

Abstract
Clostridium difficile is the leading cause of infectious nosocomial diarrhea. The pathogenesis of C. difficile infection (CDI) results from the interactions between the pathogen, intestinal epithelium, host immune system, and gastrointestinal microbiota. Previous studies of the host-pathogen interaction in CDI have utilized either simple cell monolayers or in vivo models. While much has been learned by utilizing these approaches, little is known about the direct interaction of the bacterium with a complex host epithelium. Here, we asked if human intestinal organoids (HIOs), which are derived from pluripotent stem cells and demonstrate small intestinal morphology and physiology, could be used to study the pathogenesis of the obligate anaerobe C. difficile. Vegetative C. difficile, microinjected into the lumen of HIOs, persisted in a viable state for up to 12 h. Upon colonization with C. difficile VPI 10463, the HIO epithelium is markedly disrupted, resulting in the loss of paracellular barrier function. Since similar effects were not observed when HIOs were colonized with the nontoxigenic C. difficile strain F200, we directly tested the role of toxin using TcdA and TcdB purified from VPI 10463. We show that the injection of TcdA replicates the disruption of the epithelial barrier function and structure observed in HIOs colonized with viable C. difficile.
INTRODUCTION
Clostridium difficile is an anaerobic, spore-forming bacterium that is the leading cause of infectious nosocomial diarrhea and is responsible for over 14,000 deaths annually (1). Human exposure to C. difficile results in a range of manifestations, from asymptomatic colonization, to diarrhea, to lethal toxic megacolon. Various models have been used to study C. difficile infection (CDI), including in vitro models using transformed cell lines and a variety of in vivo models (2–5). In vitro cell culture models are limited in their ability to recapitulate complexities of the human gastrointestinal tract, and detailed, real-time study of the mucosal epithelium during infection in an animal model is technically challenging.
Human intestinal organoids (HIOs) are three-dimensional spheroids of human epithelium generated through directed differentiation of human pluripotent stem cells (hPSCs), which include human embryonic stem cells (hESCs) and induced pluripotent stem cells (iPSCs). HIOs contain both mesenchymal and epithelial tissues that are structurally arranged around a central luminal cavity. The epithelial compartment of the HIO possesses an array of small intestinal cell types, including absorptive enterocytes and secretory Paneth, goblet, and enteroendocrine cells, in addition to Lgr5+ intestinal stem cells (6). HIOs have been used to model features of embryonic development, viral infection, and inflammatory bowel disease (7–9). Due to their similarity to the human gastrointestinal tract, HIOs serve as a tractable and physiologically relevant model of the human intestine.
We sought to use HIOs to study the interaction between C. difficile and complex human epithelium. We developed a real-time functional assay to demonstrate that HIOs have a robust and effective epithelial barrier, which limits paracellular diffusion. In addition, we developed microinjection techniques to introduce C. difficile into the lumen of HIOs and found that viable C. difficile persists within the HIOs. Colonization of HIOs with C. difficile strain VPI 10463 results in disruption of the organoid epithelium. These effects apparently were dependent on the primary virulence factors of C. difficile, the toxins TcdA and TcdB (10), since colonization with a nontoxigenic C. difficile strain did not disrupt the HIO epithelium while microinjection of purified TcdA recapitulated the effects mediated by toxigenic VPI 10463. These results demonstrate that HIOs can be used for detailed molecular and cellular investigation of the pathogenic interactions between C. difficile and human intestinal epithelium.
MATERIALS AND METHODS
Manuscript.
All authors had access to the study data and reviewed and approved the final manuscript.
HIO growth/propagation.
Three-dimensional HIOs were generated by directed differentiation of human pluripotent stem cells (hPSCs) as previously described (6, 9, 11). HIOs were generated from the H9 hESC line (WA09; NIH registry no. 0062), and all hESC work described was approved by the University of Michigan human pluripotent stem cell oversight committee (hPSCRO). Briefly, hESCs were differentiated into endoderm using 100 ng/ml activin A for 3 days and then further differentiated into CDX2+ intestinal tissue using 2 μM Chir99021 (04-0004-10; Stemgent) plus 500 ng/ml fibroblast growth factor 4 (FGF4) for 4 to 6 days. FGF4, used to differentiate the intestinal tissue, was either obtained from R&D Systems (235-F4) or purified in the laboratory as previously described (12). During intestinal specification, three-dimensional spheroids emerged in the culture dish. Spheroids were collected and embedded in Matrigel (354234; BD Bioscience) and expanded for 30 to 60 days in intestinal growth medium (IGM) containing advanced Dulbecco's modified Eagle medium (DMEM)-F12 medium (12634-010; Gibco by Life Technologies) supplemented with l-glutamine, 15 mM HEPES, B27 supplement (17504-044; Gibco by Life Technologies), penicillin-streptomycin, and growth factors containing 100 ng/ml Noggin (6057-NG; R&D Systems), 100 ng/ml epidermal growth factor (EGF) (236-EG; R&D Systems), and 5% R-Spondin2 conditioned medium (13). The media were replaced every 4 days. One- to 2-month-old cystic HIOs that were >1 mm in diameter were used for experiments.
Clostridium difficile isolates.
Two strains of C. difficile were used in this study, strain VPI 10463 (ATCC 43255), a toxigenic strain which produces both TcdA and TcdB, and a nontoxigenic clinical isolate, F200, described previously (4).
Growth of Clostridium difficile in vitro.
For all experiments, both strains were grown in a vinyl anaerobic chamber (Coy Laboratory Products) at 37°C in brain heart infusion (BHI) broth plus 100 mg liter−1 l-cysteine. For all microinjection experiments, overnight cultures were back diluted 1:10 and grown for 3 h at 37°C. For strain VPI 10463, after 3 h the culture was further diluted to a starting optical density at 600 nm (OD600) of 0.01. F200 did not grow well following a second dilution, so the initial 1:10 back dilution was used to start the growth curve. The OD600 of the cultures was measured in the anaerobic chamber using a handheld spectrophotometer (WPA CO8000 cell density meter no. 80-3000-45; Biochrom). After 6 h of growth from a starting OD of 0.01, the mean OD600 of VPI 10463 used to colonize the HIOs was 0.5. After 9 h from the first 1:10 dilution, the mean OD600 for F200 used to colonize the HIOs was 0.67. To prepare bacterial supernatants, the cultures described above of either strain VPI 10463 or F200 were passed through a 0.22-μm-pore-size filter to remove the bacterial cells, and then fluorescein isothiocyanate (FITC)-dextran with an average molecular mass of 4 kDa (FD4) was added to a final concentration of 1 mM.
Microinjection of HIOs.
