

Abstract

A saturation strategy focused on improving the selectivity and physicochemical properties of ATR inhibitor HTS hit 1 led to a novel series of highly potent and selective tetrahydropyrazolo[1,5-a]pyrazines. Use of PI3Kα mutants as ATR crystal structure surrogates was instrumental in providing cocrystal structures to guide the medicinal chemistry designs. Detailed DMPK studies involving cyanide and GSH as trapping agents during microsomal incubations, in addition to deuterium-labeled compounds as mechanistic probes uncovered the molecular basis for the observed CYP3A4 TDI in the series.
Keywords: ATR, structure-based drug design, Fsp3, deuterium, CYP3A4 TDI
Ataxia telangiectasia and Rad-3 related protein (ATR) is a member of the phosphatidylinositol 3-kinase-related protein kinase (PIKK) family, which also includes ataxia telangiectasia mutated (ATM) and DNA-dependent protein kinase (DNA-PK). All three are involved in a complex network of surveillance mechanisms that maintain genomic integrity in the face of various genomic insults.1−4 However, ATR is the primary responder to replication stress caused by exposure to DNA damaging agents that interfere with DNA replication, such as the antimetabolite class, including gemcitabine and hydroxyurea.5 In response to the DNA damage, ATR is activated and regulates the S and G2 checkpoint allowing for DNA repair, thus reducing the effectiveness of the cytotoxic agents. Because of mutations in p53, many cancers have a defective G1 checkpoint and, as a result, are hypothesized to be more reliant on the ATR pathway, which initiates the DNA repair.6 Thus, being able to simultaneously inhibit ATR in combination with a DNA damaging agent could potentially restore the cancer’s sensitivity to the cytotoxic agent and provide a genetic patient stratification based on p53 mutational status. Indeed, this chemosensitizer approach has been pursued in the clinic employing Chk1 inhibitors (which is immediately downstream of ATR).7 However, to date, no genetic stratification with Chk1 inhibitors has been clinically evident. Interestingly, our ATR program began as a phenotypic screen where ATR was identified as a target that synergized with cisplatin when it was inhibited in p53 null cancer cell lines. Several small molecule ATR inhibitors have been recently disclosed showing utility both as a chemosensitizer and as a monotherapy.8,9 We describe herein the discovery of novel, potent, and highly selective tetrahydropyrazolo[1,5-a]pyrazines (THPP) as ATR inhibitors, which were morphed from an initial cellular HTS hit.
Our HTS assay consisted of a gemcitabine induced pChk1 cellular assay, where specifically we were looking for compounds that inhibited the phosphorylation on S345. Hits were then run through the ATR biochemical assay to determine which were bone fide ATR inhibitors, while varying the concentration of ATP allowed for an assessment of the potential mechanism of inhibition. One of the initial hits that was deemed to be ATP competitive was the diarylimidazopyridazine 1 (Figure 1).
Figure 1.

Morphing of HTS hit 1 through saturation leads to selective THPP lead series.
As can be seen from the biochemical assays, even though 1 was more potent on PI3Kα than on ATR, with a lipophilic efficiency (LipE) of 4.6, it was deemed a reasonable starting point. Compound 1 was modeled into the ATR homology model built based on the PI3Kδ X-ray structure (Figure 2). The binding model suggested that the imidazolepyridazine core in 1 binds to the hinge region and forms a hydrogen bond with backbone NH of Val2380. The dimethoxyphenyl group sits in the affinity pocket and is aligned by Lys2327 and Asp2335. The model also suggested the possibility to pick up two hydrogen bonds to Lys2327 and Asp2335 through structure modification in this region. A scan of various moieties led to the discovery of a 7-azaindole group as having the best binding affinity, which, after subsequent exploration in the ribose pocket, led to the sub-nM compound 2, that was also very active in the cellular pChk1 target modulation assay (pChk1 cell IC50 = 44 nM). However, despite the increase in target potency, 2 still retained an unacceptable level of PI3Kα activity and had a solubility that was below the lowest level of quantification (BLLOQ) at neutral pH. To this end, to address the poor physicochemical properties as well as provide novelty, we sought to increase the fraction of sp3 hybridized carbons (Fsp3) in the molecule through saturation of one of the rings.10 Compound 3 was prepared, which disappointingly still had poor, albeit measurable, solubility. However, we were very pleased to find that it had a very clean kinase selectivity profile (as judged by hitting none of our internal panel of >60 kinases with an IC50 < 1 μM), yet maintained good ATR biochemical inhibition.
