a 105°C heated lid and were neutralized by adding 1.8 μl 1M HCl (26).
cDNAs were ethanol precipitated by bringing each reaction up to 200 μl with RNase free H2O, transferring to a clean, 1.7 ml siliconized (low-retention) microcentrifuge tube, adding 2.0 μl polyacryl carrier (Molecular Research Center), 40.0 μl 10M CH3COONH4, 3 volumes 100% ethanol, and incubating at room temperature for a period of 1 h − overnight. Following room temperature incubation, precipitate was recovered by centrifugation at 16,000 rpm for 30 min at room temperature in a microcentrifuge. The supernatant was removed and the pellet was washed with 200 μl 80% ethanol and spun for 15 min at 16,000 rpm at room temperature. The supernatant was removed, the pellet was washed with 200 μl 70% ethanol and spun for 15 min at 16,000 rpm at room temperature. The supernatant was removed and the pellet was resuspended in 10.0 μl circular ligation reaction mix (see below).
Circular ligation reactions
Ethanol precipitated cDNA reaction product (see above) was resuspended in 10.0 μl circular ligation reaction mix containing 1.0 μl 10× reaction buffer (0.33M Tris-acetate pH 7.8, 0.66M potassium acetate, 5 mM DTT), 2.5 mM MnCl2, 1M betaine and 1.25 units CircLigase II ssDNA Ligase (Epicentre). Reactions were incubated at 60°C for 2 h.
Gel fractionation, elution and isolation of biotinylated cDNA
Circularized cDNAs and circular single-stranded DNA molecular weight markers were fractionated on separate lanes of a 10% polyacrylamide/7M urea gel. The gel was stained with SYBR Gold (Life Technologies) according to manufacturer's instructions, visualized on a blue light transilluminator and circularized cDNAs migrating in the range of 80 –100 nt were excised. Each gel slice was added to a 1.7 ml siliconized (low-retention) microcentrifuge tube containing 400 μl TE + 0.3M NaCl. 5.0 μl magnetic hydrophilic streptavidin beads (New England Biolabs) were washed three times with 50 μl buffer WB (0.5M NaCl, 20 mM Tris-HCl pH 7.5, 1.0 mM EDTA), resuspended in 5.0 μl TE + 0.3M NaCl and added to each sample. Tubes were shaken overnight at 1,100 rpm at room temperature. After overnight elution, the bead-containing supernatant was transferred to a clean, low-retention 1.7 ml eppendorf tube, tubes were magnetized on a magnetic rack (Life Technologies), supernatants carefully removed, beads washed three times with 1.0 ml buffer WB (magnetizing and carefully removing supernatant between each wash step) and resuspended in 10.0 μl RNase free H2O.
For the LQ − biotin library (10D), gel fractionation and elution was done as above except magnetic streptavidin beads were excluded. After overnight shaking, buffer containing eluted material was removed, added to a 0.45 μm spin column and spun 3 min at 3,000 rpm at room temperature into a clean, 1.7 ml siliconized (low-retention) microcentrifuge tube. Circularized cDNAs were ethanol precipitated by adding 2.0 μl polyacryl carrier (Molecular Research Center), 1/10 volume 3M KAc pH 5.0, 3 volumes 100% ethanol, and incubating at -80°C for 1 h. Following -80°C incubation, precipitate was recovered by centrifugation at 16,000 rpm for 20 min at 4°C in a microcentrifuge. The supernatant was removed, the pellet was washed with 200 μl 80% ethanol and spun for 15 min at 16,000 rpm at 4°C. The supernatant was removed, the pellet was washed with 200 μl 70% ethanol and spun for 15 min at 16,000 rpm at 4°C. The supernatant was removed and the pellet was resuspended in 10.0 μl RNase free H2O.
First round PCR library amplification of cDNA library
PCR amplification was performed using 2× TaqMan Gene Expression Master Mix (Life Technologies) using 50% of the bead suspension from above in a 50.0 μl reaction with a final concentration of 0.1 μM forward and 0.1 μM reverse Round 1 PCR primers (Table 2) for up to 25 cycles removing 10.0 μl every 2 cycles starting at 12 cycles. PCR reactions conditions were: 55°C 2 min, 95°C 10 min, 12–25× (95°C s, 55°C 1 min), 10°C hold. Samples were run on an 8% non-denaturing polyacrylamide gel (29:1). The gel was stained with SYBR Gold (Life Technologies) per manufacturer's instructions and visualized on a blue light transilluminator. Appropriate 65 –70 nucleotide PCR products (Supplemental Table S1) were excised at the cycle number when first visible, gel slices cut in half vertically, and stored at -20°C.
Second round PCR amplification of cDNA library
PCR amplification was performed using 2× AmpliTaq Gold Fast PCR master mix (Life Technologies) using one-half of the first round PCR product gel slice as template in a 100 μl reaction with a final concentration of 0.125 μM forward and 0.125 μM reverse Round 2 PCR primers (Table 2) and 8 –12 cycles. Care should be taken to isolate PCR products at low cycle number, before primers are depleted and bulged PCR products appear. (Such bulged products result from denatured products that anneal at the adaptors but not at the center, and appear as a smear migrating slower than the desired PCR product.) An initial 20–30 μl PCR reaction, removing samples at 8, 10 and 12 cycles and resolving on an 8% non-denaturing polyacrylamide gel (29:1), can be performed to determine optimal cycle number. PCR reactions conditions were: 95°C 10 min, 8 –12× (95°C 15 s, 60°C 1 min), 10°C hold.
Purification and quantification of second round PCR product
Appropriate 116–138 nucleotide second round PCR products (Supplementary Table S1) were purified using BioBasic Inc. EZ-10 Spin Column PCR Purification Kit following manufacturer's recommendations. Samples were eluted in 30 μl kit-provided elution buffer and quantified using an 8% non-denaturing polyacrylamide gel (29:1) and a known DNA standard (i.e. New England Biolabs Low Molecular Weight DNA ladder) as well as on a high-sensitivity DNA BioAnalyzer chip (Agilent Technologies).
2-Linker cDNA library preparation
Libraries were generated as described in Gu et al. (13) with minor modifications. The 3′ ligation was done as described above and 5′ adapters contained the 3′ barcode as described in Table 1.
Sample preparation and high-throughput sequencing
PCR products were sequenced on the Ion Torrent PGM, Ion Torrent Proton or Illumina HiSeq2000 instrument according to manufacturer's protocols.
Computational pipeline
FastQ file formats contain the following data formats:
Ion Torrent sequences: (3′) adapter – sequence read – GNX – barcode – adapter (5′)
Illumina sequences: (5′) barcode – NXC – sequence read – adapter (3′)
For each library the following steps were applied:
- Adapters were removed using the Cutadapt method (version 1.2.1) (27): using the -e 0.25 option and the following adapter sequences:
- For Ion Torrent sequences: -g ATTGATGGTGCCTACAG and -a GATCGTTCGGACTGTAGATC
- For or Illumina sequences: -a CTGTAGGCACCATCAAT
Sequences were split into libraries according to barcode.
Randomer sequences (NX) were removed and saved in the header line of the fasta file for later PCR hotspot analysis.
Reads <17 nt were filtered and removed.
For Ion Torrent sequences, reads were reverse complemented.
Reads with identical sequences were combined and the combined count was saved.
The count of each read was corrected by the ‘PCR hotspot’ correction procedure described below.
- Plasma and mixture libraries were further processed as follows:
- For plasma libraries:
- Reads were aligned to a reference sequence (see below) using bowtie (28)
- -v 3 -f -B 1 -a --best --strata
- Alignments were then filtered based on the length of the read and the number of mismatches as follows: for sequence lengths 17, 18–19, 20–24 or >24: 0, 1, 2 or 3 mismatches were allowed, respectively.
- For miRNA mixture libraries:
- Reads were aligned to reference sequences (see below) and (2) the reference sequences were aligned to the reads using bowtie (28) -v 3 -f -B 1 -a --best --strata
A reference set for the Life Technologies synthetic mixture (LT-miRmix) libraries was constructed as follows:
All sequences that were present in mixture libraries 1-5B (Table 3) were enumerated.
All sequences with more than 10 reads in ≥7 libraries were identified. In addition, sequences with more than 50 reads in ≥1 library that were also annotated in the miRNA mixture (Life Technologies, personal communication) were collected. Sequences were combined, resulting in a total of 2,299 sequences.
Highly overlapping sequences with some 5′ and 3′ overhangs were clustered based on a 17 nt seed sequence, allowing up to two mismatches.
Each cluster was represented by a sequence spanning all sequences within the cluster, resulting in 1,047 sequences.
These final 1,047 sequences served as the miRNA mixture reference set (Supplementary Table S2).
Defining a sub-reference set of the Life Technologies synthetic mixture (denoted as LT-miRmix subset):
To perform an unbiased analysis of 5′ terminal additions in the LQ method, which could be affected by the natural heterogeneity present in the synthetic miRNA mixture, we identified a set of 154 sequences with a fixed starting point (exhibiting no heterogeneity in the 5′ end) for all the reads. These sequences served as the reference set for analysis of 5′ nt additions, 3′ end variability and biotin-introduced mismatch rate.
The reference set for the equimolar 29-miRNA synthetic mixture was as described in Zhang et al. (23) and listed in Supplementary Table S3.
Reference sets for the human blood plasma libraries were:
The human genome sequence, hg19 downloaded from UCSC (29).
Analysis of mapping results
An in-house developed code was used to analyze the results as follows:
For the synthetic miRNA mixture libraries the mapping results of (i) reads to reference sequences and (ii) reference sequences to the reads were combined. (The latter is needed in order to take into account potential 5′ or 3′ terminal additions.) A read count for each reference sequence was assigned by taking into account all reads associated with it and keeping track of the relative mapping alignment of each read for further analysis.
For plasma libraries the results of (i) mapping to the human genome sequence and (ii) mapping to small RNA sequences were combined. Reads that mapped to the genome with less mismatches than to a small RNA were assigned as non-small RNA human genome matches. To assign read counts to the miRNA sequences we considered all reads that mapped to a pre-miRNA sequence within -5 to +5 nucleotides of the annotated mature miRNA start according to miRBase. For all the other small RNA species, we included all reads that map to a small RNA reference sequence regardless of the mapping position.
For all comparisons done between libraries, a normalized read count (i.e. reads per million of aligned reads) was used.
Analyzing miRmix subset sequences
To generate the averages presented in the G/U mutation profile (Figure 5D), 5′ terminal additions profile (Figure 6A) and read length profile (Figure 6B), the synthetic miRNA sequence reference sets were used as follows:
Figure 5.