HIOs were gently removed from the Matrigel in which they were grown by cutting around each HIO using a 30-gauge needle. A cut 1,000-μl pipette tip with a large bore that did not mechanically disrupt the HIO was used to remove released HIOs from the well and to transfer them to a petri dish. HIOs were divided into experimental groups, and each group of HIOs was reembedded into fresh Matrigel in wells of a 24-well tissue culture plate. Thin-wall glass capillaries (TW100F-4; World Precision Instruments) were pulled using a Narishige PN-30 micropipette puller. The tips of the glass capillaries were cut with a scalpel, and the capillaries were passed into an anaerobic chamber. In the anaerobic chamber, the capillaries were filled with either C. difficile plus FD4 or filtered supernatant plus FD4 using Eppendorf microloader tips (5242956003; Eppendorf). The filled capillaries then were passed out of the chamber and loaded onto the microinjector (BRI XenoWorks analog microinjector; Sutter Instrument Company). FD4 was used in all microinjection experiments to aid in visualizing the injections. Even under ambient lighting, the green color of the FD4 was sufficient for us to ascertain if each injection was successful. For example, if an HIO displayed a noticeable outflow of green (FD4) immediately following injection, it was excluded from the experiment. Once all of the HIOs were injected, the wells were washed twice with DMEM-F12 medium (1263-028; Gibco by Life Technologies). Following the washes, 500 μl of IGM with growth factors was added to each well, and the HIOs were imaged using a fluorescent stereomicroscope (SZX16; Olympus) at ×1 magnification. The HIOs were incubated at 37°C in a 5% CO2 humidified incubator. Images were taken at the indicated time points postinjection. The disruption of barrier integrity was visualized by the loss of FD4 in the lumen of the HIOs.
Clostridium difficile quantitation from HIOs.
For these experiments, C. difficile was grown and individual HIOs were injected as described above. At 0, 2, and 12 h postinjection, each HIO was removed with a 1,000-μl genomic tip (2079G; Art by Molecular BioProducts) and transferred into a sterile 1.7-ml Eppendorf tube. The tube was immediately passed into an anaerobic chamber. In the chamber, 500 μl of anaerobic 1× phosphate-buffered saline (PBS) (10010-023; Gibco by Life Science) was added to each tube, and the HIO was disrupted using a 1,000-μl tip. This HIO-PBS mixture was used for subsequent dilutions. To determine the level of vegetative C. difficile, samples were plated on BHI plus 100 mg liter−1 l-cysteine. Plates were incubated in the anaerobic chamber for 24 h a 37°C, at which point colonies were counted.
Vero cell cytotoxicity assay.
The activity of the toxins used in the barrier function assay was determined using a cell rounding-based cytotoxicity assay. African green monkey kidney (Vero) cells (ATCC CCL-81) were grown to a confluent monolayer in T-75 flasks in DMEM (11965; Gibco by Life Technologies), supplemented with 10% heat-inactivated fetal bovine serum (16140; Gibco by Life Technologies) and 1% penicillin-streptomycin (15140; Gibco by Life Technologies). To remove the cells from the flask, they were washed with 1× PBS followed by treatment with 1 ml of 0.25% trypsin. The trypsin was inactivated by the addition of 10 ml of the supplemented DMEM. The cells then were transferred to a conical tube and spun at 1,000 rpm for 5 min to pellet the cells. For this assay, 1 × 105 cells in 90 μl of DMEM were seeded in each well of a 96-well plate (3596; Corning) and incubated for 4 h. Filtered culture supernatant or purified TcdA or TcdB was serially diluted 1:10 in 1× PBS. As a control, the diluted sample then was added to an equal volume of either a 1:25 dilution of anti-toxin serum (T5000; TechLab) or PBS and incubated at room temperature for 1 h. Following the incubation, 10 μl of the sample was added to the Vero cells and the plate was incubated overnight at 37°C in a 5% humidified incubator. The next day, plates were viewed at ×10 magnification for cell rounding. The cytotoxic titer was defined as the reciprocal of the highest dilution that produced rounding in 80% of the cells.
Injection of purified toxins.
HIOs were prepared and injected as described above. For these experiments, purified C. difficile toxin A (TcdA) or B (TcdB) from strain VPI 10463 was purchased (152C or 155D, respectively; List Biological Laboratories), reconstituted at a concentration of 1 μg/ml, aliquoted, and stored at −80°C. Due to lot-to-lot variation, each new lot was tested for toxin activity using the Vero cell cytotoxicity assay. Each HIO was injected with approximately 2 μl of either 12.8 ng/μl TcdA or 25.6 ng/μl TcdB with 1 mM FD4. For each experiment, a new aliquot of toxin was thawed and used.
Determining pixel intensity of FD4 in injected HIOs.
To determine the pixel intensity of the HIOs, ImageJ software was used (14). For each well of HIOs, a bright-field and fluorescent image was taken. These two images of the same well were used to determine pixel intensity. First, the fluorescent image of the well was opened in ImageJ and converted to 16-bit gray scale, and then the bright-field image of the same well was opened in ImageJ and synchronized to the gray-scale image. Using the bright-field image, the perimeter of the HIO in the well was outlined manually, and this region was used to determine the mean gray value of the HIO in the fluorescent image. These steps were repeated for each HIO in the well for all treatments. The percent pixel intensity is defined as the mean gray value of an HIO at a given time point divided by the mean gray value of that same HIO at time 0 (T = 0) multiplied by 100.
Basolateral barrier function assay.
Intact HIOs were placed into wells of a 24-well dish containing 500 μl IGM with growth factors. Purified C. difficile TcdA or TcdB was diluted in IGM. The HIOs were incubated in a well containing 400 ng/ml of either TcdA or TcdB for 2 h at 37°C. After 2 h, the HIOs were exposed to 0.1 mM FD4 for an additional 1 h at 37°C. Negative- and positive-control HIOs were processed in the same manner as toxin-treated HIOs. For positive controls, 2 mM EGTA was added with the FD4 in the final hour of incubation. Following the incubation, HIOs were washed for 5 to 10 min with 1× PBS to remove excess FD4. Following the wash, HIOs were imaged using a fluorescent stereomicroscope (SZX16; Olympus). Disruption of barrier integrity was visualized by the presence of FD4 in the lumen of the HIOs.
Immunofluorescence staining and confocal microscopy.
HIOs collected from the barrier function assays were fixed in 4% paraformaldehyde (PFA) at room temperature for 15 to 30 min and washed 3 times with PBS. HIOs were placed in optimal-cutting-temperature (OCT) compound (4583; Sakura) for 20 min and then placed at −80°C to freeze. Frozen sections were cut at 10 μm for immunostaining, followed by confocal microscopy. Immunostaining was carried out as previously described (6). Antibody information and dilutions can be found in Table S1 in the supplemental material. All immunofluorescence images were taken on a Nikon A1 confocal microscope.
Induction of apoptosis with TNF-α and IFN-γ.
To induce apoptosis, as detected by cleaved caspase 3, HIOs were incubated in 1,000 ng/ml of tumor necrosis factor alpha (TNF-α) (AF-300-01A [PeproTech] or 210-TA-010 [R&D Systems]; cytokines from either company were efficient in this assay) plus 1,000 ng/ml of gamma interferon (IFN-γ) (AF-300-02; PeproTech) for 24 h. HIOs with disrupted barrier function were fixed, embedded, and sectioned as described above.
Image processing and manipulation.
Minimal image processing was used. Image manipulation was limited to uniform changes of brightness and/or contrast and to the use of a digital zoom in the inset of Fig. 3B.