Figure 2.
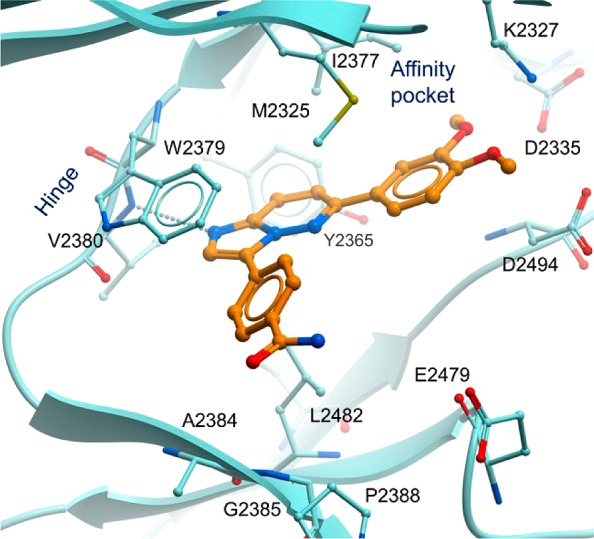
Binding model of compound 1 to ATR.
Since we had been unable to obtain ATR protein of sufficient purity and quality for crystallography, we initially utilized PI3Kα cocrystal structures with our ATR inhibitors to guide our medicinal chemistry designs. Inevitably this led to the situation where once we had dialed out PI3Kα activity, we lost the ability to obtain meaningful crystal structure information. To this end, we decided to stay with the robust PI3Kα system; however, we would mutate key residues into their ATR counterpart and use these “PI3Kα mutants” as an ATR surrogate.11 A combination of PI3Kα cocrystal structures, ATR homology modeling, medicinal chemistry structure–activity relationships (SAR), and pharmacophore models was utilized to identify key residues in the ATP active site. We initially targeted various single mutants across the adenine, affinity, and ribose sites and then followed up with multimutant combinations. One of the initial key mutants that was instrumental in driving our structure-based drug design by providing cocrystal structures of our selective THPP series was the I800M-F930V double mutant. The X-ray structure of this double mutant suggested that there is a small hydrophobic sub pocket at the adenine site, mainly due to the I800M mutation. On the basis of the crystal structure and models, the saturation of the 6-membered ring in 2 offered the correct vector to access this small pocket. Modeling predicted there was sufficient room to append a methyl group to the ring since it would prefer an axial orientation. As can be seen in the cocrystal structure of 4 in the PI3Kα I800M-F930V double mutant (Figure 3), the prediction proved correct with the axial methyl group clearly projecting up making hydrophobic contacts in what is now a deeper, less obstructed pocket. While we only observed a 2-fold increase in ATR potency, more importantly it would appear that the axial methyl group in 4 serves to both further increase selectivity toward PI3Kα due to clashes between the methyl group and I800 of PI3Kα as well as disrupt the crystal packing resulting in a substantial enhancement in solubility.
Figure 3.

Cocrystal structure of 4 with PI3Kα I800M-F930V double mutant (PDB code 4WAF).
As our THPP SAR evolved, we began to observe some disconnects between our inhibitors’ ATR biochemical potency and our cellular mechanistic assay readouts. Since we suspected this may be due to inhibition of one of the other PIKK members such as ATM or DNAPK, we developed the biochemical assays and assayed our THPP series. We found that the THPP series in general also inhibited ATM and DNAPK and had varying degrees of mTOR inhibition.