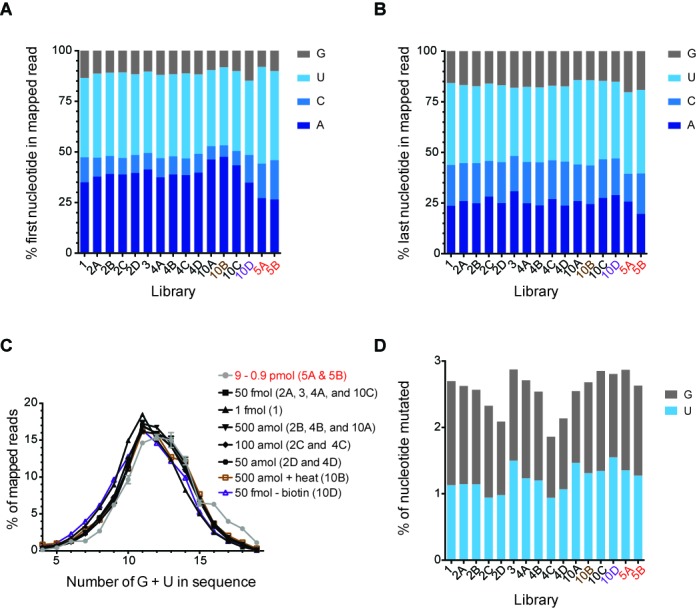
Evaluation of LQ cloning method accuracy. Histograms representing the profile of the nucleotide at the 5′ end (A) and 3′ end (B) of reads mapped to the LT-miRmix in each library using the LQ method (black text) and 2-linker method (red text). LQ library made with heat prior to the RT is shown in brown text and LQ library made without biotin is shown in purple text. (C) The frequency of reads cloned versus combined total G/U content of reads using the LQ method (black text) and the 2-linker method (red text, gray line). Library designation indicated in (). Where multiple libraries are indicated, the distribution is shown as the average of these libraries. (D) Histogram representing the percentage of Guanine (G) and Uracil (U) mutated in reads mapped to the LT-miRmix subset reference in libraries generated using either the LQ method (black text) or 2-linker method (red text).
Figure 6.
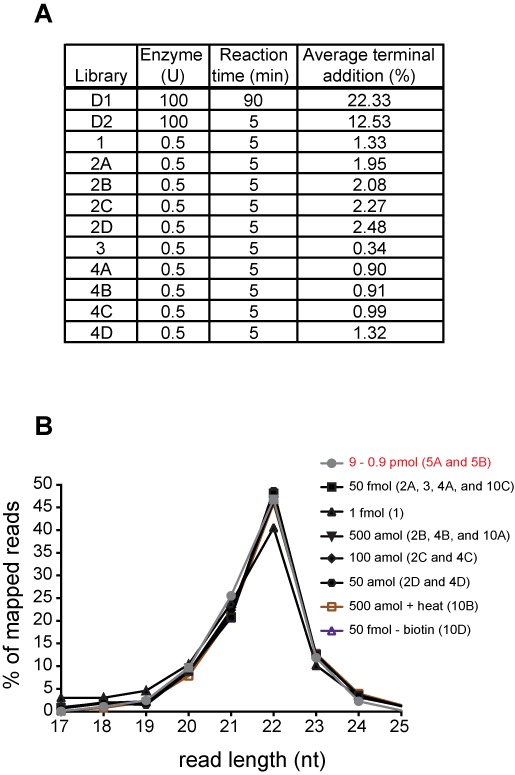
Examination of 5′ and 3′ end variation in miRNA mixture cloned sequences from the LT-miRmix. (A) Table demonstrating reverse transcription reaction conditions and corresponding average percentage of 5′ terminal additions computed on LT-miRmix subset reference (see Materials and Methods section). Libraries D1 and D2 (Development 1 and Development 2, respectively) represent libraries examined early in method development with varied RT reaction conditions as indicated. (B) The frequency of small RNA read length for those miRNAs with a fixed 5′ end indicating 3′ end variation across examined libraries. Library designation indicated in (). Where multiple libraries are indicated, the distribution is shown as the average of these libraries.
For each sequence the following were calculated:
The percent of mismatches among the mapped reads (for G or U nucleotides separately).
The percent of mapped reads that had a 5′ overhang (of up to 5 nucleotides) beyond the fixed starting point of the sequence.
The distribution of read length among the mapped reads.
The averages across all the sequences for each of the measures in points 1–3 are displayed in the figures mentioned above.
PCR hotspot correction
For each sequence in a given library, we stored the randomer sequence (NX) associated with it (Step 3 of the computational pipeline). These randomers were used for:
- Assessing the distribution of randomers from the sequencing data by:
- Calculating the distribution of randomers associated with each sequence.
- Identifying a set of sequences with random distribution of randomer sequences by:
- choosing a set of sequences that has more than 4X reads and
- identifying sequences within (a) where no randomer is represented by >5% of the reads.
- Determining the expected distribution of randomers by calculating the average percentage and the standard deviation for each randomer, based on the sequences found in A2. We denote the expected percentage for a randomer i as pi
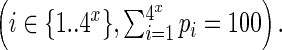
- Identification of PCR hotspots for each combined identical sequence in the library was done by:
- Identifying randomers with an observed percentage higher than the expected average plus three standard deviations.
- Let i be the randomer identified in B1, with an expected percentage pi (calculated in A3) and an observed count ci. Let n be the total read count observed for the current sequence. The new total count n′ can be corrected to
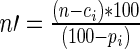 . The observed count for randomer i is corrected to
. The observed count for randomer i is corrected to 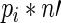 .
. - Correcting the observed distribution of randomers for all the other randomers based on their ci and n′.
- Repeating steps B2 and B3 until no corrections are needed or until the total sequence count equals the number of randomers identified for that sequence.
Note: If, for a given sequence, only a subset of randomers is observed, then the expected distribution of these randomers is scaled so that their total summation will be 100, and 0 is assigned in the pi of the other randomers. This step is needed in order to avoid collapsing of reads with underrepresentation of randomers (due to initial low abundance of the read(s)).
RESULTS
Key features of the method
Overview
Our aim was to establish a method for preparing cDNA libraries for high-throughput sequencing from a very low quantity (LQ) of input RNA. Accordingly, we developed a relatively straightforward and streamlined cloning protocol that minimizes sample loss by reducing the number of sample extraction and gel purification steps compared to conventional cloning protocols while enabling cloning from significantly less material compared to commercially available streamlined methods. We also incorporated provisions for sample multiplexing and future development of high-throughput applications. Our protocol involves sequential linker ligation and RT reactions in a single tube (Figure 1A), where a single adapter is ligated to the 3′ end of the RNA, followed by generation of biotin-containing reverse transcribed cDNAs. cDNAs are then circularized, the biotin-containing cDNAs are isolated (Figure 1B) and libraries are amplified by PCR (Figure 1C). This streamlined method allows for preparation of libraries ready for quantification and sequencing in 2–3 days (Figure 1). The steps of the protocol are diagrammed in Figure 2 and described in detail in the Materials and Methods section. Specific features of the protocol that contribute to its enhanced sensitivity and simplicity are presented below.
Figure 1.
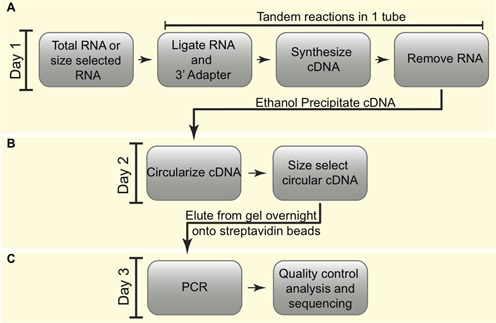
Overview of 3-day generation of cDNA libraries. (A) On the first day, total RNA is ligated to a 3′ adapter and cDNA is generated by reverse transcription by tandem reactions in a single tube, RNA is degraded and cDNAs are isolated by ethanol precipitation. (B) On the second day, cDNAs are circularized, size selected by gel fractionation and eluted overnight in the presence of streptavidin beads. (C) PCR is done on bead-bound purified cDNAs to generate templates ready for high-throughput sequencing.
Figure 2.
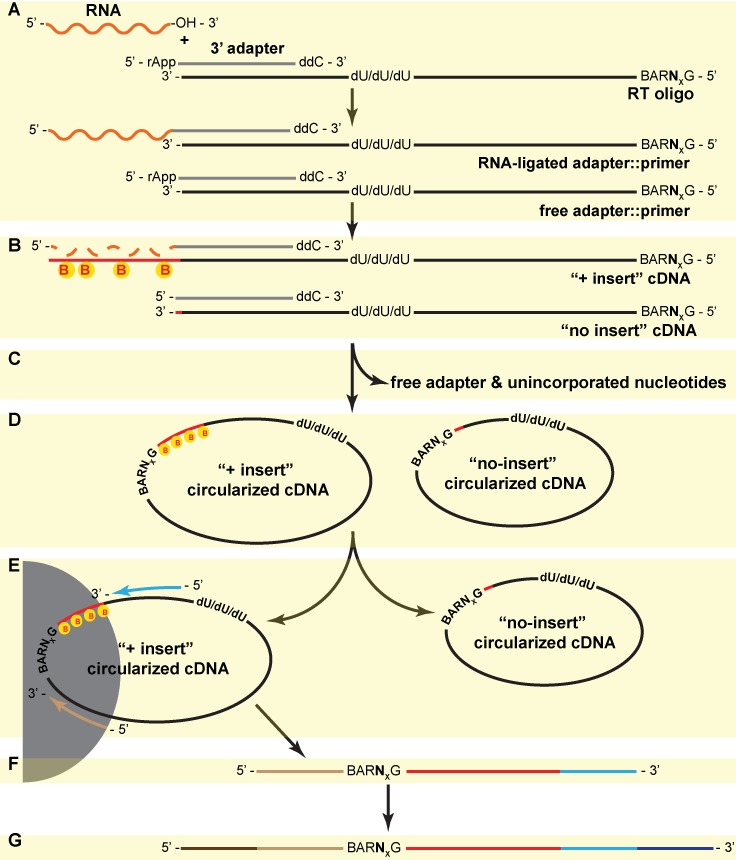
Detailed LQ cloning method. (A) A pre-adenylated (rApp) 3′-terminal dideoxy-C (ddC) blocked adapter (gray) is annealed to a ssDNA reverse transcription (RT) oligo (black) in a 1:1 molar ratio. The annealed adapter is ligated to 3′-hydroxyl-containing RNA (orange) using T4 RNA Ligase 2 (truncated K227Q) without ATP. Each RT oligo contains a 5′ Guanine (G) followed by a 4 or 6 nucleotide randomer (NX), a 3–6 nucleotide barcode (BAR) and 3 internal deoxyUridine (dU) nucleotides. The adapter::RT oligo hybrid is in excess over RNA, resulting in free adapter::primer material present in the completed reaction. (B) Reverse transcription of ligated RNA is carried out in the same tube as the ligation reaction generating ‘+ insert’ and ‘no insert’ cDNA products (red and black line) using dGTP, dTTP, dATP, dCTP as well as biotinylated dATP and dCTP (yellow ‘B’-containing circles). The RNA template is degraded (dashed orange line) by base hydrolysis and cDNA is ethanol precipitated with ammonium acetate to facilitate maximum removal of free adapter and unincorporated nucleotides (C). Ethanol precipitated cDNAs are circularized (D) and resolved on a 10% denaturing polyacrylamide gel. ‘+ insert’ circularized cDNAs are isolated by excising and eluting them from the gel overnight in the presence of magnetic streptavidin beads (E). Bead-bound ‘+ insert’ cDNAs serve as templates in the first round of PCR. Amplification is done using a mix containing uracil-N-deglycosylase (UNG) to remove dU nucleotides, thereby generating a linear template through strand scission, and with primers complimentary to the 3′ adapter (blue) and 5′ end of the RT oligo (tan) (F). First round PCR products are resolved on an 8% native polyacrylamide gel, the 60–70 nucleotide products are excised and a portion is used as the template for second round PCR. Second round PCR products are generated using primers complimentary to the 3′ adapter (dark blue) and 5′ end of the RT oligo (brown) that contain the full Illumina or Ion Torrent adapter sequences (dark blue and brown) (G).
