

Abstract
This article reports the design, synthesis, and evaluation of a novel class of molecules of intermediate size (approximately 7000 Da), which possess both the targeting and effector functions of antibodies. These compounds—called synthetic antibody mimics targeting prostate cancer (SyAM-Ps)—bind simultaneously to prostate-specific membrane antigen and Fc gamma receptor I, thus eliciting highly selective cancer cell phagocytosis. SyAMs have the potential to combine the advantages of both small-molecule and biologic therapies, and may address many drawbacks associated with available treatments for cancer and other diseases.
Introduction
Targeted protein-based therapeutics, or “biologics”, have revolutionized cancer treatment during the past decade.1,2 Agents in this class exert their actions selectively on pathological cells, and include many varieties of monoclonal and bispecific antibodies. Overall, biologics have exhibited high clinical success rates against both hematologic and solid malignancies, as well as against non-neoplastic diseases ranging from bacterial infection to autoimmune disease.3
Monoclonal antibodies possess excellent specificity and affinity for cancer cell targets, and often function by hijacking native immune effector mechanisms.4 Bispecific antibodies, diabodies, and other next-generation protein-based biologics specifically target cell-surface markers on diseased cells and then activate receptors on cytotoxic immune cells such as Fc gamma receptors (FcγRs)5−8 and CD3.9,10 Despite the many virtues of these biologics, they suffer from limitations that relate primarily to their high molecular weights and/or peptidic structures.11,12 These potential disadvantages include the possibility for life-threatening allergic reactions, poor tissue penetration, immunogenicity (even for “humanized” proteins),13 lack of oral bioavailability, requirement for low-temperature storage, difficulties in their large-scale preparation, and high cost.14
Here we report novel synthetic molecules that possess both the targeting and effector-cell-activating functions of antibodies, while being less than 1/20th (5%) of their molecular weight. We term these compounds synthetic antibody mimics (SyAMs, Figure 1A). The class of SyAMs reported here—called SyAM-Ps—have been designed to target prostate cancer cells. SyAM-Ps contain a glutamate-urea-based targeting domain15,16 that binds prostate-specific membrane antigen (PSMA)—a cell surface protein that is highly overexpressed on prostate cancer cells17−19—and an effector domain that associates with the IgG receptor type I (FcγRI)—a cell surface receptor found on immune cells responsible for initiating pro-inflammatory responses against antibody-opsonized targets.20−22 Notably, FcγRI activates immune cells to initiate phagocytosis or release cytotoxic contents upon multivalent recognition of cell-surface-immobilized ligand. In direct analogy to antibodies, SyAM-Ps induce the formation of three-component complexes between effector and target cells, thus leading to multivalent cross-linking and activation of Fc receptors, followed by destruction of cancer cells (Figure 1A).
Figure 1.

Design and structures of synthetic antibody mimics targeting prostate cancer (SyAM-Ps). (A) Schematic depiction of SyAM-P’s proposed mechanism of action. (B) Docking of SyAM-P into PSMA binding pocket and FcγRI binding surface to determine the linker length needed to template a ternary complex. (C) Schematic illustration of the evolution of SyAM-P’s design from a monoclonal antibody template. (D) Molecules discussed herein. CP33 is an FcγRI-targeting motif. The first-generation construct (SyAM-P1, 1) displays single FcγRI- and PSMA-binding motifs linked with aminocaproic units. The second-generation construct (SyAM-P2, 2) displays a pair of PSMA-targeting motifs linked to a single CP33 motif. The third-generation construct (SyAM-P3, 3) displays a pair of PSMA-targeting motifs tethered to a pair of CP33 motifs.
Although several examples of synthetic or semi-synthetic molecules capable of performing targeting or effector functions of antibodies have appeared in the literature, these molecules mediate their effects with the assistance of endogenous antibody proteins5 or protein-based appendages,23−25 or they lack immune effector function altogether.26−29 Also, while recombinant proteins with both targeting and effector functions have been reported—including scFv molecules,30−34 diabodies,33 camelids,34 and others—these molecules are very large (generally >50 kDa) and likely to suffer from the limitations outlined above for protein-based agents.