Interestingly, further saturation of the phenyl ring in compounds such as 5 led to a new subseries such as 6 (Figure 4), which dialed out the other PIKKs (Table 1). The binding model of 6 to ATR (Figure 5) suggested that the methylsulfonylpiperidine group binds in the lower hinge/ribose site region, which is formed by multiple nonconserved residues (e.g., G2385/P/T/T/T, L2482/L/M/M/M, A2384/V/V/D/H, P2388/E/D/A/Q, and E2479/Q/N/S/S; the residues are listed as in ATR/ATM/DNAPK/mTOR/PI3Kα). ATR has a unique small G2385 in this region, which possibly allows the formation of a hydrogen bond from the backbone NH to the sulfonamide oxygen. Unlike ATR, G2385 is replaced by bulky Pro or Thr in other PIKK/PIK family members. G2385 together with other nonconserved residues likely causes this area to have a different shape, size, and electrostatic properties including the hydrogen bonding capacity in ATR vs other members in the PIKK family and hence contribute to the observed selectivity.9 As can been seen from the data in Table 1, in addition to improving the selectivity, this double saturation (where Fsp3 had now increased substantially from 0.1 for HTS hit 1, to 0.5 for 7) was also consistent in suppressing hERG ion channel inhibition as well as improving our physicochemical properties further.
Figure 4.

Selectivity and hERG addressed through double saturation.
Table 1. Selectivity and Property Data for Compounds 5–7.
| compd | ATR IC50 (μM) | ATM IC50 (μM) | DNA-PK IC50 (μM) | mTOR cell IC50 (μM)a | pChk1 IC50 (μM)b | hERG IC50 (μM)c | solubility (μM)d | LogD, pH 7.4e | Caco-2 A–B/B–Af |
|---|---|---|---|---|---|---|---|---|---|
| 5 | 0.0003 | 0.13 | 0.14 | 1.1 | 0.08 | 1.5 | 122 | 2.3 | 9/47 |
| 6 | 0.001 | 14 | 6.5 | >2.3 | 0.16 | >30 | 965 | 2.4 | 10/44 |
| 7 | 0.0004 | 4.3 | 1.6 | 4.4 | 0.037 | 12.5 | 37 | 3.4 | 18/30 |
Subconfluent TSC1 −/– MEF cells were harvested by trypsinization and cultured overnight at 37 °C, 5% CO2. Test compounds were added to the cells for 1 h at 37 °C, 5% CO2. Samples were further processed using the Alpha Screen SureFire p70 S6K (p-Thr389) Assay Kit.
Inhibition of pChk1 Ser-345 in HeLa S3 cells exposed to 1 μM gemcitabine and test compound for 4 h.
Determined using automated whole cell electrophysiology.
Measured using miniaturized shake flask equilibrium solubility assay with PBS buffer at pH7.0.
Measured using direct logD (octanol/water) with PBS buffer at pH 7.4.
Papp(×10–6 cm/s), pH 7.4
Figure 5.

Binding of compound 6 in ATR homology model.
However, as can be seen from the Caco-2 data, compound 6 did suffer from efflux. We found that the addition of a 6-chloro to the azaindole consistently shut down the efflux and increased our cell activity, resulting in the potent and highly selective compound 7 that had an excellent in vitro and sufficient in vivo profile (rat PK of 7 administered at 2.5 mg/kg; CL 57 mL/min/kg, Vss 3.3 L/kg, t1/2 0.86 h, 64% F) to allow us to probe and further understand ATR biology. To this end, compound 7 was deemed a key tool compound, the synthesis of which is shown in Scheme 1.
Scheme 1.

Alaninol-derived benzyl amine 8 was joined with aldehyde 9 through reductive amination to provide intermediate 10. The tetrahydropyrazolopyrazine was forged through a two-step process of chloride formation and then cyclization, leading to key intermediate 11. The piperidine ribose pocket moiety was introduced through Suzuki coupling with vinylboronic ester 12; a subsequent transfer hydrogenation fully saturated this group and cleaved the benzylamine protecting group, providing 13. Introduction of the azaindole was accomplished through Buchwald cross-coupling conditions.12 Finally, a two-step protocol served to transform Boc-protected intermediate 15 to the key compound 7.