FIG 3.
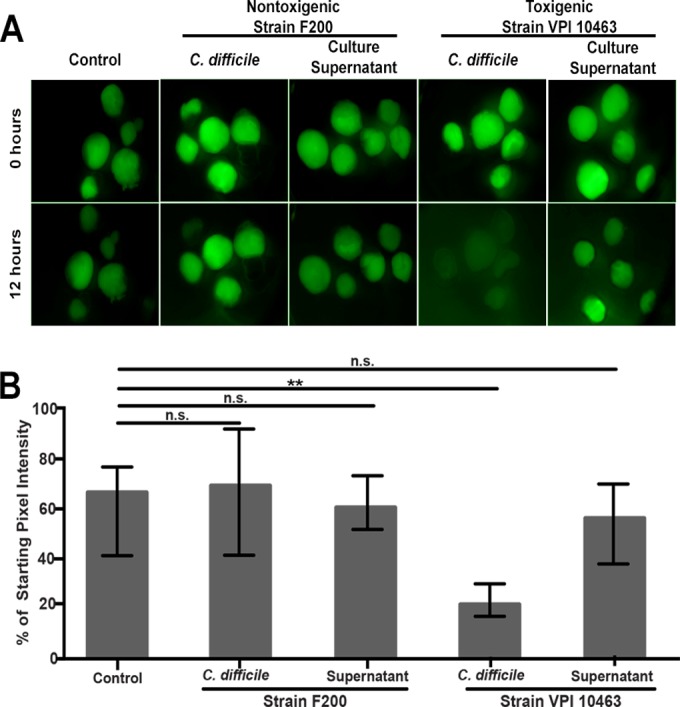
Injection of toxigenic but not nontoxigenic C. difficile results in loss of HIO barrier function. (A) HIOs were injected with FD4 alone (control) (n = 5), a nontoxigenic strain (F200) of C. difficile (n = 5), filtered culture supernatant from that strain (n = 6), a toxigenic C. difficile strain (VPI 10463) (n = 5), or filtered culture supernatant from the toxigenic strain (n = 5). Only VPI 10463-injected HIOs lost barrier function. Images are representative of three independent experiments using 5 to 6 HIOs per group. (B) Quantification of epithelial barrier disruption by measuring retention of injected FD4 after 12 h. Bars represent the medians and the interquartile ranges. Only the HIOs colonized with the toxigenic strain (VPI 10463) lost a significantly greater amount of FD4 from the lumen, indicating significant epithelial damage (P = 0.0079 by Mann-Whitney test).
Statistical analysis.
Statistical analysis was performed using Prism 6 GraphPad software. The Mann-Whitney test was used to compare each group to the control. Statistical significance was set at a P value of <0.05.
RESULTS
HIOs have a functional epithelium with an intact paracellular barrier.
Beyond nutrient absorption, a key function of the gastrointestinal epithelium is as a barrier that prevents free passage of antigens from the lumen of the gastrointestinal tract to the rest of the body (15). To assess if HIOs possess a functional epithelial barrier, we microinjected FD4 into the lumen of HIOs and monitored fluorescence over the course of 18 h. FD4 is used to examine the permeability of the epithelial paracellular barrier in a variety of in vitro-polarized monolayers and in vivo assays (16, 17). In this assay, an intact barrier confines the FD4 in the HIO lumen, maintaining green fluorescence, while the loss of paracellular barrier integrity results in the measurable loss of fluorescence due to the diffusion of FD4 out of the lumen. We observed that FD4-injected HIOs maintained fluorescence over the course of 18 h (n = 5) (Fig. 1A).
FIG 1.
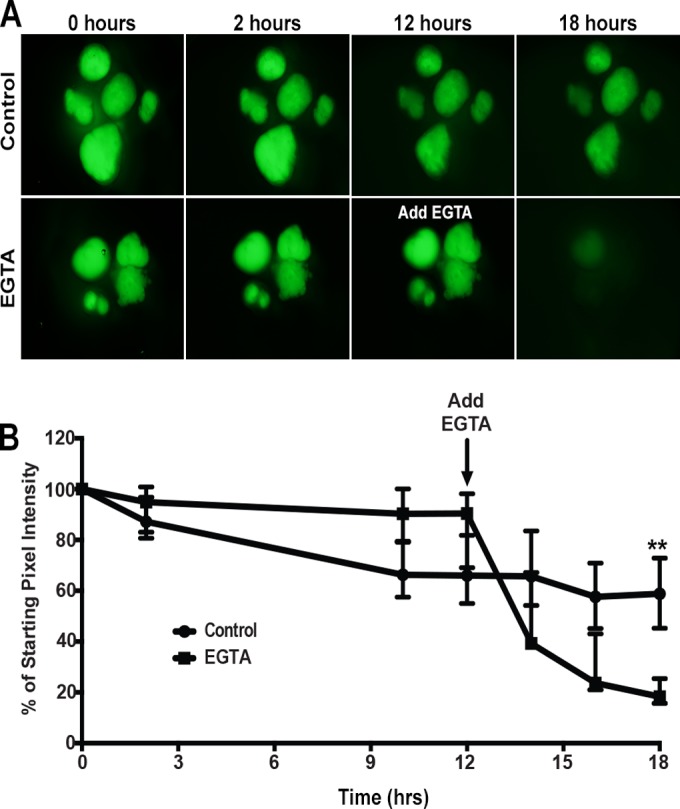
Assessment of HIO epithelial barrier function. (A) Representative images of FD4 dynamics in HIOs following injection. HIOs (n = 5 per treatment) were injected with FD4 and imaged at 0, 2, 10, 12, 14, 16, and 18 h postinjection. HIOs retained the majority of injected FD4 within the lumen over 18 h. The addition of EGTA to the media 12 h after injection resulted in the rapid loss of FD4 from the lumen, indicating the loss of epithelial paracellular barrier function. Images represent the results from at least three independent experiments. (B) Quantitation of barrier disruption by determination of the fraction of initial FD4 fluorescence retained over time. Points represent the medians and bars represent the interquartile ranges. Eighteen hours after injection, control HIOs retained significantly more FD4 than the EGTA-treated HIOs (P = 0.0079 by Mann-Whitney test).
To quantitate barrier function over time, we measured the fluorescence of each HIO throughout the assay and compared the fluorescence at each time point to the initial fluorescence at time zero (T = 0). The fluorescence of each HIO was defined as the pixel intensity per HIO, normalized to the area of each HIO (pixel intensity/area). Eighteen hours following the injection of FD4, HIOs maintained a median of 58.8% of the fluorescence observed at T = 0. As a control, we added 2 mM EGTA, a calcium chelator known to disrupt tight junctions (TJs) and adherens junctions, to the culture media 12 h after injecting HIOs (n = 5) with FD4. HIOs in the EGTA treatment group maintained a median of 90.4% of the fluorescence at 12 h post-FD4 injection. However, the addition of EGTA at this point resulted in a rapid loss of fluorescence such that the median fluorescence of the HIOs at T = 18 was 18.3% of the intensity at T = 0 (Fig. 1B). By the conclusion of the experiment at 18 h post-FD4 injection, there was a statistically significant loss of pixel intensity in the EGTA-treated HIOs compared to control HIOs. These results strongly support the notion that HIOs have a robust paracellular epithelial barrier that can be disrupted by chemical means and also demonstrate that FD4 can be used to measure paracellular barrier function over time.