The general strategy reported herein has the potential to combine the beneficial attributes of biologics while overcoming their most significant disadvantages. Although SyAM-Ps still possess some of the structural liabilities of biologics (i.e., they possess peptide motifs), they are non-recombinant, readily prepared by organic synthesis, and, due to their relatively small size (∼7 kDa), likely to have favorable tumor penetration properties compared to high-molecular-weight agents. We therefore believe that SyAMs have the potential to represent a useful new direction for treating cancer and other diseases.35,36
Material and Methods
Docking of Protein and SyAM Binding Partners
Crystal structures were visually docked in the program Chimera to generate a composite overlay, with approximate distances measured in Chimera. For SyAM modeling, the linker was drawn in ChemDraw and docked into PSMA and FcγRI using Chimera. As a comparison, the ARM-P8–PSMA (PDB 2XEF) complex was docked with IgG1 (PDB 1IGY) and FcγRIa (PDB 3RJD), also using Chimera (Supporting Information (SI), Figure S1).
Binding of SyAMs to PSMA: Bead and Cell-Based Assays
PSMA-coated beads were generated by incubating 6 μm streptavidin-labeled polystyrene beads (Polysciences) with recombinant avi-tagged-PSMA protein (kindly provided by Dr. Cyril Barinka and Dr. Jan Konvalinka) for 30 min. Beads were washed twice with PBS, blocked with 1 mg/mL biotin for 30 min, and then washed twice with TBS + 1.5% BSA. A total of 105 PSMA-positive beads were incubated with SyAMs plus Streptavidin-AlexaFluor 647 (Invitrogen; final concentration 3.3 μM). Samples were kept on ice for 30 min, washed twice, and evaluated on an Accuri C6 flow cytometer. Cell-based assays were performed under conditions identical to those described above, substituting PSMA-positive LNCaP cells for beads.
Binding of SyAMs to FcγRI: Cell-Based Assay
IIA1.6 cells, either stably transfected with FcγRIA/γ-chain or non-transfected (isogenic negative control), were suspended in Assay Media (phenol-free RPMI 1640 medium plus 10% ultra-low IgG FBS). A total of 105 cells were mixed with the indicated compound on ice. After a 1 h incubation on ice, 10 μg of streptavidin conjugated to AlexaFluo 488 or AlexaFluo 647 was added. After an additional hour, cells were washed three times with 1 mL of cold Assay Media before flow cytometric analysis.
Ternary Complex Binding Assay
Detection of FcγRI Recruitment
LNCaP PSMA+ cells were detached with enzyme-free detachment solution (0.5 mM EDTA and EGTA) and resuspended in TBS + 1.5% BSA at a concentration of 2 × 106 cells/mL. Next, 100 μL aliquots were dispensed into Eppendorf tubes on ice. Incubation mixtures of 10 μL were prepared; the small volume was used to maximize protein concentration. These mixtures contained 1 μL of a desired concentration of 1 plus 5 μL of soluble recombinant FcγRI (R&D Systems, 200 μg/mL) and 4 μL of an anti-FcγRI antibody conjugated to phycoerythrin (R&D Systems). Eppendorf tubes with LNCaP cells were spun down, and all supernatant was removed. The 10 μL incubation mixtures were transferred onto the cells, and the pellet was resuspended gently. Incubations were left on ice for 1 h before washing 3 × 1 mL with TBS + 1.5% BSA.
Detection of PSMA Recruitment
Experiments performed were identical to those in the previous section describing the detection of FcγRI recruitment, except that (1) FcγRI + IIa1.6 cells were substituted for LNCaPs, (2) soluble, recombinant his-tagged PSMA (R&D Systems) was substituted for soluble recombinant FcγRI, and (3) an anti-his antibody conjugated to phycoerythrin (R&D Systems) was substituted for the anti-FcγRI antibody.