Further profiling revealed that the THPP series as a whole had a strong CYP3A4 time-dependent inhibition (TDI) signal (compound 7 CYP3A4 TDI kinact/KI = 0.028 μL/min/pmol). Since new CYP enzyme must be synthesized to restore metabolic capability, it could lead to clinically relevant drug–drug interactions (DDI) if the inactivated CYP enzyme is involved in the metabolism of a coadministered drug. Hence, in order for the THPP series to progress further than mere tool compounds, the TDI would need to be addressed. Since our THPP series harbored an indole (a well-known CYP TDI liability),13,14 we suspected the azaindole was responsible for the observed CYP TDI. Given that electron rich aromatic rings are more susceptible to CYP oxidation, our strategy was to decrease the electron density on the azaindole or to find a suitable replacement. One of the well-known indole replacements that can mitigate CYP TDI is the indazole. While indazole 17 was TDI negative, it resulted in a 30-fold loss in ATR potency (Figure 6). An extensive screen of other azaindole replacements did not reveal a moiety that was both TDI negative and had acceptable levels of ATR activity. Interestingly, when we replaced the indole with other moieties that are not known CYP TDI liabilities such as the azabenzimidazole 18 or the aminopyridine 19, the compounds were still TDI positive, indicating that the (aza)indole is not the sole driver of the TDI. It is possible that the lack of TDI of 17 is driven by low lipophilicy (cLogP = 0.9) as opposed to the removal of the azaindole structural alert.
Figure 6.

Modification to the indole moiety.
In order to gain understanding of the mechanism(s) causing the TDI, we ran microsomal incubations in the presence of various trapping agents to try and pinpoint the site of metabolism. We were surprised to find that metID was unable to detect any glutathione (GSH) adducts, so we hypothesized reactive imine metabolites might be forming on the piperizino part of the core, and if so, this reactive hard electrophile could be trapped with a hard nucleophile such as cyanide. In this instance, metabolite identification revealed cyanide adducts were formed during microsomal incubations with cyanide. Unfortunately, the mass spectrum fragmentation patterns did not allow pinpointing the exact site of metabolism, but we were able to determine it was somewhere on the piperazine ring. With this information in hand, we focused our efforts on modifying the core.
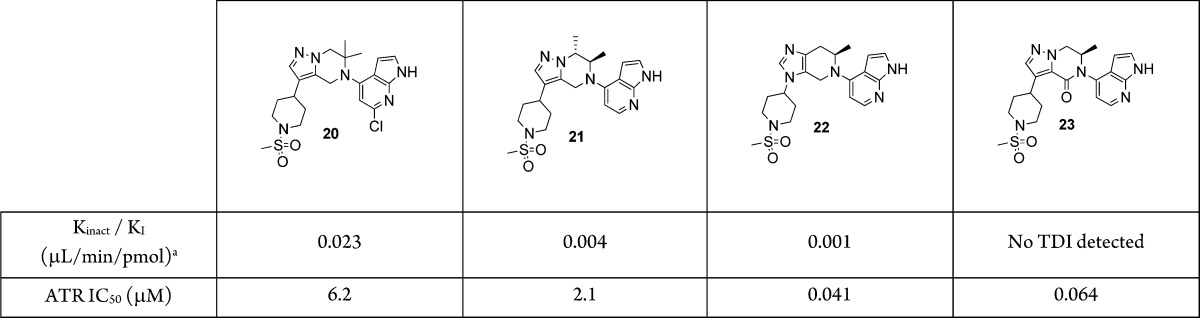
Sterically shielding the piperazine core (20 and 21) or electronically modifying the core (22) still resulted in TDI positive compounds (Table 2). Interestingly, introduction of a carbonyl as in the lactam 23 eliminated the TDI; however, with an ATR IC50 of 64 nM it was detrimental to the on-target potency. We were unsure if the carbonyl was merely blocking the site of oxidation or if it was more of an electronic deactivating effect. In order to further pinpoint the site of metabolism, we turned to deuterium-labeled compounds as a mechanistic probe. We made the mono (24), bis (25), and tris (26) deuterated compounds (Table 3) and found them all to be TDI positive. In the case of 24, just like its undeuterated counterpart 7, although no GSH adducts were observed in the metID study, a cyanide adduct on the saturated ring was observed. However, the deuterium was still intact, inferring that the metabolism must be on the methylene carbon next to the amine nitrogen. Interestingly, for the doubly labeled compound 25, cyanide adducts were no longer observed. Instead it appears there is metabolism switching from the piperazine to the azaindole since the compounds were still TDI positive, and we were able to trap and identify GSH adducts on the azaindole ring. This was also the case for the tris deuterated compound 26.