To confirm these results, we also performed a second outside-in barrier function experiment (see Fig. S1 in the supplemental material). In this experiment, we added FD4 to the tissue culture media and reasoned that if the paracellular barrier was disrupted, a leak would occur from the basal-to-apical (luminal) direction in addition to the apical-to-basal leak demonstrated in Fig. 1. Indeed, when control HIOs (n = 10) were incubated in FD4, an intact barrier did not permit FD4 to diffuse into the HIO lumen. In contrast, when EGTA was added to the media, 100% of HIOs had a disrupted epithelial barrier, and FD4 was observed in the lumen after 1 h (n = 12 individual HIOs) (see Fig. S1 in the supplemental material). These results indicate that HIOs have a robust paracellular barrier that restricts movement across the epithelium in both the luminal/basolateral and basolateral/luminal direction.
Viable C. difficile persists in the lumen of HIOs and damages the epithelium.
To investigate the interaction between C. difficile and human epithelium, we microinjected C. difficile strain VPI 10463 into the lumen of HIOs. To assess the viability of vegetative C. difficile in HIOs, injected organoids were mechanically disrupted and cultured under anaerobic conditions. Immediately following the injection (T = 0), the mean vegetative CFU per HIO (CFU/HIO) was 2 × 104 (Fig. 2A). By 2 h postinjection, the mean CFU/HIO ratio had decreased, as some of the initial inoculum died. Twelve hours postinjection, the mean CFU/HIO ratio was 1 × 102, suggesting that viable, vegetative C. difficile was able to persist within the HIOs for at least 12 h.
FIG 2.

Vegetative C. difficile persists in the HIO lumen. (A) HIOs injected with either C. difficile strain VPI 10463 or F200 were collected at 0, 2, and 12 h postinjection and plated on BHI with cysteine to quantitate the number of vegetative CFU per HIO. C. difficile was able to persist in the lumen of HIOs for 12 h. Points on the graph represent individual HIOs, and the dashed line represents the limit of detection. Bars indicate the means and SEM. (B) Twelve hours after colonization, HIOs injected with C. difficile were fixed and stained using hematoxylin and eosin. The epithelium of the HIO colonized with strain VPI 10463 is severely disrupted, and large rods with what appear to be subterminal spores are visible (inset) within the HIO (inset is a 3.25× digital zoom of the boxed region). (C) An HIO colonized with the nontoxigenic strain F200 has an intact epithelium despite the presence of large rod-shaped bacteria in the lumen of the HIO.
Histopathologic examination of HIOs 12 h postinjection revealed the epithelium was markedly disrupted, with sloughing of cells with pyknotic nuclei into the lumen (Fig. 2B). In addition, there were numerous rod-shaped bacteria in the lumen with what appear to be subterminal bacterial spores.
Epithelial damage is the hallmark of C. difficile infection in the intestine, and this is mediated by the toxin A and B (TcdA and TcdB, respectively) virulence factors. To investigate if the epithelial damage observed in HIOs colonized with VPI 10463 was due to toxin, we microinjected a nontoxigenic C. difficile strain (F200). As with VPI 10463, viable F200 persisted within the HIO for 12 h. However, the epithelium of HIOs colonized with F200 appeared intact (Fig. 2C).
These results suggest that VPI 10463 produces toxin in the HIO lumen. However, it is known that C. difficile can produce and release toxin into the culture media during in vitro growth. Using the barrier function assay as a readout of epithelial damage, we determined if the toxigenic strain VPI 10463 was producing toxin within the HIOs. HIOs injected with VPI 10463 lost barrier integrity by 12 h postinjection, resulting in 22% of the starting fluorescence (Fig. 3A and B). Importantly, barrier function was maintained in HIOs injected with the nontoxigenic F200 isolate of C. difficile. Similar to our previous observations (Fig. 1), 12 h after the injection, the control HIOs (n = 5) maintained barrier integrity as visualized by the presence of green fluorescence, retaining 66% of the initial fluorescence. As a control for the release of toxins into culture supernatant during in vitro growth, HIOs were injected with a volume of filter-sterilized bacterial culture supernatant equal to that used for the injection of intact bacteria. Filtered supernatant from VPI 10463 or F200 culture media had no effect on HIO barrier function (Fig. 3A and B). We did, however, measure detectable cytotoxicity in VPI 10463 supernatant using an in vitro Vero cell cytotoxicity assay, suggesting that this strain released toxin into the supernatant but that quantities were not sufficient to disrupt barrier function within the 12-h time frame of our assay (Fig. 3; also see Fig. S2 in the supplemental material).
These results suggest that viable C. difficile persists within the lumen of HIOs and that the epithelial damage associated with VPI 10463 is due to the in situ production of toxin.
Purified C. difficile toxins disrupt HIO paracellular barrier function.
Epithelial damage and loss of barrier function in HIOs injected with C. difficile was dependent on the ability of the strain to produce toxin. Since F200 is a naturally occurring nontoxigenic strain and not an isogenic mutant of VPI 10463, we directly determined the effect of TcdA or TcdB purified from VPI 10463 on HIO barrier function. FD4 was combined with purified TcdA or TcdB, and the mixture was injected into the lumen of HIOs. Compared to controls in which 100% of HIOs (n = 5) retained barrier function, TcdA had a strong effect that resulted in a loss of barrier function in 100% of HIOs (n = 5), while TcdB had a less potent effect on the HIOs and did not appear to robustly disrupt the epithelial barrier (n = 5) (Fig. 4A). Importantly, at the same concentration, TcdA and TcdB had similar cytotoxicity on Vero cells in vitro (see Fig. S3 in the supplemental material). When we quantitated the relative fluorescence over time, HIOs injected with TcdA lost a greater amount of fluorescence than HIOs injected with either TcdB or FD4 alone (Fig. 4B).
FIG 4.