Measurement of PSMA Density on Target Surfaces
PSMA-positive beads with a biotin loading capacity of 2.0 μg/mL were prepared as described above. A phycoerythrin-labeled anti-PSMA antibody (Abcam) was then added for 30 min on ice. After being washed two times with PBS + 5% BSA, beads were analyzed for increased mean fluorescence intensity by flow cytometry. PSMA expression on RM1.PGLS cells (obtained from Dr. Michael Sadelain, MSKCC) was determined using staining conditions identical to those used for the beads. R-Phycoerythrin Quantitation beads from Bangs Laboratories were employed as calibration standards in these assays, per the manufacturer’s instructions.
ROS Production Assay
U937 cells were primed for 72 h with IFN-γ (final concentration 2 ng/mL; media changed daily). A total of 3 × 105 cells/mL U937 cells in Assay Media were mixed with 105 PSMA-coated beads/mL and SyAMs in a 96 well plate with a total volume of 90 μL. To each well, 10 μL of 2.5 mM lucigenin (Tokyo Chemicals) solution was added. The plate was centrifuged at 200 rcf for 2 min. Chemiluminescence was then measured every 2 min by plate reader (Biotek Synergy 2) for 60–90 min.
Compound Cytotoxicity Assay
A total of 12 500 RM1-hPSMA cells (obtained from Dr. J. Mathis, LSU) were plated per well in a volume of 100 μL of media (DMEM 4.5 g/L glucose + 10% FBS + 1% penicillin/streptomycin + 2 mM glutamine + 1 mM sodium pyruvate) on an Xcelligence E-Plate-16 (ACEA Biosciences) 36 h before the start of the experiment. One hundred microliters of media per well without cells was used to obtain background cell index readings on the E-Plate 16. Adherence and growth of the cells were monitored by obtaining cell index readings every 15 min using the Xcelligence system (model RTCA-DP, ACEA Biosciences), maintained inside an incubator (37 °C, 5% CO2). At time 0, the old media was carefully aspirated from the E-Plate, and fresh media containing various concentrations of SyAM-P3, vehicle only (1% DMSO; negative control), or 3% Triton detergent (positive control) was added to the wells. The plate was returned to the Xcelligence port, and cell index readings were obtained every 2 min for 3 h and then every 10 min for the remaining 21 h.
Bead and Cell Phagocytosis Assay
IFN-γ-primed U937 cells were stained with DiD dye (final concentration 1.9 μM) for 30 min at 37 °C. PSMA-positive beads were prepared as described above using 6 μm FL-1 fluorescent beads modified with an AlexaFluor 488 derivative (Streptavidin Fluoresbrite YG Microspheres, Polysciences). RM1.PGLS target cells were prepared by staining adherent cells with DiO dye (Invitrogen; final concentration 1.9 μM), and then non-enzymatically detaching the cells with 0.5 mM EDTA and EGTA. Cells were counted with trypan blue staining to verify cell viability. To measure phagocytosis, we combined 4 × 104 beads or 1.25 × 104 target cells with 105 U937 cells in Assay Media, either in the presence or absence of molecule, as indicated (final volume 100 μL). This experimental setup yielded effector-to-target ratios (E:T ratios) of 2.5:1 and 8:1 for bead and cell targets, respectively. Eppendorf tubes were centrifuged at 200 rcf for 2 min and then incubated at 37 °C for 1 h. Phagocytosis was halted by placing the tubes on ice. Flow cytometric measurements were then made using an Accuri C6 flow cytometer. For data presented in Figure 3 (below), percent phagocytosis was calculated by the formula % targets phagocytosed = [(double-positive cells)/(remaining target cells + double-positive cells) × 100%] – background phagocytosis. Data presented in the Supporting Information were also processed via two alternate formulas, as indicated: (1) % effectors phagocytosing = [(double-positive cells)/(non-participating effectors + double-positive cells) × 100%] – background phagocytosis,37 and (2) phagocytic score = (% effectors phagocytosing × mean fluorescence intensity of phagocytosing effectors) – background phagocytic score.38 Background phagocytosis is defined as phagocytosis measured in the absence of compound. For confirmation of the viability of cells analyzed as shown in SI, Figure S10, propidium iodide (2 μg/mL final concentration) was added to samples while on ice after 1 h of phagocytosis.