Table 2. Effects of Core Modifications on TDI.
Test compound (50 μM with serial dilutions) is incubated with human liver microsomes (0.5 mg/mL) in phosphate buffer (100 mM) with NADPH (1 mM) for 0, 5, 15, and 30 min. The incubation mixture is diluted 20-fold then incubated with CYP3A4 substrate midazolam (20 μM) to determine residual CYP3A4 enzyme activity. The enzyme activity vs incubation time is plotted to obtain kinetic parameters for TDI.
Table 3. THPP Core Deuteration Effects on CYP3A4 TDI.

Cyanide trapping assay: Compounds (20 μM) were incubated for 0, 30, and 60 min in rat liver microsomes (1 mg/mL) fortified with KH2PO4 (0.1 M; pH7.4) buffer, NADPH (2 mM), MgCl2 (5 mM), KCN, and stable label K13C15N (1 mM; 70:30) at 37 °C.
GSH trapping assay: Compounds (10 μM) were incubated for 1 h in human liver microsomes (1 mg/mL) fortified with KH2PO4 (0.1 M; pH: 7.4) buffer, NADPH (2 mM), MgCl2 (3 mM), GSH, and stable label GSH (4 mM; 50:50) at 37 °C.
In conclusion, a saturation strategy focused on increasing the Fsp3 in the HTS hit led to the novel and selective THPP series. Use of PI3Kα mutants as ATR crystallography surrogates were instrumental in providing relevant cocrystal structures that added confidence to the ATR homology modeling efforts. The use of trapping agents in conjunction with deuterium labeling of compounds revealed the molecular basis for the observed CYP3A4 TDI within the series. Ultimately, while 7 contained a TDI liability, potential safety concerns need to be assessed in context, taking dosing regimen and free efficacious exposure into consideration. To that end, since relatively large (∼1 μM) and long (18–24 h) free efficacious exposures would be required when utilizing 7 as a single agent, the CYP3A4 TDI would present too much of a liability to develop 7 as a monotherapy. However, the required exposure and duration of an ATR inhibitor is greatly reduced when utilized as chemosensitizers, whereby the cells have already been sensitized to ATR inhibition through the coadministration of a cytotoxic agent that interferes with DNA replication and arrests the cells in S-phase.15 Thus, despite the TDI liability, 7 possesses the required potency, selectivity, and physicochemical properties to find utility as a chemosensitizer in a variety of oncology indications.
Acknowledgments
We thank Shengtian Yang for running 2D NMR structure elucidation of 21 and for collecting 13C NMR data as well as Amin Kamel and Mithat Gunduz for running the metID microsomal incubations in the presence of cyanide. We are grateful to William Forrester, Mary Ellen Digan, and Debra Burdick for initiating the ATR project and the initial phenotypic screen.
Supporting Information Available
Synthetic procedures, characterization data, and assay information. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
The atomic coordinates and structure factors for the X-ray crystal structures of the PI3Kα I800M-F930V double mutant in complex with 4 have been deposited in the Protein Data Bank RSCB PDB (PDB code 4WAF).
Author Present Address
† Nurix, Inc., 1700 Owens Street, Suite 290, San Francisco, California 94158, United States.
The authors declare no competing financial interest.