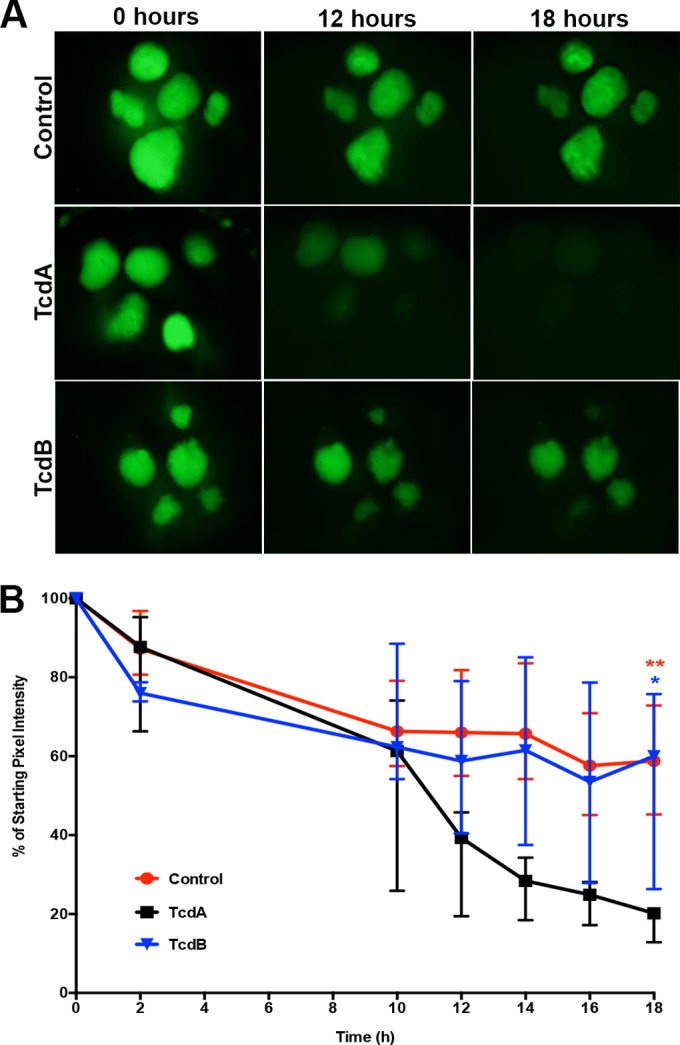
Purified TcdA and TcdB injected into the lumen of HIOs disrupt paracellular barrier function. (A) Representative images of FD4 leakage from the lumen of HIOs treated with C. difficile toxin. HIOs (n = 5 per treatment) were injected with FD4 alone (top), with purified TcdA (middle), or with TcdB (bottom). In this system, TcdA is more potent than TcdB. Images are representative of at least three independent experiments using 5 to 6 HIOs per group. (B) Quantitation of fluorescence of each HIO relative to time zero. Injection of purified TcdA into HIOs causes significantly greater loss of paracellular barrier function than injection with either TcdB (blue asterisk, P = 0.0159) or FD4 alone (red asterisks, P = 0.0079). Points represent the median percentages, and bars represent the interquartile ranges. The data were analyzed using the Mann-Whitney test. Control data presented in panels A and B are the same as those used in Fig. 1, as these assays were preformed at the same time.
C. difficile toxins are known to inactivate the Rho family GTPases, leading to the disruption of the cytoskeleton and cellular junctions. To determine if these effects were seen in the epithelium of HIOs treated with C. difficile toxins, we examined several junctional and cytoskeletal proteins. These include proteins of the adherens junctions and tight junctions, which are crucial for the maintenance of epithelial barrier function (18). In controls, E-cadherin, a cellular transmembrane adherens junction protein, is localized to the basolateral surfaces of epithelial cells and is absent from the apical surface (Fig. 5A, top). In HIOs injected with TcdA, E-cadherin is redistributed and can be seen on the apical surface of the epithelium, where, as in HIOs injected with TcdB, E-cadherin localization does not appear to be different from that of controls (Fig. 5A, middle and bottom). In addition to E-cadherin, we also examined the cellular localization of zonula occludens protein 1 (ZO-1) and occludin (OCLN), both components of cellular TJs. In control HIOs, ZO-1 is present at the TJ near the apical surface of the epithelium, whereas OCLN is seen at the TJ and along the lateral surface of the cell (Fig. 5B, top). Similar to what was observed with E-cadherin, TcdA-treated HIOs had dramatically redistributed tight junction proteins, whereas TcdB-treated HIOs were not different from controls (Fig. 5B, middle and bottom). Reports by others have demonstrated that OCLN is internalized via endocytosis upon disruption of the TJ and can be visualized in endocytic vesicles. We did not observe obvious endocytic vesicles containing OCLN; however, this likely is due to differences in tissue processing conditions that are required to visualize vesicles (19). Lastly, we used phalloidin staining to assess the organization of F-actin and acetylated alpha tubulin (AcTub) immunofluorescence to visualize stabilized microtubules within the cell (Fig. 5C). In controls, F-actin was strongly localized to the apical surface of the epithelium, and weak staining was seen along the lateral and basal surface of the epithelium (Fig. 5C, top). Similarly, AcTub staining was strongest on the apical side of the cell but also was weakly present throughout the cell and on the basal surface (Fig. 5C, top). Compared to FD4-injected controls, HIOs treated with TcdA had a marked reduction of F-actin with areas where staining was undetectable. In addition, HIOs treated with TcdA displayed a severe disruption of AcTub at the apical border of the epithelial cells (Fig. 5, middle), consistent with previous reports (20). In contrast, TcdB-treated HIOs showed F-actin distribution and apical AcTub staining that was similar to that of the controls, whereas AcTub immunofluorescence on the basal side of the cells appeared to be disrupted, indicating that TcdB had a mild effect on the basal side of the HIO epithelium (Fig. 5C, bottom). In conclusion, the examination of the cellular effects of purified C. difficile toxin on HIO epithelium demonstrated that this model recapitulates the hallmark effects of toxin host epithelium.
FIG 5.
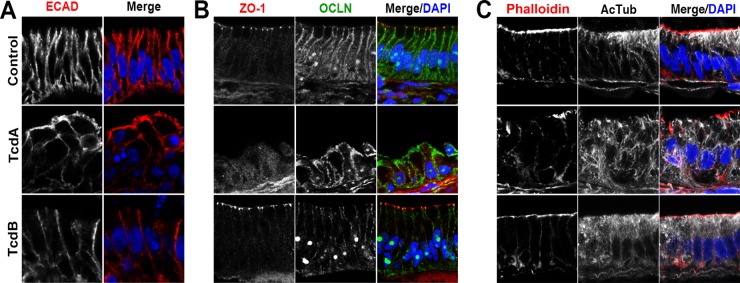
Cellular effects of injection of HIOs with TcdA or TcdB. HIOs were injected with FD4 alone (control), TcdA, or TcdB and monitored for disruption of barrier function. At 18 h postinjection, the HIOs were fixed and stained. (A) The normal basolateral distribution of the adherens junction protein E-cadherin (ECAD) is disrupted following injection of TcdA. In these HIOs, ECAD is redistributed and can be seen on the apical surface of the epithelium. HIOs injected with TcdB maintain a basolateral distribution of ECAD similar to that of the controls. (B) Altered localization of TJ proteins ZO-1 and OCLN following injection with TcdA. In control HIOs, ZO-1 is present at the apical surface of the epithelium, whereas OCLN is seen at the lateral surface of the cell. In HIOs injected with TcdA, apical ZO-1 at TJs is lost and OCLN no longer is restricted to the lateral surface, while TcdB-injected HIOs have ZO-1 and OCLN immunofluorescence similar to those of the control. DAPI, 4′,6-diamidino-2-phenylindole. (C) HIOs were stained with phalloidin to assess the organization of F-actin and acetylated alpha tubulin (AcTub) to visualize stabilized microtubules. In control HIOs, F-actin is strongly localized to the apical surface of the epithelium, while AcTub immunofluorescence is strongest on the apical and basal sides of the cell. Compared to control HIOs injected with TcdA, epithelial cells displayed a reduction of F-actin staining, with areas where staining was undetectable, and showed a severe disruption of AcTub at the apical border. In contrast, in HIOs injected with TcdB, phalloidin staining was similar to that of the controls and AcTub immunofluorescence was mostly similar to that of the controls, with a mild reduction of immunofluorescence on the basal side of the cells.