Figure 3.
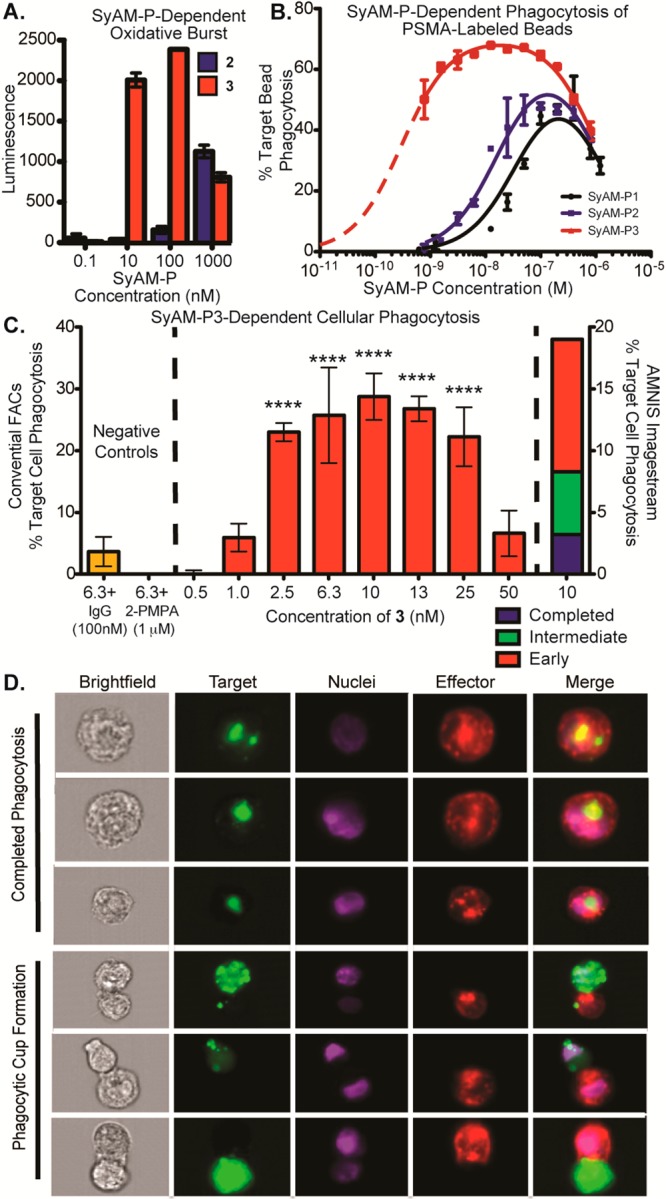
Functional assays using SyAM-P2 and SyAM-P3. (A) Superoxide burst assays. Primed FcγRI-expressing U937 effector cells and PSMA-coated beads were incubated in the presence of 2 and 3 at the indicated concentrations, and peak superoxide burst was measured as a function of reaction with the chemiluminescent compound lucigenin. (B) Phagocytosis of PSMA-labeled beads by primed FcγRI-expressing effector cells was measured using flow cytometry following exposure to the indicated concentrations of 1, 2, or 3. Phagocytosis was calculated as % target cells phagocytosed, with background phagocytosis in the absence of compound subtracted. (C) Cellular phagocytosis assays. Phagocytosis of PSMA-positive cells by FcγRI-expressing U937 effector cells was measured using flow cytometry following exposure to the indicated concentrations of 3, in the presence or absence of inhibitors, as shown. The left y-axis reports % targets phagocytosed = [(double-positive cells)/(remaining target cells + double-positive cells) × 100%] – background phagocytosis in the absence of compound, as measured by conventional flow cytometry. The right y-axis reports phagocytic events on a separate day analyzed by Amnis Imagestream flow cytometry. For Amnis experiments, double positives were calculated from dot-plot analyses of all events, as with conventional FACS. Double positive events with corresponding in-focus images were further evaluated for initiated (formation of a phagocytic cup) or completed phagocytosis. Calculations for each event type can be found in SI, Table S3. Data points shown in panels A–C represent the mean of at least duplicate experiments plus/minus standard deviation, and the reported trends were reproduced on at least three separate occasions. P-values represent the probabilities that pairwise differences in mean values (no compound versus the indicated concentration of SyAM-P3) could have arisen by chance alone, as determined by one-way ANOVA with Dunnett’s multiple-comparisons test. ****, P < 0.0001. (D) Amnis flow cytometry imaging of phagocytic events. Depicted are representative images of completed phagocytosis as compared to phagocytic cup formation. Channels shown are brightfield, target (stained with DiO), nuclei (stained with Hoechst), effector cell (stained with DiD, anti-CD14-APC, and anti-CD11b-APC), and merged image.