Single-tube, sequential ligation and reverse transcription reactions
Ligation reactions are performed with truncated and mutated T4 RNA Ligase 2 (T4Rnl2tr K227Q) at 30°C for 6 h. T4Rnl2tr K227Q carries out a more specific and efficient ligation between RNA and 3′ adapter compared to wild-type T4Rnl2 and the 30°C ligation reaction temperature reduces sequence biases that may be introduced by RNA secondary structure such as stem-loops (33,34).
To promote efficient first-strand cDNA synthesis, the RT primer oligonucleotide is pre-annealed to the 3′ adapter (adapter::RT oligo; Figure 2A) prior to the ligation reaction. This provides for maintaining equimolar stoichiometry of the adapter and RT oligo throughout the ligation and, importantly, pairs ligated RNA with a barcoded RT oligo. Moreover, since no extraction or precipitation steps occur after ligation, the pre-annealed RT primer serves as a substrate for RT of ligated RNA in sequential single-tube ligation and RT reactions. Accordingly, immediately following ligation, the thermostable reverse transcriptase SuperScript III (Invitrogen), RT buffer and reaction components are added directly to the ligation mixture. This direct transition from the ligation to the RT reaction avoids a gel purification step necessary in some other methods and hence reduces sample losses. Finally, by generating cDNAs at 45°C, efficient extension is achieved while maintaining annealing of the 3′ adapter::RT oligo hybrid. Importantly, removal of a denaturing step prior to the RT reaction did not inhibit our ability to isolate sequences with high GC content (Supplementary Figure S1) and libraries generated with (10B) or without (10A) a 65°C heat step for 5 min prior to the RT correlated very well (Figure 8).
Figure 8.
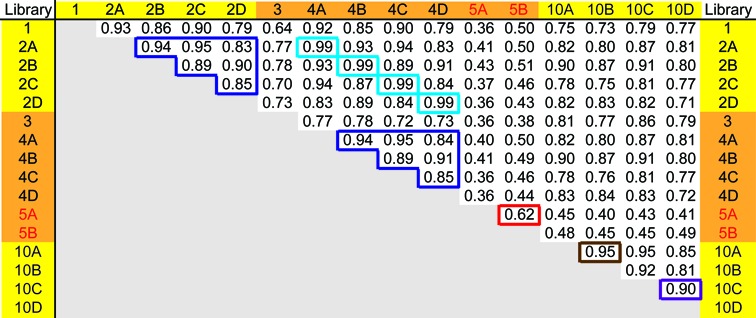
Comparison between cDNA libraries generated from the synthetic miRNA mixture. Correlation coefficients (R) were computed using read counts associated with each sequence in the LT-miRmix reference set. Libraries were sequenced on the Ion Torrent (yellow) or Illumina (orange) platform. Dark blue boxes highlight correlations between libraries generated from decreasing amount of RNA and sequenced on either the Ion Torrent or Illumina platform. Light blue boxes highlight correlations between libraries made from identical input material and sequenced on both the Ion Torrent and Illumina sequencing platforms. Brown box highlights correlations between libraries made from identical input material with (10A) or without heat (10B) prior to the RT. Purple box highlights correlations between libraries made from identical input material with (10C) or without (10D) Biotin incorporation. Correlations between libraries made with 9.1 or 0.9 pmol of input using the 2-linker cloning method (red text) are highlighted in the red box.
Novel features introduced into cDNAs
Notable features of the RT oligo include a 5′ terminal Guanine (G) to minimize nucleotide bias inherent to the circular ligase, a barcode to enable sample multiplexing, a randomer to identify PCR hotspots (see Materials and Methods) and internal deoxyuracil (dUTP) nucleotides to enable linearization of circular cDNAs by Uracil-D-glycosylase (UDG, aka Uracil-N glycosylase, UNG) in the first round of PCR (Table 1 and Figure 2). Finally, to promote efficient recovery of purified cDNA (Figure 2A), biotinylated dCTP and biotinylated dATP are included in the cDNA reaction (Figure 2B and Materials and Methods section). Importantly, biotinylated nucleotides are required for recovery of cDNAs resulting from successful ligation reactions. In the absence of biotin, only cDNAs reflective of RT oligos are recovered as evidenced by short (44–49 nt) first round PCR products (Supplementary Figure S2).
Circularization and isolation of biotinylated cDNA
Following first-strand cDNA synthesis, RNA is removed by base hydrolysis and cDNAs are ethanol precipitated with 10M ammonium acetate at room temperature, facilitating removal of free 3′ adapter and unincorporated nucleotides (35,36) (Figure 2C). All recovered material is circularized using CircLigase II (Epicenter) (Figure 2D) and fractionated on a 10% denaturing gel to obtain circular single-stranded DNA of the desired length: 69–74 nucleotides for cloned small RNAs (Supplementary Table S1). This is the only gel purification step in the protocol prior to sample amplification, hence this step allows for size selection of circularized ‘+ insert’ products and also serves as a first step in separation of ‘+ insert’ material away from ‘no insert’ material. Excised ‘+ insert’ cDNAs are eluted from the gel overnight in the presence of streptavidin beads, allowing for selective binding and hence a full isolation of biotin containing ‘+ insert’ cDNAs away from remaining ‘no insert’ (non-biotinylated) material. Beads are washed and the streptavidin bead-bound ‘+ insert’ cDNAs are amplified directly from the beads in the first round PCR.
Utilization of deoxyuracil in PCR amplification of libraries
In the first round of PCR, primers complementary to sequences flanking the cloned sequence (Table 2, Figure 2E and F) and Taqman Gene Expression Master Mix (Life Technologies) are added directly to streptavidin bead-bound ‘+ insert’ material. UNG in the master mix enables excision of dUTP from template molecules, promoting strand scission and generation of a linear PCR template. The master mix also contains a blend of dTTP/dUTP nucleotides: incorporation of dUTP into new amplicons serves to minimize carry-over PCR contamination between first round products. First round PCR products are resolved on an 8% non-denaturing polyacrylamide gel and appropriate 65–70 nt products are excised. Primers complementary to the 5′ and 3′ sequences of first round PCR products that include adapters specific for sequencing on either the Ion Torrent or Illumina platform (Figure 2G and Table 2) are then used in the second round of PCR for final library amplification prior to sequencing.
Computational analysis of cDNA libraries
In order to identify cloned sequences, we use an in-house computational pipeline (Figure 3 and Materials and Methods section). In this pipeline, first, 5′ and 3′ adapter sequences (Ion Torrent-derived libraries) or 3′ adapter sequences (Illumina-derived libraries) are removed. Second, by using the unique 3–6 nucleotide (nt) barcode sequence incorporated into each library, multiplexed samples are split by barcode. Next, the randomer sequences are removed and saved for later PCR hotspot analysis. Reads are then filtered for length. All sequences shorter than 17 nt are discarded, as this limited read length cannot be mapped with high confidence. All remaining ≥17 nt reads having an identical sequence are combined and counted. Identical reads are checked for PCR hotspots by comparing the distribution of the randomers associated with each sequence to the expected distribution of randomers (determined from the sequencing data; see Materials and Methods section) and filtered accordingly. Remaining reads are aligned to the reference sequences using the Bowtie alignment tool (28) followed by a final filtering of reads based on length and mismatch cut-offs as described in Materials and Methods section.
Figure 3.
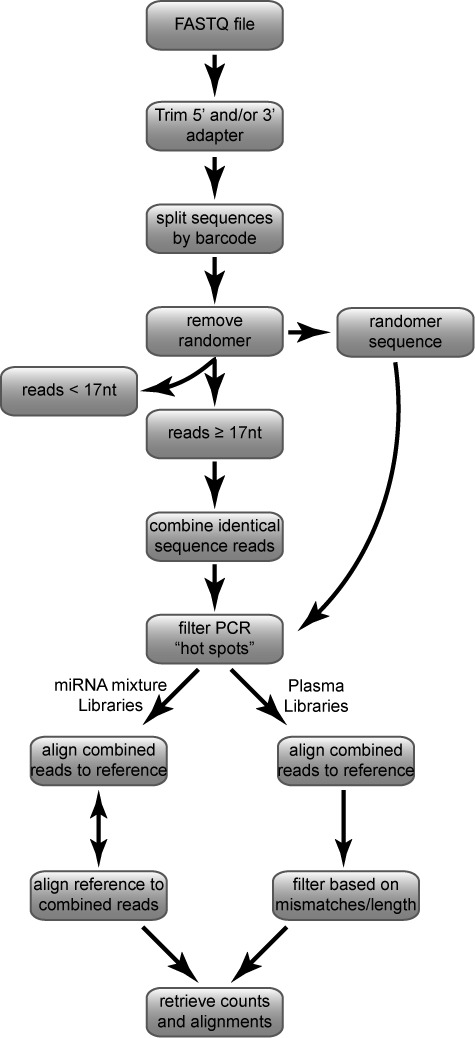
Computational pipeline for analysis of deep sequencing libraries. See Materials and Methods section for detailed explanation.