Basolateral addition of purified toxins disrupts HIO barrier function.
Studies have suggested that TcdA acts to disrupt the localization of tight-junction proteins, which then allows for TcdB to act on the basolateral side of the cell (21). Moreover, a fence-and-gate model recently has been proposed whereby C. difficile toxins cause a redistribution of basal-lateral proteins to the apical surface so that the bacteria can bind to the cell surface (2). However, the receptors for TcdA and TcdB have not been definitively identified, and it has not been shown conclusively that toxins preferentially affect the apical versus basal surfaces of the epithelium. Therefore, to test a differential effect of apical (Fig. 5) versus basal exposure to toxin, we added FD4 along with TcdA or TcdB to the tissue culture media in order to expose HIOs to toxin on the basal side of the epithelium (Fig. 6). In this experiment, since FD4 was added outside the HIO, inward leak and fluorescence inside the HIO lumen indicates a disrupted epithelial barrier. Consistent with apical exposure (Fig. 5) and compared to controls, TcdA again was more potent than TcdB at disrupting barrier function (Fig. 6). While barrier function remained intact in 100% of control HIOs (n = 10), TcdA disrupted barrier function in 100% of HIOs (n = 13) and the barrier was disrupted in only 23% of TcdB-treated HIOs (n = 22). Importantly, barrier disruption in toxin-treated HIOs was not due to apoptosis (see Fig. S4 in the supplemental material).
FIG 6.
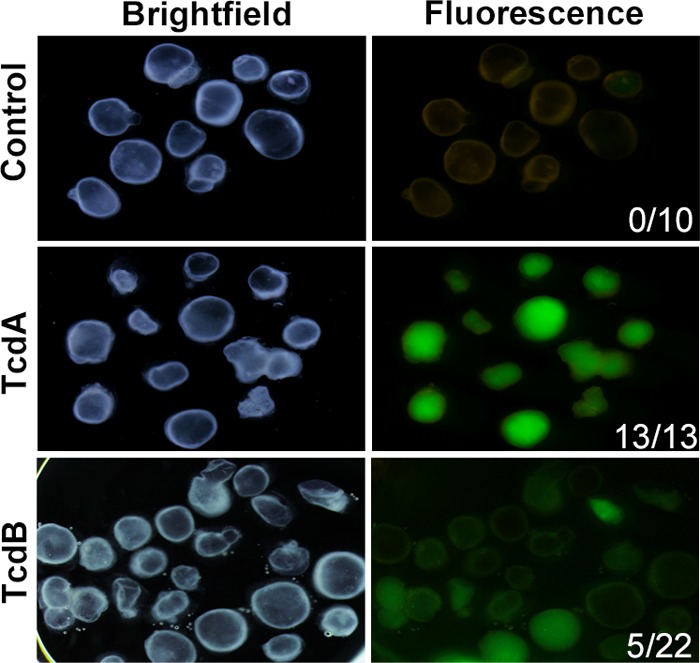
Basolateral exposure to purified toxins causes loss of barrier function. Purified TcdA or TcdB was added to tissue culture media containing HIOs, followed by the addition of FD4. In this assay, the loss of paracellular barrier function was indicated by diffusion of FD4 into the lumen of HIOs, while HIOs with an intact epithelium excluded FD4. Bright-field images show the location of the HIOs. Images taken under fluorescent light indicate barrier function status. None (0/10) of the untreated HIOs lost barrier function, whereas 100% (13/13) of HIOs treated with TcdA lost barrier function, while only 23% of HIOs treated with TcdB lost barrier function. The results shown are representative of five independent experiments.
Taken together, these results indicate that the C. difficile toxins TcdA and TcdB have the ability to interact with both the apical and basolateral aspects of the epithelium of human intestinal organoids.
DISCUSSION
The pathogenesis of CDI is multifaceted and involves interactions between the host, the gut microbiota, and C. difficile (22–24). In order to study the effect of C. difficile colonization on human epithelium, we developed techniques to colonize the HIO lumen with viable vegetative C. difficile. The vegetative cell is sensitive to oxygen, leading to the concept that the environmentally stable spore is responsible for transmission (25). Our data suggest that the vegetative cell can be tolerant of oxygen in the lumen of HIOs, as viable C. difficile persists despite cultivating the HIOs in ambient oxygen conditions. While it may be surprising that C. difficile can persist in HIOs grown at ambient oxygen levels, many anaerobes can tolerate oxygen under conditions that limit the toxicity of reactive oxygen species (26, 27). Preliminary measurements using an oxygen microsensor indicate that the lumen of HIOs has lower than ambient oxygen levels, with the luminal oxygen concentrations ranging from 5 to 15% (data not shown). A recent report suggests that some strains of C. difficile are able to grow even at these high levels of oxygen (28).
Recently, others have utilized mouse-derived enteroids (epithelium-only organoids) to study host-microbe interactions (29, 30). A key difference between those reports and the data presented here is that our system utilizes organoids derived from human cells rather than mouse intestinal crypts. In addition, this is the first report demonstrating that organoids can be colonized with an obligate anaerobe.
A caveat of our model is that while HIOs are more similar to the human small intestine, CDI manifests clinically as colitis. However, the small intestinal epithelium is relevant to the study of C. difficile pathogenesis, as patients with ileal pouch anal anastomoses will develop clinical CDI. Additionally, animal ileal loop models historically have been used to understand the pathogenesis of this infection with respect to the activity of the toxins on the host.
Many previous studies of C. difficile virulence and pathogenesis have focused on the glucosyltransferase toxins TcdA and TcdB. Depending on which system has been used to study these toxins, the relative activities of TcdA and TcdB differ. Previous studies in cell culture lines demonstrate that TcdB has greater cytotoxicity than TcdA (31, 32). However, TcdA has been reported to be more potent than TcdB in animal models, which recapitulate the diverse cell types and structure of the gastrointestinal tract (33, 34). The greater activity of TcdA in the organoid may reflect the fact that the organoid epithelium responds more like the intestinal epithelium in situ instead of like in vitro-cultured cell lines. One caveat to consider in light of our observations is the growth conditions for the HIOs, which include media containing EGF, Noggin, and R-Spondin2. Previous studies have shown the EGF can reduce TcdA- and TcdB-induced damage in the colonic mucosa (35). Thus, it is possible that EGF present in the media is able to attenuate the effect of one toxin (TcdB) more than another (TcdA), a possibility that will be tested in the future. However, our finding that TcdA caused greater disruption of HIO epithelial barrier function than TcdB is consistent with the finding that TcdA (but not TcdB) can inactivate the Ras family GTPase Rap, which regulates cell-cell junctions (36). Our FD4-based assay measures the disruption of intercellular junctions, which allows paracellular leak and is consistent with the described activity of TcdA. Finally, our results showing that TcdA can disrupt barrier function when added apically and that both TcdA and TcdB can disrupt barrier function when exposed to the basolateral surface demonstrate that toxins can interact with receptors on both surfaces. As yet, the definitive receptor for either of these toxins is not known; however, the basolateral activity of toxins have been reported previously, so it is possible that receptors exist on both apical and basolateral cell surfaces (37). The HIO system may represent a new avenue to search for this important molecular target.