Amnis Imagestream Imaging
Phagocytosis experiments were conducted as described above, and then cells were fixed in 3% formaldehyde for 30 min on ice. Cells were washed once in PBS, and then stained with anti-CD14-APC and anti-CD11b-APC antibodies (Biolegend) plus Hoechst dye (Invitrogen) for 30 min on ice. Addition of the APC-conjugated antibodies was found to be necessary after fixation reduced the fluorescence of the DiD membrane dye; CD14 and CD11b are monocyte surface markers commonly used in flow cytometric phagocytosis assays.39−41 The cells were washed once in PBS and then analyzed on an Amnis Imagestream X flow cytometer, where images and data for 30 000 events/sample were collected. Data were analyzed using Amnis IDEAS software. Double-positive events were manually scored for phagocytic cup formation or complete engulfment of the target.
Results and Discussion
In constructing SyAM-Ps, we first identified chemical motifs capable of occupying the targeting and effector domains. Several small-molecule ligands—including 2-PMPA and the glutamate ureas—have been developed that bind PSMA selectively and with high affinity.15,16,42 Glutamate-ureas have also been used to image PSMA-positive tumors in mice, with low background in normal tissues, which express the highly homologous murine PSMA protein.43,44 Our laboratory has utilized these ligands to develop antibody-recruiting molecules (termed ARMs) directed against prostate cancer (ARM-P8). ARM-P8 has been shown to bind PSMA, recruit endogenous anti-dinitrophenyl (anti-DNP) antibodies to cancer cell surfaces, and then induce immune-mediated cytotoxic responses.45 We incorporated the PSMA-binding glutamate urea motif of ARMs into SyAM-Ps (Figure 1D). Conversely, for immune-cell targeting, we chose the CP33 cyclic peptide. CP33 was discovered using phage display screening, and has demonstrated highly selective binding to FcγRI over other FcγR subtypes.46
In constructing our first-generation synthetic antibody mimic (SyAM-P1, 1), we began by modeling its ternary complex with target and effector proteins. We combined co-crystal structures of the ARM-P8–PSMA complex (PDB 2XEF)47 with a docked structure of the CP33–FcγRI complex (PDB 3RJD),48 to design a linker domain with sufficient length, solubility, and rigidity to enable simultaneous binding of targeting and effector termini to their respective protein targets, and also to prevent steric clashes between components (Figure 1B). Interestingly, these measurements predicted that SyAMs should be able to function adequately at lengths significantly shorter than those separating antigen-binding (Fab) and FcγR-binding (Fc) domains in antibodies, in part due to the superficial location of the predicted CP33 binding site on FcγRI. Whereas Fab and Fc domains in antibodies are separated by approximately 140 Å (Figure 1C; SI, Figure S1), our modeling studies suggested that flexible, linear functionality approximately 22 Å in length could connect the hydrophobic interior of PSMA to bulk solvent, while access to the ligand binding surface of FcγRI would require an additional 20 Å. Overall, we therefore estimated that a linker of approximately 40 Å in length would be required to enable efficient ternary complex formation in SyAM-Ps. Translating these distance estimates into a molecular construct led us to choose a series of aminocaproic acid spacers to separate the PSMA- and FcγRI-binding functionality in SyAM-P1 (Figure 1D).