Method evaluation and application
An ideal method for generating cDNA libraries from small RNAs should be sensitive, reproducible over a wide range of input RNA quantity and should accurately represent the sequence profile of the input RNA. Therefore, we performed rigorous assessments of these criteria by first creating libraries from the Life Technologies synthetic miRNA mixture (LT-miRmix) using both our LQ method and the established 2-linker cloning method (13). While the LT-miRmix is complex and better represents sequences that may be encountered in biological samples, limited sequence annotation and unknown molarity for the contents of the LT-miRmix led us to further examine the LQ method using a well-characterized miRNA mixture (29-miRmix) comprised of 29 synthetic miRNAs with known stoichiometry and sequence context (23). We also assessed the potential applicability of our method in a clinical setting by cloning cDNA libraries from small quantities of total RNA isolated from human blood plasma.
Libraries generated using the synthetic miRNA mixtures were used to assess the accuracy of the method in recovering sequences in a standardized set of RNA. Sequences obtained from cDNA libraries generated from the mixtures were mapped to the LT-miRNA mixture reference set of sequences (Materials and Methods section and Supplementary Table S2) or the set of 29-miRNAs (Supplementary Table S3). The accuracy of our method was then assessed in terms of read length distribution, 5′ end identity and heterogeneity (particularly from potential terminal transferase activity of the RT enzyme), 3′ end identity and heterogeneity, and potential sequence bias or mutagenesis introduced through the use of biotinylated nucleotides. Central to this assessment was a comparison of libraries generated using the LQ method with and without biotin as well as to those made with the biotin-free 2-linker approach.
cDNA library sequence overview
We generated libraries from two synthetic miRNA mixtures, one containing approximately 1,000 miRNA sequences provided by Life Technologies (personal communication and Supplementary Table S2) and a second containing 29 miRNAs (Supplementary Table S3) (23), as well as total RNA isolated from normal human blood plasma. The average number of reads obtained from libraries sequenced on the Ion Torrent and Illumina platforms ranged from 190,000 to >1,000,000 depending on the number of samples multiplexed and the platform used (Table 3).
As expected, we found that as RNA input quantity decreased, the number of PCR cycles required to yield detectable PCR product increased (Table 3). In general, increasing the number of PCR cycles affected the percentage of hotspots detected in our libraries, as evidenced by clusters of identical reads containing an identical randomer sequence. We found that our libraries that required relatively more PCR amplification steps could contain up to 14% of total reads associated with hotspot amplification (Table 3). These results emphasize the importance of including randomer sequence tags in the cloning oligo backbone for the identification and compression of hotspot reads. We also examined the impact of using a 6 versus a 4 nt randomer. Interestingly, we did not see any particular advantage to using a longer randomer (Supplementary Figure S3).
After compression of hotspot sequence reads, remaining reads were mapped to the reference sequence set corresponding to the source RNA: the synthetic mixture reference set for LT-miRNAmix libraries, the 29 miRNA sequences for 29-miRmix libraries, or the small RNA reference and the human genome for plasma-derived libraries (Materials and Methods section). We observed the percentage of sequences that mapped to the respective reference sequences varied from 51% to 96%, somewhat dependent upon the quantity of input RNA. For example, in libraries generated from the synthetic miRNA mixtures, greater quantities of input RNA tended to result in libraries with higher percent mapping to the reference. However, for libraries generated from lower amounts of input RNA, the reference sequence coverage was still substantial (at least 93%) (Table 3).
For libraries generated from human blood plasma RNA, we identified sequences corresponding to a broad repertoire of small RNAs matching the human genome. The majority of sequences mapped to human miRNAs, while the remainder mapped to ribosomal RNA (rRNA), transfer RNA (tRNA) fragments, other circulating RNAs such as Y RNA (37), and also sequences annotated as Piwi-interacting RNAs (piRNAs) (38,39). Future study is required to assess the potential biological relevance of these various circulating RNA populations.
For libraries made from synthetic RNA input or from plasma RNA input, a fraction of reads (from 49% down to 4%) did not map to the respective reference sequence set. Prominent among these non-mapping sequences were apparent plasmid vector sequences (data not shown), indicating that small amounts of laboratory nucleic acid contamination from unrelated experiments can enter the workflow.
Read length distribution
The expected length distribution of cDNA sequences obtained by the LQ method should depend upon the length of input material and on the size of circular ligated material excised from the gel. In this analysis, we sought to clone small RNAs in the range of 18–24 nt from either synthetic miRNA mixtures or from total RNA isolated from human blood plasma. We found the length distribution of RNAs cloned from synthetic mixtures using the LQ method was consistent regardless of input RNA concentration, the addition to heat prior to the RT step, or the absence of biotin. Interestingly, the cDNA length distribution of the LQ method included a 17–20 nt fraction that was significantly reduced in the length distribution isolated from the 2-linker method (Figure 4A), perhaps owing to the additional size-specific gel purification step included in the 2-linker method.
Figure 4.
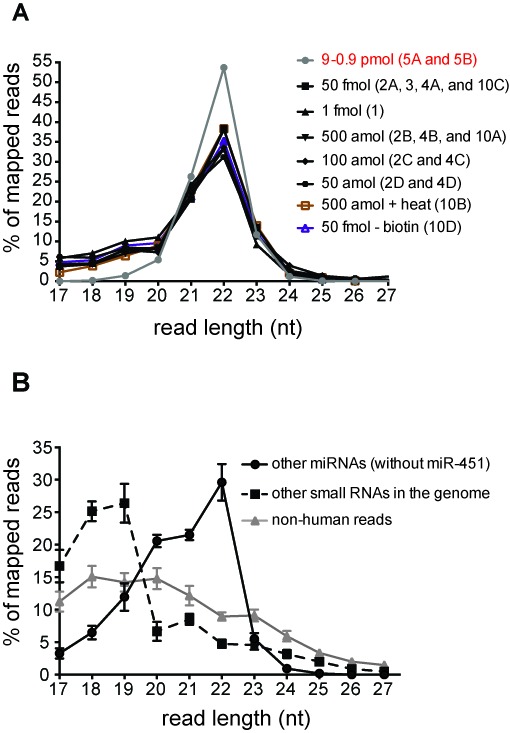
LQ and 2-linker cloning methods isolate small RNAs from a synthetic miRNA mixture and total RNA from human blood plasma. (A) The frequency of small RNA read lengths obtained from libraries generated with decreasing amounts of the synthetic miRNA mixture using the LQ method (black text) and the 2-linker method (gray line, red text). Library designation indicated in (). Where multiple libraries are indicated, the distribution is shown as the average of these libraries. (B) The frequency of small RNA read lengths in libraries generated using total RNA isolated from human blood plasma is represented as the average across all plasma libraries with error bars indicating the standard deviation.
To assess length distribution of RNAs cloned from total plasma RNA, we divided reads into three groups: (i) human miRNAs (excluding the highly abundant miR-451a; see below for explanation), (ii) other small RNAs mapping to the human genome and (iii) reads that did not match the human genome. For miRNAs, the majority of reads (72%) range in length from 20 to 22 nt. Similar to what we observed with the miRNA mixtures, shorter reads (22%) include small RNA fragments that map to miRNAs and reflect mainly truncation at the 3′ ends (data not shown), and the remaining reads are sequences longer than 22 nt. Moreover, the size distribution for these sequences, as well as other human and non-human small RNAs, falls within the size range expected from the size selection step of circularized cDNA in our protocol (Figures 2 and 4B; Materials and Methods section).
5′ and 3′ end nucleotide bias
In order to asses potential nucleotide-generated bias in either the ligation of RNA to the 3′ adapter or in the circular ligation of generated cDNAs, we examined the 5′ and 3′ nucleotides of sequences cloned from the sequence-diverse LT-miRNA mixture. Overall, we observed a relative increase in 5′ Adenine (A) and a decrease in 5′ Cytosine (C) in the LQ method versus the 2-linker method (Figure 5A). This bias may be attributed to the use of different enzymes in the 5′ end ligation reactions. In the LQ method, CircLigase II is used to ligate the 3′ end of the cDNA (corresponding to the 5′ nt of the RNA) to the 5′ end of the RT primer, whereas the 2-linker method utilizes T4 RNA ligase I to ligate the 5′ end of RNA to a 5′ end adapter. T4 RNA ligase I exhibits sequence dependent ligation preferences (33) that very likely differ from those of CircLigase II. Interestingly, the CircLigase II enzyme exhibits a preference for ligation of 3′ Thymine (T) to 5′ Guanine (G) (Epicentre personal communication). Our LQ RT oligo contains a 5′ G, and so this intrinsic T-to-G ligation bias for CircLigase II could explain our observed enrichment in LQ libraries for cDNA sequences corresponding to RNAs containing a 5′ A (Figure 5A). Additionally, the 2-linker method employs 5′ end adapters with different nucleotides at the 3′ end (Table 1). Observed sequence bias among the two libraries generated by the 2-linker method is likely due to the sequence variation at the 3′ end (33,40,41). In contrast, the 3′ end ligations in both methods utilize the same enzyme, truncated and mutated T4 RNA ligase 2 (T4 Rnl2tr K227Q). Therefore, no 3′ nucleotide (3′ nt) bias was expected in libraries generated from the two different methods, and indeed no significant difference was observed (Figure 5B).
Affect of biotin incorporation on sequence accuracy
To our knowledge, this is the first use of biotinylated nucleotide incorporation into cDNAs generated from linker-ligated RNA. Moreover, at the outset, it was not clear how competent SuperScript III (Invitrogen) would be at incorporating biotinylated nucleotides or whether the fidelity of Amplitaq Gold DNA Polymerase (Invitrogen) would be impacted by the presence of biotinylated nucleotides in the cDNA template. Therefore, we sought to determine whether incorporation of biotinylated A and biotinylated C could lead to preferential isolation of RNAs with a high Guanine (G) and/or Uracil (U) content, and/or whether biotinylated nucleotides would be mutagenic in this context. We compared the G/U content of mapped reads from the 2-linker method to the mapped reads from the LQ method and found no significant difference between the two populations (Figure 5C). As a more direct comparison, using the LQ method, 50 fmol of LT-miRmix was cloned with and without biotin incorporation. Consistent with comparison of the LQ and 2-linker methods, we found no significant difference in the G/U content of sequences isolated in the presence or absence of biotin (Figure 5C). Additionally, we examined the relationship between read count (as measured by proportion of reads) and G/U content and found that G/U content did not directly correlate with read count when either high (2.5 pmol) or low (500 amol) amounts of 29-miRmix RNA was cloned (Supplementary Figure S4). Together, these results indicate that incorporation of biotinylated A and C does not appreciably affect the base composition of cDNAs recovered in the biotin-containing LQ procedure. Note that the RT reaction contains a mixture of biotinylated and non-biotinylated dCTP and dATP in ratios of 0.54:1 and 0.43:1, respectively. These ratios were optimized for cloning of small RNA cDNAs; it is possible that the proportion of biotinylated nucleotides in the RT reaction would need to be adjusted for applications involving the generation of appreciably longer cDNAs.