The gastrointestinal epithelium is an interface between the host and the environment and is crucial for many aspects of health, including nutrient absorption, maintenance of immune homeostasis, and forming a selective barrier against antigens (38, 39). Defects in intestinal epithelial barrier function have been associated with the pathogenesis of inflammatory bowel diseases, celiac disease, and enteric infections (40). Here, we demonstrated that HIOs have an intact polarized epithelium with paracellular barrier function, which can be used for detailed, real-time studies of both normal physiological barrier function as well as barrier dysfunction in the context of chemical perturbations or infection. As such, this system represents the first robust three-dimensional, nontransformed, primary human intestinal system to study the effects of C. difficile infection and will be a valuable tool to study epithelial barrier defects in a variety of injury and disease contexts.
Supplementary Material
ACKNOWLEDGMENTS
We thank Thomas Schmidt and Dishari Mukherjee for assistance with measuring oxygen in the lumen of the HIOs. In addition, we are grateful to Matthew Jenior for helpful discussions.
This work was funded by NIH grants 5T32AI007528 (J.L.L.), K01DK091415 (J.R.S.), P30DK034933 (Michigan Gastrointestinal Peptide Research Center), and U19 AI090871 and R01GM0099549 (V.B.Y.).
J.R.S. is the coinventor on a patent for generating intestinal organoids from human pluripotent stem cells. All other authors have no disclosures.
J.L.L., S.H., J.S.O., M.S.N., V.B.Y., and J.R.S. contributed to the design and analysis of experiments. M.K. generated critical reagents required to carry out experiments. J.L.L., S.H., J.S.O., and M.S.N. performed the experiments. J.L.L., S.H., V.B.Y., and J.R.S. wrote the manuscript.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.02561-14.
REFERENCES
- 1.Hall AJ, Curns A, McDonald L, Parashar U, Lopman B. 2012. The roles of Clostridium difficile and norovirus among gastroenteritis-associated deaths in the United States, 1999-2007. Clin Infect Dis 55:216–223. doi: 10.1093/cid/cis386. [DOI] [PubMed] [Google Scholar]
- 2.Kasendra M, Barrile R, Leuzzi R, Soriani M. 2013. Clostridium difficile toxins facilitate bacterial colonization by modulating the fence and gate function of colonic epithelium. J Infect Dis 209:1095–1104. doi: 10.1093/infdis/jit617. [DOI] [PubMed] [Google Scholar]
- 3.Goulding D, Thompson H, Emerson J, Fairweather NF, Dougan G, Douce GR. 2009. Distinctive profiles of infection and pathology in hamsters infected with Clostridium difficile strains 630 and B1. Infect Immun 77:5478–5485. doi: 10.1128/IAI.00551-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Theriot CM, Koumpouras CC, Carlson PE, Bergin II, Aronoff DM, Young VB. 2011. Cefoperazone-treated mice as an experimental platform to assess differential virulence of Clostridium difficile strains. Gut Microbes 2:326–334. doi: 10.4161/gmic.19142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lizer JT, Madson DM, Schwartz KJ, Harris H, Bosworth BT, Kinyon JM, Ramirez A. 2013. Experimental infection of conventional neonatal pigs with Clostridium difficile: a new model. J Swine Health Prod 21:22–29. [Google Scholar]
- 6.Spence JR, Mayhew CN, Rankin SA, Kuhar MF, Vallance JE, Tolle K, Hoskins EE, Kalinichenko VV, Wells SI, Zorn AM, Shroyer NF, Wells JM. 2011. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature 470:105–109. doi: 10.1038/nature09691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Du A, McCracken KW, Walp ER, Terry NA, Klein TJ, Han A, Wells JM, May CL. 2012. Arx is required for normal enteroendocrine cell development in mice and humans. Dev Biol 365:175–188. doi: 10.1016/j.ydbio.2012.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Finkbeiner SR, Zeng X-L, Utama B, Atmar RL, Shroyer NF, Estes MK. 2012. Stem cell-derived human intestinal organoids as an infection model for rotaviruses. mBio 3:e00159-12. doi: 10.1128/mBio.00159-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xue X, Ramakrishnan S, Anderson E, Taylor M, Zimmermann EM, Spence JR, Huang S, Greenson JK, Shah YM. 2013. Endothelial PAS domain protein 1 activates the inflammatory response in the intestinal epithelium to promote colitis in mice. Gastroenterology 145:831–841. doi: 10.1053/j.gastro.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuehne SA, Cartman ST, Heap JT, Kelly ML, Cockayne A, Minton NP. 2010. The role of toxin A and toxin B in Clostridium difficile infection. Nature 467:711–713. doi: 10.1038/nature09397. [DOI] [PubMed] [Google Scholar]
- 11.McCracken KW, Howell JC, Wells JM, Spence JR. 2011. Generating human intestinal tissue from pluripotent stem cells in vitro. Nat Protoc 6:1920–1928. doi: 10.1038/nprot.2011.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sugawara S, Ito T, Sato S, Sato Y, Kasuga K, Kojima I, Kobayashi M. 2014. Identification of site-specific degradation in bacterially expressed human fibroblast growth factor 4 and generation of an aminoterminally truncated, stable form. Appl Biochem Biotechnol 172:206–215. doi: 10.1007/s12010-013-0544-0. [DOI] [PubMed] [Google Scholar]
- 13.Bell SM, Schreiner CM, Wert SE, Mucenski ML, Scott WJ, Whitsett JA. 2008. R-spondin 2 is required for normal laryngeal-tracheal, lung and limb morphogenesis. Development 135:1049–1058. doi: 10.1242/dev.013359. [DOI] [PubMed] [Google Scholar]
- 14.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peterson LW, Artis D. 2014. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol 14:141–153. doi: 10.1038/nri3608. [DOI] [PubMed] [Google Scholar]
- 16.Bergstrom KS, Kissoon-Singh V, Gibson DL, Ma C, Montero M, Sham HP, Ryz N, Huang T, Velcich A, Finlay BB, Chadee K, Vallance BA. 2010. Muc2 protects against lethal infectious colitis by disassociating pathogenic and commensal bacteria from the colonic mucosa. PLoS Pathog 6:e1000902. doi: 10.1371/journal.ppat.1000902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sappington PL, Yang R, Yang H, Tracey KJ, Delude RL, Fink MP. 2002. HMGB1 B box increases the permeability of Caco-2 enterocytic monolayers and impairs intestinal barrier function in mice. Gastroenterology 123:790–802. doi: 10.1053/gast.2002.35391. [DOI] [PubMed] [Google Scholar]