Access to SyAM-P1 (1) was achieved through a straightforward, convergent synthetic sequence starting with known azide 4(45) (Scheme 1). This compound was employed as a substrate in a Sharpless–Meldal modified Huisgen [3+2] cycloaddition, or “click” reaction,49−51 to afford carboxylic acid 5. This product was then subjected to a four-step protocol for coupling to the linker and CP33 fragments. This sequence involved acylation of resin-bound intermediate 6 (derived from 7, synthesized entirely using Fmoc solid-phase peptide protocols), Fmoc deprotection, resin cleavage and global deprotection, and air oxidation to afford SyAM-P1 in 1.1% yield over four steps. Notably, SyAM-P1 also contains a biotin motif to enable orthogonal tracking of the conjugate during cellular experiments.
Scheme 1.

With SyAM-P1 in hand, we first explored the compound’s ability to interact with cells expressing either FcγRI (IIA1.6-FcγRI) or PSMA (LNCaP). Thus, SyAM-P1 was incubated with the indicated cell type, and binding was monitored using flow cytometry, detecting with a fluorescently labeled streptavidin conjugate. These experiments demonstrated concentration-dependent association of SyAM-P1 with both FcγRI- and PSMA-expressing cells (Figure 2A,B; SI, Figure S2B). No binding was observed between SyAM-P1 and a control cell line that does not express either FcγRI or PSMA up to 10 μM (SI, Figure S2A). These data indicate that SyAM-P1 is capable of binding to FcγRI and PSMA in cellular contexts.
Figure 2.

Binding and functional properties of SyAM-P1 (1). (A) SyAM-P1 binds FcγRI-expressing cells in a concentration-dependent manner. Binding was measured by probing with fluorescently labeled streptavidin, which associates with the biotin function in 1. (B) SyAM-P1 binds PSMA-expressing cells in a concentration-dependent manner. Binding was measured by probing with fluorescently labeled streptavidin, which associates with the biotin function in 1. (C) Ternary complex formation between 1, PSMA-positive LNCaP cells, and soluble recombinant FcγRI, probing with a fluorescent anti-FcγRI antibody. (D) Ternary complex formation between 1, FcγRI-positive cells, and soluble recombinant human PSMA (his-tagged), probing with an anti-his antibody. (E) Superoxide burst generation from primed FcγRI-positive cells, induced by 1 bound to PSMA-labeled beads. Superoxides were detected by their reactivity with the chemiluminescent compound lucigenin. Peak luminescence generated during an 80 min time course is shown. (F) Phagocytosis of fluorescent PSMA-labeled beads by primed FcγRI-positive cells induced by compound 1. Phagocytosis was calculated as percent of targets phagocytosed minus background phagocytosis with 0 nM compound. (G) Effect of PSMA density on target cells on SyAM-P1-induced phagocytic response. The red dot indicates the PSMA level measured on RM1.PGLS cells, while black dots indicate PSMA level on labeled beads. All data points reflect the phagocytic response induced using a 50 nM concentration of SyAM-P1. The x-intercept was determined to be 606 PSMA/μm2 by linear regression analysis using Prism 5 (GraphPad). For functional assays in panels E and F, data points represent the mean of at least duplicate samples plus/minus standard deviation, and the reported trends were reproduced on at least three separate occasions.
We next performed several additional flow cytometry-based binding experiments to evaluate SyAM-P1’s capacity to form ternary complexes at both targeting and effector termini. Thus, we incubated PSMA-expressing cancer cells (LNCaP cells) with 1 and soluble FcγRI in the presence of a fluorescently labeled anti-FcγRI antibody probe. Increases in fluorescence were observed only in the presence of SyAM-P1 (Figure 2C, red and purple curves), indicative of the molecule’s ability to recruit soluble FcγRI in a concentration-dependent fashion. Conversely, exposure of 1 to FcγRI-expressing cells, followed by detection with soluble his-tagged PSMA and a fluorescent anti-his antibody, demonstrated a significant increase in mean fluorescence intensity only for FcγRI-positive cells incubated with both 1 and soluble PSMA (Figure 2D, orange curve). No fluorescence increase was observed in the absence of 1 (green curve) or soluble PSMA (gray curve). Taken together, these data indicate that SyAM-P1 (1) is capable of interacting with both cellular FcγRI and PSMA simultaneously, thus validating our proposed structural model.