To assess possible mutagenic affects of biotinylated nucleotides, the LT-miRmix subset reference (Materials and Methods section) was used to compare any apparent mutation rates at G and U positions among sequences generated from each cloning method as well as from the LQ method with and without biotin incorporation. We did not observe any significant differences in nucleotide representation, indicating that the incorporation of biotinylated nucleotides was not appreciably mutagenic (Figure 5D).
5′ additions, 3′ variations and miRNA isoforms
Our LQ method involves generation of cDNAs without a 5′ linker, offering the advantage of cloning material independent of the presence of a 5′ monophosphate structure. However, absence of a linker sequence at the 5′ end of the template RNA introduces the potential for confounding effects of terminal transferase (TdT) activity of the reverse transcriptase. Although Superscript III reverse transcriptase has minimal TdT activity, we nevertheless identified significant 5′ nt additions during the early stages of method development (libraries D1 and D2, Figure 6A). We were able to reduce these 5′ additions from >22% (in libraries D1 and D2) to <3% (in libraries 1-4D) by reducing the quantity of RT enzyme to 0.5 units in each reaction, and by shortening the reaction time to 5 min (Figure 6A).
Because miRNA 3′ end heterogeneity has been characterized in biological samples (42,43), we sought to verify the ability of our LQ cloning method to identify these sequence variations. To do so, we generated a profile of sequences with length variability in the LT-miRmix subset reference as described in the Materials and Methods section. Sequence length profiles identified in the LQ versus 2-linker method indicate a similar ability for both methods to clone a wide range of miRNAs including those with 3′ end heterogeneity (Figure 6B). Due to the 5′ independent nature of our protocol, it was not surprising that we isolated miRNAs with 5′ end truncations from both the synthetic miRNA mixtures analyzed (data not shown). It is likely that these truncated molecules are due to heterogeneity arising at oligo synthesis or are generated by spurious base hydrolysis. Importantly, these sequence variations are missed by cloning methods employing a 5′ linker ligation that is dependent upon a 5′ phosphate and their identification highlights the importance of analyzing replicate libraries and employing differential analysis of the same method.
Interestingly, when plasma-derived miRNAs were compared to miRBase sequence annotations we identified a number of 5′ and 3′ differences between the annotated sequences and those obtained in our libraries (data not shown). Such apparent miRNA isoforms (isomiRs) could reflect alternative processing of miRNA transcripts or incorrect sequence annotation. IsomiRs are of interest, since alterations in the 5′ end of a miRNA would change the seed sequence, and therefore alter target recognition, while 3′ end modifications may affect miRNA stability and/or function (44–46).
Sensitivity and reproducibility
To test method sensitivity, we generated libraries across 3–4 orders of magnitudes using the synthetic miRNA mixtures (from 50 amol or 500 amol to 50 fmol) and across two orders of magnitude from human blood plasma (10–110 plasma equivalents corresponding to ∼90 amol–1,000 amol miRNA). From the LT-miRNA mixture, using the LQ method we recovered >93% of the reference miRNA sequences and >91% miRNA reference sequence coverage using the 2-linker method. In order to rigorously examine the ability to clone miRNAs in a sequence independent manner, libraries generated from decreasing input concentrations of the 29-miRmix were analyzed. Importantly, 100% of miRNA sequences in the mixture were isolated (Table 3 and Figure 7). Compared to methods developed by Zhang et al. (23) and Heyer et al. (personal communication), we observed underrepresentation of some sequences using our method. This apparent sequence bias became more pronounced with lower input concentrations. However, we were unable to identify 5′ end, 3′ end or general sequence context features predictive of this underrepresentation. It is possible that incorporation of additional modifications to the LQ method may enhance uniformity of sequence recovery. Possible modifications could include: heating RNA samples prior to 3′ adapter ligation, the use of PEG-8000 in ligation reactions, a degenerate 5′ end of the RT oligo and further optimization of enzymatic steps including buffer compositions (23,33,41,47–49). Additionally, it is important to note that use of the LQ method enables generation of libraries from significantly less material than these other methods as well as those commercially available.
Figure 7.
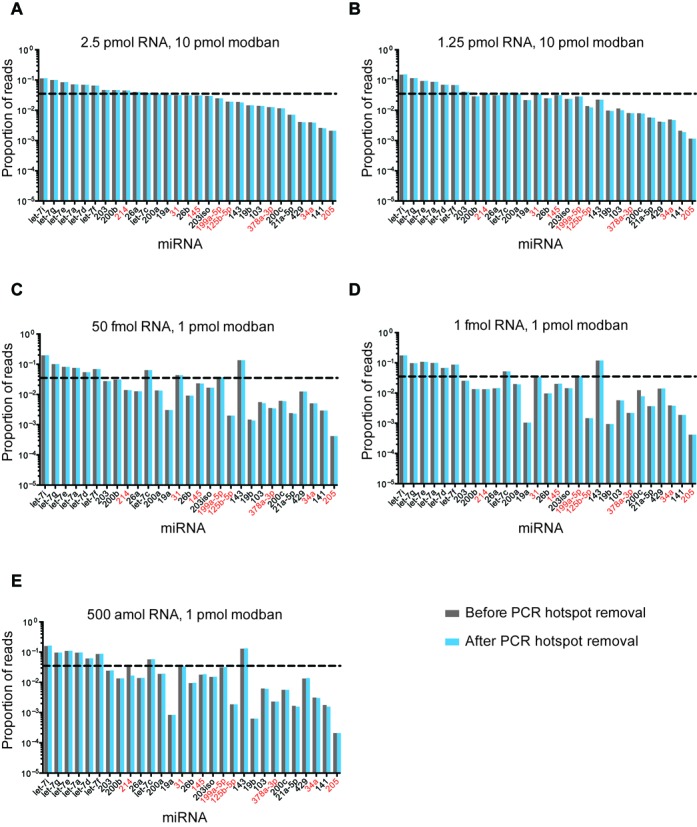
Analysis of synthetic miRNAs captured by the LQ cloning method. Deep sequencing data of an equimolar mixture of 29 synthetic miRNAs (29-miRmix) (23) before (gray bars) and after (blue bars) removal of sequences generated by PCR hotspots. The proportion of total reads (Y axis) for each miRNA (X axis) is plotted. The amount of input RNA (2.5 pmol–500 amol) and 3′ adapter (10 pmol or 1 pmol) are indicated. All 3′ adapters were pre-annealed to an equal concentration of RT oligo (see Materials and Methods section) and all RT oligos have a 4 nt randomer sequence. Dashed line represents the expected result for equimolar sequence representation and miRNAs with ≥50% G/C content are indicated with red text.
Cloning from human blood plasma demonstrates recovery of a range of 127–223 mature miRNA sequences that had ≥10 reads per million for those reads that mapped the human genome. This range is dependent upon the depth of the library examined and the extent of hemolysis that could be indicated by miR-451a representation (see below for explanation of miR-451a) (Table 3). Together, these data demonstrate significant sequence coverage and identification using our LQ cloning method.
To assess the consistency of sequences recovered by the LQ approach, we examined the correlation of sequence read counts among libraries from the same source RNA. Read counts from synthetic miRNA-derived libraries correlated well whether from the same amount or varying amounts of input RNA (Figure 8, dark blue boxes). Comparison of reads from two libraries generated from the LQ method demonstrates good correlation (Figure 9A). Not unexpectedly, there were some non-correlating outliers for certain low abundance sequences. Generally, libraries generated from the LQ method were more highly correlated (Figure 8, dark blue boxes) than those generated using the 2-linker method (Figure 8, red box, and Figure 9B). Finally, we found that while libraries generated by the LQ method and 2-linker method had similar read coverage, read counts for individual sequences were not well correlated between the two methods (Table 3 and Figure 9C).
Figure 9.
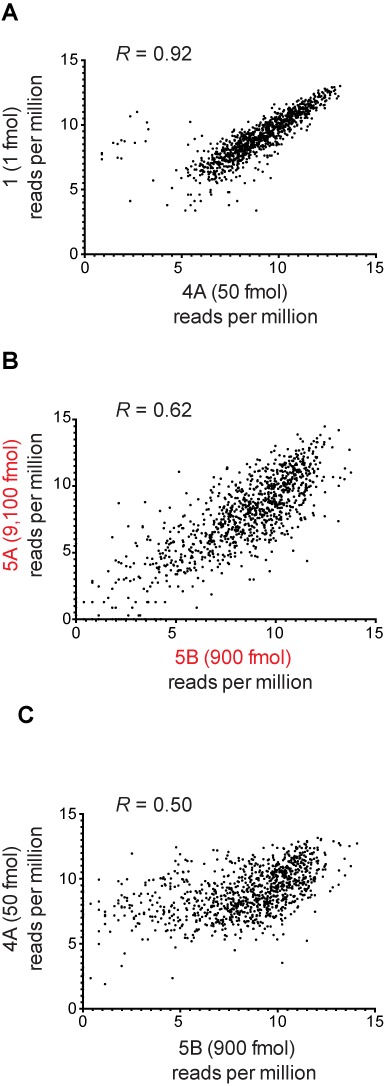
Scatter plots comparing miRNA reads from different library preparations and sequencing methods. (A) Comparison of miRNA reads generated by LQ cloning method originating from 50-fold difference in input RNA concentration. (B) Comparison of miRNA reads generated by 2-linker cloning method originating from 10-fold dilution of input RNA concentration. (C) Comparison of miRNA reads generated by LQ method versus 2-linker method. R = correlation coefficient between the two compared libraries.
To assess reproducibility between sequencing platforms, we generated a single library and then amplified the library with either Ion Torrent (Libraries 2A–2D) or Illumina (Libraries 4A–4D) specific adapters at the second round of PCR. Sequencing on the Ion Torrent or Illumina platform yielded similar percentages of reference sequence covered (Table 3) and high correlation of sequence read counts in corresponding libraries (generated from the same input material) (Figure 8, light blue boxes).
Analysis of miRNA read counts isolated from human blood plasma showed correlation above 0.98 for all library comparisons (data not shown). This substantiates the view that our LQ method is sufficiently sensitive and reproducible for application in the context of scarce clinical samples containing very low quantities of RNA.
miRNA repertoire of human blood plasma
Identification of miRNAs in blood plasma (4) has led to the characterization and profiling of circulating miRNA populations in a variety of disease contexts where the miRNA expression has been shown to indicate tissue damage and disease status (for examples see 2,50–53). A confounding issue to the circulating miRNA profile is the presence of blood-cell-associated miRNAs (54). In particular, the miRNA repertoire detected in plasma RNA is highly sensitive to the degree of hemolysis that can occur during sample isolation and processing (54,55). Remarkably, even traces (0.031% (v/v)) of red blood cells in plasma can alter the miRNA expression profile compared to non-hemolyzed samples (56).