- 18.Bruewer M, Hopkins AM, Hobert ME, Nusrat A, Madara JL. 2004. RhoA, Rac1, and Cdc42 exert distinct effects on epithelial barrier via selective structural and biochemical modulation of junctional proteins and F-actin. Am J Physiol Cell Physiol 287:C327–335. doi: 10.1152/ajpcell.00087.2004. [DOI] [PubMed] [Google Scholar]
- 19.Shen L, Turner JR. 2005. Actin Depolymerization Disrupts Tight Junctions via Caveolae-mediated Endocytosis. Mol Biol Cell 16:3919–3936. doi: 10.1091/mbc.E04-12-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nam HJ, Kang JK, Kim S-K, Ahn KJ, Seok H, Park SJ, Chang JS, Pothoulakis C, Lamont JT, Kim H. 2010. Clostridium difficile toxin A decreases acetylation of tubulin, leading to microtubule depolymerization through activation of histone deacetylase 6, and this mediates acute inflammation. J Biol Chem 285:32888–32896. doi: 10.1074/jbc.M110.162743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Du T, Alfa MI. 2004. Translocation of Clostridium difficile toxin B across polarized Caco-2 cell monolayers is enhanced by toxin A. Can J Infect Dis 15:83–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Madan R, Guo X, Naylor C, Buonomo EL, Mackay D, Noor Z, Concannon P, Scully KW, Pramoonjago P, Kolling GL, Warren CA, Duggal P, Petri WA. 2013. Role of leptin-mediated colonic inflammation in defense against Clostridium difficile colitis. Infect Immun 82:341–349. doi: 10.1128/IAI.00972-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reeves AE, Theriot CM, Bergin IL, Huffnagle GB, Schloss PD, Young VB. 2010. The interplay between microbiome dynamics and pathogen dynamics in a murine model of Clostridium difficile infection. Gut Microbes 2:145–158. doi: 10.4161/gmic.2.3.16333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Britton RA, Young VB. 2012. Interaction between the intestinal microbiota and host in Clostridium difficile colonization resistance. Trends Microbiol. 20:313–319. doi: 10.1016/j.tim.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deakin LJ, Clare S, Fagan RP, Dawson LF, Pickard DJ, West MR, Wren BW, Fairweather NF, Dougan G, Lawley TD. 2012. The Clostridium difficile spo0A gene is a persistence and transmission factor. Infect Immun 80:2704–2711. doi: 10.1128/IAI.00147-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rolfe RD, Hentges DJ, Campbell BJ, Barrett JT. 1978. Factors related to the oxygen tolerance of anaerobic bacteria. Appl Environ Microbiol 36:306–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brusa T, Canzi E, Pacini N, Zanchi R, Ferrari A. 1989. Oxygen tolerance of anaerobic bacteria isolated from human feces. Curr Microbiol 19:39–43. doi: 10.1007/BF01568901. [DOI] [Google Scholar]
- 28.Green L, Worthington T, Hilton A, Lambert P. 2012. The effect of oxygen on the growth characteristics and virulence of Clostridium difficile, abstr R2411 Abstr 22nd Eur. Cong. Clin. Microbiol. Infect. Dis. [Google Scholar]
- 29.Lukovac S, Belzer C, Pellis L, Keijser BJ, de Vos WM, Montijn RC, Roeselers G. 2014. Differential modulation by Akkermansia muciniphila and Faecalibacterium prausnitzii of host peripheral lipid metabolism and histone acetylation in mouse gut organoids. mBio 5:e01438-14. doi: 10.1128/mBio.01438-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilson SS, Tocchi A, Holly MK, Parks WC, Smith JG. 13 August 2014. A small intestinal organoid model of non-invasive enteric pathogen-epithelial cell interactions. Mucosal Immunol doi: 10.1038/mi.2014.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chaves-Olarte E, Weidmann M, Eichel-Streiber C, Thelestam M. 1997. Toxins A and B from Clostridium difficile differ with respect to enzymatic potencies, cellular substrate specificities, and surface binding to cultured cells. J Clin Investig 100:1734–1741. doi: 10.1172/JCI119698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Riegler M, Sedivy R, Pothoulakis C, Hamilton G, Zacherl J, Bischof G, Cosentini E, Feil W, Schiessel R, LaMont JT. 1995. Clostridium difficile toxin B is more potent than toxin A in damaging human colonic epithelium in vitro. J Clin Investig 95:2004–2011. doi: 10.1172/JCI117885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hirota SA, Iablokov V, Tulk SE, Schenck LP, Becker H, Nguyen J, Al Bashir S, Dingle TC, Laing A, Liu J, Li Y, Bolstad J, Mulvey GL, Armstrong GD, MacNaughton WK, Muruve DA, MacDonald JA, Beck PL. 2012. Intrarectal instillation of Clostridium difficile toxin A triggers colonic inflammation and tissue damage: development of a novel and efficient mouse model of Clostridium difficile toxin exposure. Infect Immun 80:4474–4484. doi: 10.1128/IAI.00933-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Triadafilopoulos G, Pothoulakis C, O'Brien MJ, LaMont JT. 1987. Differential effects of Clostridium difficile toxins A and B on rabbit ileum. Gastroenterology 93:273–279. [DOI] [PubMed] [Google Scholar]
- 35.Riegler M, Sedivy R, Sogukoglu T, Castagliuolo I, Pothoulakis C, Cosentini E, Bischof G, Hamilton G, Teleky B, Feil W, Lamont JT, Wenzl E. 1997. Epidermal growth factor attenuates Clostridium difficile toxin A- and B-induced damage of human colonic mucosa. Am J Physiol 273:G1014–G1022. [DOI] [PubMed] [Google Scholar]
- 36.Pruitt RN, Chumbler NM, Rutherford SA, Farrow MA, Friedman DB, Spiller B, Lacy DB. 2012. Structural determinants of Clostridium difficile toxin A glucosyltransferase activity. J Biol Chem 287:8013–8020. doi: 10.1074/jbc.M111.298414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nusrat A, von Eichel-Streiber C, Turner JR, Verkade P, Madara JL, Parkos CA. 2001. Clostridium difficile toxins disrupt epithelial barrier function by altering membrane microdomain localization of tight junction proteins. Infect Immun 69:1329–1336. doi: 10.1128/IAI.69.3.1329-1336.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Groschwitz KR, Hogan SP. 2009. Intestinal barrier function: molecular regulation and disease pathogenesis. J Allerg Clin Immunol 124:3. doi: 10.1016/j.jaci.2009.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Artis D. 2008. Epithelial-cell recognition of commensal bacteria and maintenance of immune homeostasis in the gut. Nat Rev Immunol 8:411–420. doi: 10.1038/nri2316. [DOI] [PubMed] [Google Scholar]
- 40.Catalioto RM, Maggi CA, Giuliani S. 2011. Intestinal epithelial barrier dysfunction in disease and possible therapeutical interventions. Curr Med Chem 18:398–426. doi: 10.2174/092986711794839179. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.