Supplementary Material
References
- Weinert T. A. DNA damage checkpoint meets the cell cycle engine. Science 1997, 277, 1450–1451. [DOI] [PubMed] [Google Scholar]
- Hartwell L. H.; Weinert T. A. Checkpoints: controls that ensure the order of cell cycle events. Science 1989, 246, 629–634. [DOI] [PubMed] [Google Scholar]
- Kastan M. B.; Bartek J. Cell-cycle checkpoints and cancer. Nature 2004, 432, 316–323. [DOI] [PubMed] [Google Scholar]
- Bartek J.; Lukas C.; Lukas J. Checking on DNA damage in S phase. Nat. Rev. Mol. Cell Biol. 2004, 5, 792–804. [DOI] [PubMed] [Google Scholar]
- Cimprich K. A.; Cortez D. ATR: an essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 2008, 9, 616–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo L. I.; Murga M.; Fernandez-Capetillo O. Targeting ATR and Chk1 kinases for cancer treatment: a new model for new (and old) drugs. Mol. Oncol. 2011, 5, 368–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a recent review on Chk1 clinical compounds see:McNeely S.; Beckmann R.; Bence Lin A. K. CHEK again: revisiting the development of Chk1 inhibitors for cancer therapy. Pharmacol. Ther. 2014, 142, 1–10. [DOI] [PubMed] [Google Scholar]
- Foote K. M.; Blades K.; Cronin A.; Fillery S.; Guichard S. S.; Hassall L.; Hickson I.; Jacq X.; Jewsbury P. J.; McGuire T. M.; Nissink J. W. M.; Odedra R.; Page K.; Perkins P.; Suleman A.; Tam K.; Thommes P.; Broadhurst R.; Wood C. Discovery of 4-{4-[(3R)-3-methylmorpholin-4-yl]-6-[1-(methylsulfonyl)cyclopropyl]pyrimidin-2-yl}-1H-indole (AZ20): a potent and selective inhibitor of ATR protein kinase with monotherapy in vivo antitumor activity. J. Med. Chem. 2013, 56, 2125–2138. [DOI] [PubMed] [Google Scholar]
- Charrier J. D.; Durrant S. J.; Golec J. M. C.; Kay D. P.; Knegtel R. M. A.; MacCormick S.; Mortimore M.; O’Donnell M. E.; Pinder J. L.; Reaper P. M.; Rutherford A. P.; Wang P. S. H.; Young S. C.; Pollard J. R. Discovery of potent and selective inhibitors of ataxia telangiectasia mutated and Rad3 related (ATR) protein kinase as potential anticancer agents. J. Med. Chem. 2011, 54, 2320–2330. [DOI] [PubMed] [Google Scholar]
- Lovering F.; Bikker J.; Humblet C. Escape from flatland: increasing saturation as an approach to improving clinical success. J. Med. Chem. 2009, 52, 6752–6756. [DOI] [PubMed] [Google Scholar]
- Barsanti P. A.; Crawford K.; Doyle M. V.; Elling R.; Knapp M.; Lu Y.; Sim J.; Warne B.; Yan K.; Zhang L. Engineering of chimeric PI3Kα proteins sensitive to small molecule inhibitors targeting ATR. Manuscript in preparation.
- Surry D. S.; Buchwald S. L. Dialkylbiaryl phosphines in Pd-catalyzed aminatino: a user’s guide. Chem. Sci. 2011, 2, 27–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenberg P. F.; Kent U. M.; Bumpus N. N. Mechanism-based inactivation of human cytochromes P450s: experimental characterization, reactive intermediates, and clinical implications. Chem. Res. Toxicol. 2008, 21, 189–205. [DOI] [PubMed] [Google Scholar]
- Wong S. J.; Fan P. W.; Subramanian R.; Tonn G. R.; Henne K. R.; Johnson M. G.; Lohr M. T.; Wong B. K. Bioactivation of a novel 2-methylindole-containing dual chemoattractant receptor-homologous molecule expressed on T-helper type-2 cells/D-prostanoid receptor antagonist leads to mechanism-based CYP3A inactivation: glutathione adduct characterization and prediction of in vivo drug-drug interaction. Drug Metab. Dispos. 2010, 38, 841–850. [DOI] [PubMed] [Google Scholar]
- Menezes D. L.; Holt J.; Tang Y.; Feng J. F.; Barsanti P.; Pan Y.; Ghoddusi M.; Zang W.; Holash J.; Lees E.; Taricani L.. Mol. Cancer Res.; DOI: 10.1158/1541-7786.MCR-14-0240. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.