Examination of cDNA libraries prepared from human blood plasma identified a range (127–223) of significantly expressed mature miRNAs. Analysis of miRNA read counts done on all cDNA libraries revealed significant representation of blood-cell-independent miRNAs as well as a number of miRNAs that have not been carefully studied in the context of hemolysis (Table 4). Consistent with previous findings, we also identified hemolysis-associated miRNAs. For example, we observed that miR-451a accounted for 58–82% of all reads that map to the human genome (Figure 10), indicating that these blood plasma samples had suffered significant hemolysis. Because miR-451a reads constituted a predominant and variable fraction of our libraries, we excluded miR-451a reads for certain aspects of the analysis of the other miRNAs represented in the libraries.
Table 4. Top-40 miRNAs isolated from human blood plasma.
| miRNA | Average reads per million | Hemolysis associated |
|---|---|---|
| hsa-miR-451a | 685,118 | yes56,55,65,54 |
| hsa-miR-144-3p | 33,490 | yes65 |
| hsa-miR-16-5p | 26,367 | yes64,56,55,54 |
| hsa-miR-15a-5p | 10,525 | |
| hsa-miR-22-3p | 6,994 | |
| hsa-miR-20a-5p | 4,833 | yes55 |
| hsa-miR-142-3p | 3,852 | no55 |
| hsa-let-7g-5p | 3,387 | |
| hsa-miR-29c-3p | 3,326 | |
| hsa-miR-486-5p | 3,267 | yes54 |
| hsa-miR-223-3p | 2,950 | no64,56,55,65,54 |
| hsa-miR-93-5p | 2,561 | no55 |
| hsa-miR-15b-5p | 2,330 | yes64,56 |
| hsa-miR-103a-3p | 2,301 | yes55 |
| hsa-miR-21-5p | 2,264 | yes55 |
| hsa-let-7i-5p | 2,191 | |
| hsa-miR-26a-5p | 1,979 | |
| hsa-miR-101-3p | 1,915 | |
| hsa-miR-107 | 1,911 | |
| hsa-miR-92a-3p | 1,682 | yes56,54 |
| hsa-miR-142-5p | 1,568 | no55 |
| hsa-miR-126-3p | 1,448 | no55 |
| hsa-miR-23a-3p | 1,348 | no63 |
| hsa-miR-29a-3p | 1,275 | no55 |
| hsa-miR-30e-5p | 1,261 | |
| hsa-miR-27a-3p | 1,215 | |
| hsa-miR-185-5p | 1,156 | |
| hsa-let-7a-5p | 1,089 | no54 |
| hsa-miR-29b-3p | 1,001 | |
| hsa-miR-26b-5p | 943 | |
| hsa-miR-25-3p | 928 | |
| hsa-let-7b-5p | 902 | yes55 |
| hsa-miR-19b-3p | 870 | |
| hsa-miR-122-5p | 852 | no64,54,66, yes55 |
| hsa-miR-425-5p | 767 | yes55 |
| hsa-miR-32-5p | 757 | |
| hsa-miR-18a-5p | 722 | |
| hsa-let-7f-5p | 701 | |
| hsa-miR-24-3p | 658 | no55, slightly64 |
| hsa-miR-146a-5p | 512 | no55 |
The average (in reads per million) of indicated mature miRNAs in libraries 6A-8E. Hemolysis association, where clearly identified, is referred to and reference(s) are indicated.
Figure 10.
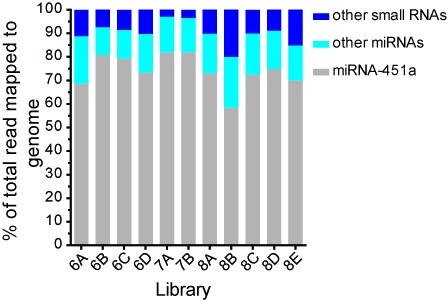
Small RNAs identified in human blood plasma. Total small RNA content in cDNA libraries generated from human blood plasma.
DISCUSSION
Deep sequencing of small RNA cDNA libraries from biological samples enables the discovery and quantitation of RNA transcripts, including miRNAs and other small noncoding RNAs, associated with distinct developmental, physiological or pathological states. Ideally, it is desirable to resolve transcriptional profiles on the level of individual cells, or in small samples of experimental material, such as tissue biopsies or biofluids. Single-cell RNA-seq methods have been developed for cloning mRNA (18,20–22), but not yet for small noncoding RNAs. cDNA library preparation has been reported using samples of small RNA isolated from human blood plasma, where the yield of RNA was in the nanogram range (14). However, the quantity of RNA available from clinical samples, especially biofluids, is often far less than a nanogram, even in the sub-picogram range. Accordingly, we developed a robust, sensitive and technically straightforward protocol (Low Quantity, LQ, method) through which very small quantities of input RNA can be used to generate cDNA libraries for deep sequencing. The LQ method permits the preparation of cDNA libraries from sub-picogram amounts of RNA − at least 1,000-fold less input than for existing approaches (for examples see (57) and (58)).
We developed and tested the LQ cloning protocol using two synthetic miRNA mixtures as input, followed by analysis of sequence data obtained from the LQ libraries along with that generated using a well-established 2-linker cloning method (13). The coverage of sequences in the reference set was >93% for LQ libraries, somewhat greater than that of libraries generated by the 2-linker method. The length distributions of insert sequences were similar for the two methods, although there was greater representation of shorter (17–20 nt) inserts for the LQ method perhaps reflecting the multiple size-selection steps utilized by the 2-linker method. Importantly, the 2-linker method requires dramatically more input material than the LQ method, as each of the size selection steps contributes to material loss.
Because the two methods utilized different enzymes in 5′ end ligation reactions, we examined any ligation-based biases through analysis of both 5′ and 3′ end nt profiles. The increase in 5′ A and decrease in 5′ C observed in the LQ method can be attributed to the use of CircLigase II in our method versus T4 RNA ligase I in 2-linker method. Additionally, different ligation reaction temperatures (60°C for CircLigase II and ≤15°C for T4 RNA ligase I) are also known to affect ligation in a sequence dependent manner and are likely to contribute to the observed 5′ nt differences as well. As expected, we saw no difference with 3′ end nt profiles, likely due to the fact that the same enzyme and reaction conditions were used in both methods.
We demonstrate systematic isolation of biotin-containing cDNAs using streptavidin beads. Because this is, to our knowledge, the first use of biotinylated nucleotides in this manner, we analyzed our data for evidence of sequence bias or mutagenesis that might have been introduced by the use of biotinylated nucleotides. We could discern no evident effect of biotin incorporation on either the base composition of the sequences obtained, or on the frequency of apparent nucleotide substitutions.
We did detect effects of cloning method on the frequency of non-templated nucleotides at the 5′ end of the RNA sequences. We traced these events to likely residual terminal transferase (TdT) activity of the Superscript III reverse transcriptase. We were able to minimize TdT activity by optimizing the RT reaction conditions such that the frequency of untemplated additions is a manageable 2–3% of sequences. The knowledge of expected 5′ addition rates can be used to identify additions that are not due to enzyme activity and a correction can be incorporated into future computational pipelines. Those sequences with 5′ addition rates significantly higher than that expected from RT enzyme terminal transferase activity could be assessed further to determine their biological significance. However, identification of ≤3% 5′ terminal additions cannot be distinguished from background terminal transferase activity. Sequence length variation at the 3′ end of sequences was similar in both the LQ method and 2-linker method.
Finally, to examine the sensitivity and reproducibility of the LQ method, we compared libraries generated from a broad range of quantities of input RNA. In doing so, we identified a high correlation of the sequence content between libraries generated from RNA content ranging over four orders of input material. Examination of the equimolar 29-miRmix revealed that, for some miRNAs, such as members of the let-7 family, read counts are reflective of relative sequence abundance while for other sequences (e.g. miR-205), read counts do not necessarily reflect relative abundance. This phenomenon became more pronounced as RNA input concentrations decreased (Figure 7). These observations reveal one limitation in the described method. As such, read counts are not necessarily reflective of absolute sequence abundance and cannot be used to compare different small RNAs within the same library or across libraries prepared from different input concentrations of RNA. However, the method is appropriate for cloning cDNAs derived from very small amounts of starting material, and for quantifying the levels of a specific sequence across different samples, provided that libraries are prepared from similar amounts of input RNA.
Although we identified similar reference sequence coverage for the LQ method compared to conventional 2-linker cloning, the read count representation for individual sequences varied between the two methods. These differences could reflect the differing conditions for ligation and RT reactions between the methods, or sequence-dependent sample loss. Moreover, the LQ method permits correction for PCR hotspot reads whereas the 2-linker method did not incorporate such provisions in this implementation. Importantly, we devised a method that uses the randomer sequence incorporated into the RT oligo to identify PCR hot spots and correct for them. Several works have explored the use of random sequences to correct for PCR-induced artifacts (18,59–62). These methods rely on the assumptions that (i) the number of randomer sequences is more than the number of distinct molecules and/or (ii) each randomer has an equal probability of being observed. The first assumption is needed in order to avoid saturation of a single sequence with the full randomer pool and to enable collapsing the observed read count to the number of observed randomer sequences. Our method is designed to detect and correct hot spots in both saturated and unsaturated sequences. Thus a 4 nt randomer was sufficient and use of a longer, 6 nt, randomer did not show any significant change in terms of read collapsing. In regard to the second assumption, our data show that randomer sequences are not equally distributed. Rather, in some cases there is a 1,000-fold difference between the least and most abundant randomers. As a result, we assess the distribution of randomers in each library separately, by identifying a set of sequences that do not show a sharp bias to any particular randomer sequences. Therefore, we found that for small RNA libraries, a 4 nt randomer is necessary and sufficient to detect and correct for PCR hotspots.
When applying the LQ to human plasma RNA we were able to reproducibly clone cDNA from RNA isolated from 10 –100 μl of human blood plasma (estimated 90 amol –1,000 amol miRNA). We found that miRNAs constitute the major fraction of sequences cloned from human blood plasma, and our sequences represented from 127 to 223 distinct miRNA species with read counts >10 reads per million, similar to other analysis of human blood plasma performed with more material (5,63). Consistent with previous findings that heamolysis can significantly affect circulating miRNA profiles (54–56,63–65), miRNA-451a reads were dramatically predominant in our libraries. As the biological relevance of miRNA-451a and other potentially heamolysis-associated miRNAs in samples is determined, sequences identified as background can be removed prior to library generation using hybridization/bead capture or hybridization/digestion techniques. Importantly, the circulating miRNA profiles that we identified by deep sequencing include numerous plasma-borne miRNAs that are known to not originate in blood cells (Table 4) (54,55,63–66).
As expected, we identified other RNA sequences (besides miRNAs) that would be anticipated from a method that does not select for 5′-phosphate containing RNAs. These non-miRNA sequences included rRNA fragments, tRNA fragments, Y RNA (37) and sequences annotated as piRNA (67–70). Together, these data from libraries made from human blood plasma confirm the highly sensitive and reproducible nature of the method as well as the ability to clone a broader repertoire of small RNA sequences than any single method previously published. Thus, this method has broad applications in a clinical setting, making it feasible to generate cDNA libraries from human biofluids as well as from small and scarce tissue specimens including biopsy tissue blocks and small numbers of individual cells isolated from laser capture microdissection of heterogeneous cell populations (71,72).
We have developed a novel, robust method to clone small amounts of RNA. While the limited amounts of input material we have cloned is unprecedented, our findings do reveal possibilities for further improvement and minimization of sequence biases (23,33,41,47–49). Importantly, features of the LQ method, including utilization of barcode and randomer sequences, provide the opportunity to pool samples early in library generation thereby permitting exploration of cloning from even more limiting sample concentrations. Additionally, further development toward removal of precipitation as well as gel and column purification steps will enable the LQ method to be applied to high-throughput, robotic, applications.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Erin Heyer, Emiliano Ricci and Melissa Moore for helpful discussions throughout method development, and Keith Tomaszewicz and Lloyd Hutchinson for sequencing on the Ion Torrent PGM. Jason Potter, Stephen Hendricks and Caifu Chen at Life Technologies were helpful in resolving terminal transferase activity. Fred Hyde and Hank Daum at Epicentre provided helpful insight into the circularization reaction.
FUNDING
ALS Therapy Alliance [to C.S.]; National Institutes of Health [GM034028-28 to C.S., GM034028-25A2 to I.V-L.]; Translational Cancer Biology Training Program [CA130807-2 to C.S.].
Conflict of interest statement. None.
REFERENCES
- 1.Bartel D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 2.Mitchell P.S., Parkin R.K., Kroh E.M., Fritz B.R., Wyman S.K., Pogosova-Agadjanyan E.L., Peterson A., Noteboom J., O'Briant K.C., Allen A., et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. U.S.A. 2008;105:10513–10518. doi: 10.1073/pnas.0804549105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lawrie C.H., Gal S., Dunlop H.M., Pushkaran B., Liggins A.P., Pulford K., Banham A.H., Pezzella F., Boultwood J., Wainscoat J.S., et al. Detection of elevated levels of tumour-associated microRNAs in serum of patients with diffuse large B-cell lymphoma. Br. J. Haematol. 2008;141:672–675. doi: 10.1111/j.1365-2141.2008.07077.x. [DOI] [PubMed] [Google Scholar]
- 4.Chim S.S.C., Shing T.K.F., Hung E.C.W., Leung T.-Y., Lau T.-K., Chiu R.W.K., Lo Y.M.D. Detection and characterization of placental microRNAs in maternal plasma. Clin. Chem. 2008;54:482–490. doi: 10.1373/clinchem.2007.097972. [DOI] [PubMed] [Google Scholar]
- 5.Chen X., Ba Y., Ma L., Cai X., Yin Y., Wang K., Guo J., Zhang Y., Chen J., Guo X., et al. Characterization of microRNAs in serum: a novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008;18:997–1006. doi: 10.1038/cr.2008.282. [DOI] [PubMed] [Google Scholar]
- 6.Weber J.A., Baxter D.H., Zhang S., Huang D.Y., Huang K.H., Lee M.J., Galas D.J., Wang K. The microRNA spectrum in 12 body fluids. Clin. Chem. 2010;56:1733–1741. doi: 10.1373/clinchem.2010.147405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu J., Getz G., Miska E.A., Alvarez-Saavedra E., Lamb J., Peck D., Sweet-Cordero A., Ebert B.L., Mak R.H., Ferrando A.A., et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 8.He L., Thomson J.M., Hemann M.T., Hernando-Monge E., Mu D., Goodson S., Powers S., Cordon-Cardo C., Lowe S.W., Hannon G.J., et al. A microRNA polycistron as a potential human oncogene. Nature. 2005;435:828–833. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taylor D.D., Gercel-Taylor C. MicroRNA signatures of tumor-derived exosomes as diagnostic biomarkers of ovarian cancer. Gynecol. Oncol. 2008;110:13–21. doi: 10.1016/j.ygyno.2008.04.033. [DOI] [PubMed] [Google Scholar]
- 10.Pfeffer S., Lagos-Quintana M., Tuschl T. Cloning of small RNA molecules. Curr. Protoc. Mol. Biol. 2005 doi: 10.1002/0471142727.mb2604s72. Chapter 26, Unit 26.4. [DOI] [PubMed] [Google Scholar]
- 11.König J., Zarnack K., Rot G., Curk T., Kayikci M., Zupan B., Turner D.J., Luscombe N.M., Ule J. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat. Struct. Mol. Biol. 2010;17:909–915. doi: 10.1038/nsmb.1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morin R.D., O'Connor M.D., Griffith M., Kuchenbauer F., Delaney A., Prabhu A.-L., Zhao Y., McDonald H., Zeng T., Hirst M., et al. Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells. Genome Res. 2008;18:610–621. doi: 10.1101/gr.7179508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gu W., Lee H.-C., Chaves D., Youngman E.M., Pazour G.J., Conte D., Mello C.C. CapSeq and CIP-TAP identify Pol II start sites and reveal capped small RNAs as C. elegans piRNA precursors. Cell. 2012;151:1488–1500. doi: 10.1016/j.cell.2012.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Williams Z., Ben-Dov I.Z., Elias R., Mihailović A., Brown M., Rosenwaks Z., Tuschl T. Comprehensive profiling of circulating microRNA via small RNA sequencing of cDNA libraries reveals biomarker potential and limitations. Proc. Natl. Acad. Sci. U.S.A. 2013;110:4255–4260. doi: 10.1073/pnas.1214046110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kwon Y.-S. Small RNA library preparation for next-generation sequencing by single ligation, extension and circularization technology. Biotechnol. Lett. 2011;33:1633–1641. doi: 10.1007/s10529-011-0611-y. [DOI] [PubMed] [Google Scholar]
- 16.Mendell J.T., Olson E.N. MicroRNAs in stress signaling and human disease. Cell. 2012;148:1172–1187. doi: 10.1016/j.cell.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Farazi T.A., Hoell J.I., Morozov P., Tuschl T. MicroRNAs in human cancer. Adv. Exp. Med. Biol. 2013;774:1–20. doi: 10.1007/978-94-007-5590-1_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Islam S., Zeisel A., Joost S., La Manno G., Zajac P., Kasper M., Lönnerberg P., Linnarsson S. Quantitative single-cell RNA-seq with unique molecular identifiers. Nat. Methods. 2014;11:163–166. doi: 10.1038/nmeth.2772. [DOI] [PubMed] [Google Scholar]
- 19.Adiconis X., Borges-Rivera D., Satija R., DeLuca D.S., Busby M.A., Berlin A.M., Sivachenko A., Thompson D.A., Wysoker A., Fennell T., et al. Comparative analysis of RNA sequencing methods for degraded or low-input samples. Nat. Methods. 2013;10:623–629. doi: 10.1038/nmeth.2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ramsköld D., Luo S., Wang Y.-C., Li R., Deng Q., Faridani O.R., Daniels G.A., Khrebtukova I., Loring J.F., Laurent L.C., et al. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat. Biotechnol. 2012;30:777–782. doi: 10.1038/nbt.2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Picelli S., Faridani O.R., Björklund Å.K., Winberg G., Sagasser S., Sandberg R. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 2014;9:171–181. doi: 10.1038/nprot.2014.006. [DOI] [PubMed] [Google Scholar]
- 22.Picelli S., Björklund Å.K., Faridani O.R., Sagasser S., Winberg G., Sandberg R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods. 2013;10:1096–1098. doi: 10.1038/nmeth.2639. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Z., Lee J.E., Riemondy K., Anderson E.M., Yi R. High-efficiency RNA cloning enables accurate quantification of miRNA expression by deep sequencing. Genome Biol. 2013;14:R109. doi: 10.1186/gb-2013-14-10-r109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ho C.K., Wang L.K., Lima C.D., Shuman S. Structure and mechanism of RNA ligase. Structure. 2004;12:327–339. doi: 10.1016/j.str.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 25.Ho C.K., Shuman S. Bacteriophage T4 RNA ligase 2 (gp24.1) exemplifies a family of RNA ligases found in all phylogenetic domains. Proc. Natl. Acad. Sci. U.S.A. 2002;99:12709–12714. doi: 10.1073/pnas.192184699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ingolia N.T., Ghaemmaghami S., Newman J.R.S., Weissman J.S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 2011;171:10–12. [Google Scholar]
- 28.Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karolchik D., Hinrichs A.S., Furey T.S., Roskin K.M., Sugnet C.W., Haussler D., Kent W.J. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004;32:D493–D496. doi: 10.1093/nar/gkh103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sai Lakshmi S., Agrawal S. piRNABank: a web resource on classified and clustered Piwi-interacting RNAs. Nucleic Acids Res. 2008;36:D173–D177. doi: 10.1093/nar/gkm696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kin T., Yamada K., Terai G., Okida H., Yoshinari Y., Ono Y., Kojima A., Kimura Y., Komori T., Asai K. fRNAdb: a platform for mining/annotating functional RNA candidates from non-coding RNA sequences. Nucleic Acids Res. 2007;35:D145–D148. doi: 10.1093/nar/gkl837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Griffiths-Jones S., Saini H.K., van Dongen S., Enright A.J. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36:D154–D158. doi: 10.1093/nar/gkm952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hafner M., Renwick N., Brown M., Mihailović A., Holoch D., Lin C., Pena J.T.G., Nusbaum J.D., Morozov P., Ludwig J., et al. RNA-ligase-dependent biases in miRNA representation in deep-sequenced small RNA cDNA libraries. RNA. 2011;17:1697–1712. doi: 10.1261/rna.2799511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Viollet S., Fuchs R.T., Munafo D.B., Zhuang F., Robb G.B. T4 RNA ligase 2 truncated active site mutants: improved tools for RNA analysis. BMC Biotechnol. 2011;11:72–86. doi: 10.1186/1472-6750-11-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Okayama H., Berg P. High-efficiency cloning of full-length cDNA. Mol. Cell. Biol. 1982;2:161–170. doi: 10.1128/mcb.2.2.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Crouse J., Amorese D. Ethanol precipitation: ammonium acetate as an alternative to sodium acetate. Focus. 1996;18:17–20. [Google Scholar]
- 37.Dhahbi J.M., Spindler S.R., Atamna H., Boffelli D., Mote P., Martin D.I.K. 5′-YRNA fragments derived by processing of transcripts from specific YRNA genes and pseudogenes are abundant in human serum and plasma. Physiol. Genomics. 2013;45:990–998. doi: 10.1152/physiolgenomics.00129.2013. [DOI] [PubMed] [Google Scholar]
- 38.Cheng J., Guo J.-M., Xiao B.-X., Miao Y., Jiang Z., Zhou H., Li Q.-N. piRNA, the new non-coding RNA, is aberrantly expressed in human cancer cells. Clin. Chim. Acta. 2011;412:1621–1625. doi: 10.1016/j.cca.2011.05.015. [DOI] [PubMed] [Google Scholar]
- 39.Lu Y., Li C., Zhang K., Sun H., Tao D., Liu Y., Zhang S., Ma Y. Identification of piRNAs in Hela cells by massive parallel sequencing. BMB Rep. 2010;43:635–641. doi: 10.5483/BMBRep.2010.43.9.635. [DOI] [PubMed] [Google Scholar]
- 40.Alon S., Vigneault F., Eminaga S., Christodoulou D.C., Seidman J.G., Church G.M., Eisenberg E. Barcoding bias in high-throughput multiplex sequencing of miRNA. Genome Res. 2011;21:1506–1511. doi: 10.1101/gr.121715.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun G., Wu X., Wang J., Li H., Li X., Gao H., Rossi J., Yen Y. A bias-reducing strategy in profiling small RNAs using Solexa. RNA. 2011;17:2256–2262. doi: 10.1261/rna.028621.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee L.W., Zhang S., Etheridge A., Ma L., Martin D., Galas D., Wang K. Complexity of the microRNA repertoire revealed by next-generation sequencing. RNA. 2010;16:2170–2180. doi: 10.1261/rna.2225110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ameres S.L., Zamore P.D. Diversifying microRNA sequence and function. Nat. Rev. Mol. Cell Biol. 2013;14:475–488. doi: 10.1038/nrm3611. [DOI] [PubMed] [Google Scholar]
- 44.Ebhardt H.A., Fedynak A., Fahlman R.P. Naturally occurring variations in sequence length creates microRNA isoforms that differ in argonaute effector complex specificity. Silence. 2010;1:12–18. doi: 10.1186/1758-907X-1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Neilsen C.T., Goodall G.J., Bracken C.P. IsomiRs—the overlooked repertoire in the dynamic microRNAome. Trends Genet. 2012;28:544–549. doi: 10.1016/j.tig.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 46.Kim Y.-K., Heo I., Kim V.N. Modifications of small RNAs and their associated proteins. Cell. 2010;143:703–709. doi: 10.1016/j.cell.2010.11.018. [DOI] [PubMed] [Google Scholar]
- 47.Jayaprakash A.D., Jabado O., Brown B.D., Sachidanandam R. Identification and remediation of biases in the activity of RNA ligases in small-RNA deep sequencing. Nucleic Acids Res. 2011;39:e141. doi: 10.1093/nar/gkr693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhuang F., Fuchs R.T., Sun Z., Zheng Y., Robb G.B. Structural bias in T4 RNA ligase-mediated 3′-adapter ligation. Nucleic Acids Res. 2012;40:e54. doi: 10.1093/nar/gkr1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sorefan K., Pais H., Hall A.E., Kozomara A., Griffiths-Jones S., Moulton V., Dalmay T. Reducing ligation bias of small RNAs in libraries for next generation sequencing. Silence. 2012;3:4–15. doi: 10.1186/1758-907X-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kosaka N., Iguchi H., Ochiya T. Circulating microRNA in body fluid: a new potential biomarker for cancer diagnosis and prognosis. Cancer Sci. 2010;101:2087–2092. doi: 10.1111/j.1349-7006.2010.01650.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang G.-K., Zhu J.-Q., Zhang J.-T., Li Q., Li Y., He J., Qin Y.-W., Jing Q. Circulating microRNA: a novel potential biomarker for early diagnosis of acute myocardial infarction in humans. Eur. Heart J. 2010;31:659–666. doi: 10.1093/eurheartj/ehq013. [DOI] [PubMed] [Google Scholar]
- 52.Wang K., Zhang S., Weber J., Baxter D., Galas D.J. Export of microRNAs and microRNA-protective protein by mammalian cells. Nucleic Acids Res. 2010;38:7248–7259. doi: 10.1093/nar/gkq601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Recchioni R., Marcheselli F., Olivieri F., Ricci S., Procopio A.D., Antonicelli R. Conventional and novel diagnostic biomarkers of acute myocardial infarction: a promising role for circulating microRNAs. Biomarkers. 2013;18:547–558. doi: 10.3109/1354750X.2013.833294. [DOI] [PubMed] [Google Scholar]
- 54.Pritchard C.C., Kroh E., Wood B., Arroyo J.D., Dougherty K.J., Miyaji M.M., Tait J.F., Tewari M. Blood cell origin of circulating microRNAs: a cautionary note for cancer biomarker studies. Cancer Prev. Res. (Phila) 2012;5:492–497. doi: 10.1158/1940-6207.CAPR-11-0370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kirschner M.B., Edelman J.J.B., Kao S.C.-H., Vallely M.P., van Zandwijk N., Reid G. The impact of hemolysis on cell-free microRNA biomarkers. Front. Genet. 2013;4:94–107. doi: 10.3389/fgene.2013.00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kirschner M.B., Kao S.C., Edelman J.J., Armstrong N.J., Vallely M.P., van Zandwijk N., Reid G. Haemolysis during sample preparation alters microRNA content of plasma. PLoS ONE. 2011;6:e24145. doi: 10.1371/journal.pone.0024145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hafner M., Renwick N., Farazi T.A., Mihailović A., Pena J.T.G., Tuschl T. Barcoded cDNA library preparation for small RNA profiling by next-generation sequencing. Methods. 2012;58:164–170. doi: 10.1016/j.ymeth.2012.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Burgos K.L., Javaherian A., Bomprezzi R., Ghaffari L., Rhodes S., Courtright A., Tembe W., Kim S., Metpally R., Van Keuren-Jensen K. Identification of extracellular miRNA in human cerebrospinal fluid by next-generation sequencing. RNA. 2013;19:712–722. doi: 10.1261/rna.036863.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kivioja T., Vähärautio A., Karlsson K., Bonke M., Enge M., Linnarsson S., Taipale J. Counting absolute numbers of molecules using unique molecular identifiers. Nat. Methods. 2012;9:72–74. doi: 10.1038/nmeth.1778. [DOI] [PubMed] [Google Scholar]
- 60.Shiroguchi K., Jia T.Z., Sims P.A., Xie X.S. Digital RNA sequencing minimizes sequence-dependent bias and amplification noise with optimized single-molecule barcodes. Proc. Natl. Acad. Sci. U.S.A. 2012;109:1347–1352. doi: 10.1073/pnas.1118018109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Casbon J.A., Osborne R.J., Brenner S., Lichtenstein C.P. A method for counting PCR template molecules with application to next-generation sequencing. Nucleic Acids Res. 2011;39:e81. doi: 10.1093/nar/gkr217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fu G.K., Hu J., Wang P.-H., Fodor S.P.A. Counting individual DNA molecules by the stochastic attachment of diverse labels. Proc. Natl. Acad. Sci. U.S.A. 2011;108:9026–9031. doi: 10.1073/pnas.1017621108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Blondal T., Jensby Nielsen S., Baker A., Andreasen D., Mouritzen P., Wrang Teilum M., Dahlsveen I.K. Assessing sample and miRNA profile quality in serum and plasma or other biofluids. Methods. 2013;59:S1–S6. doi: 10.1016/j.ymeth.2012.09.015. [DOI] [PubMed] [Google Scholar]
- 64.McDonald J.S., Milosevic D., Reddi H.V., Grebe S.K., Algeciras-Schimnich A. Analysis of circulating microRNA: preanalytical and analytical challenges. Clin. Chem. 2011;57:833–840. doi: 10.1373/clinchem.2010.157198. [DOI] [PubMed] [Google Scholar]
- 65.Rasmussen K.D., Simmini S., Abreu-Goodger C., Bartonicek N., Di Giacomo M., Bilbao-Cortes D., Horos R., Lindern Von M., Enright A.J., O'Carroll D. The miR-144/451 locus is required for erythroid homeostasis. J. Exp. Med. 2010;207:1351–1358. doi: 10.1084/jem.20100458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cheng H.H., Yi H.S., Kim Y., Kroh E.M., Chien J.W., Eaton K.D., Goodman M.T., Tait J.F., Tewari M., Pritchard C.C. Plasma processing conditions substantially influence circulating microRNA biomarker levels. PLoS ONE. 2013;8:e64795. doi: 10.1371/journal.pone.0064795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Aravin A., Gaidatzis D., Pfeffer S., Lagos-Quintana M., Landgraf P., Iovino N., Morris P., Brownstein M.J., Kuramochi-Miyagawa S., Nakano T., et al. A novel class of small RNAs bind to MILI protein in mouse testes. Nature. 2006;442:203–207. doi: 10.1038/nature04916. [DOI] [PubMed] [Google Scholar]
- 68.Girard A., Sachidanandam R., Hannon G.J., Carmell M.A. A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature. 2006;442:199–202. doi: 10.1038/nature04917. [DOI] [PubMed] [Google Scholar]
- 69.Grivna S.T., Beyret E., Wang Z., Lin H. A novel class of small RNAs in mouse spermatogenic cells. Genes Dev. 2006;20:1709–1714. doi: 10.1101/gad.1434406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Watanabe T., Takeda A., Tsukiyama T., Mise K., Okuno T., Sasaki H., Minami N., Imai H. Identification and characterization of two novel classes of small RNAs in the mouse germline: retrotransposon-derived siRNAs in oocytes and germline small RNAs in testes. Genes Dev. 2006;20:1732–1743. doi: 10.1101/gad.1425706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nakamura N., Ruebel K., Jin L., Qian X., Zhang H., Lloyd R.V. Laser capture microdissection for analysis of single cells. Methods Mol. Med. 2007;132:11–18. doi: 10.1007/978-1-59745-298-4_2. [DOI] [PubMed] [Google Scholar]
- 72.Redmond L.C., Pang C.J., Dumur C., Haar J.L., Lloyd J.A. Laser capture microdissection of embryonic cells and preparation of RNA for microarray assays. Methods Mol. Biol. 2014;1092:43–60. doi: 10.1007/978-1-60327-292-6_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.