We next evaluated whether SyAM-P1 (1) could induce effector responses from FcγRI-positive immune cells. In these assays, U937 cells were first primed with IFN-γ, which induces an up-regulation of FcγRI (SI, Figure S2C,D), and stimulates pro-inflammatory responses.52 SyAM-P1 exhibited high-level binding to IFN-γ-primed U937 cells in a concentration-dependent fashion (SI, Figure S2F). Only very low levels of binding to unprimed cells were detected (SI, Figure S2E), consistent with low basal levels of FcγRI expression on these cells (SI, Figure S2C). SyAM-P1 (1) was also found to elicit robust, target-dependent responses in superoxide burst experiments (Figure 2E), as assessed with a ROS-sensitive chemiluminescent probe.53 PSMA-coated beads were chosen as targets in these experiments because they enabled tight control over PSMA density. In experiments performed in the absence of target beads altogether, SyAM-P1 failed to induce ROS production, indicating that multivalent templating on PSMA-expressing surfaces is required for compound function (SI, Figure S2G).
Finally, we analyzed SyAM-P1-dependent phagocytic responses of primed U937 monocytic cells against PSMA-coated beads using a two-color flow cytometry assay.54 In this system, SyAM-P1 (1) was found to enhance both the phagocytosis of PSMA-coated beads (Figure 2F) and the phagocytic activity of U937 effector cells (SI, Figure S3F,G) in a dose-dependent manner. All of these metrics yielded EC50 values for SyAM-P1 of approximately 20–30 nM (Figure 2F; SI, Figure S3F,G). Internalization of PSMA-coated beads by U937 effector cells was confirmed by fluorescence quenching of non-internalized beads (SI, Figure S3A–C) and by microscopy (SI, Figure S3D,E). These effector responses were abrogated by either a competitive small-molecule ligand to PSMA (2-PMPA)15 or an anti-FcγRI antibody (Figure 2F; SI, Figure S3F,G). Unprimed U937 cells were unable to mediate phagocytosis (SI, Figure S2H), as expected from their low FcγRI expression (SI, Figure S2C). Follow-up experiments indicated that PSMA loading density directly correlates with phagocytic responses (Figure 2G; SI, Table S2). Robust effector responses were observed at PSMA densities ranging from 840 ± 88 to 2375 ± 250 molecules per μm2. These results establish a minimal PSMA density necessary for SyAM-P1 to elicit an immune response, and a linear dependence on the level of phagocytosis with PSMA densities above that threshold. Together these data indicate that SyAM-P1 is capable of mediating immunological responses—including both superoxide burst and phagocytosis—in a PSMA- and FcγRI-specific manner. More broadly, these proof-of-principle studies also provide essential support for the hypothesis that both targeting and effector responses of an antibody can be effectively mimicked in vitro by a fully synthetic bifunctional molecule.
Although initial studies with SyAM-P1 (1) were encouraging, attempts to repeat phagocytosis results using PSMA-expressing cancer cells in lieu of PSMA-labeled beads were unsuccessful (SI, Figure S4D). Because PSMA expression levels on RM1.PGLS cells were measured to be 720 ± 139 molecules per μm2, below the threshold observed in Figure 2G, we hypothesized that this low level of PSMA expression on the surface of target cells was insufficient to enable SyAM-P1 (1)-mediated immune responses.
We therefore pursued a second-generation strategy. We hypothesized that a bivalent display of PSMA-binding motifs would increase the degree of SyAM-P binding to PSMA at equilibrium, in a manner analogous to IgG avidity.55 This analysis led to the design of SyAM-P2 (Figure 1D, 2), which we were able to synthesize using a straightforward convergent sequence. As illustrated in Scheme 2, azide 4 was converted to the corresponding triazole through a microwave-assisted [3+2] cycloaddition with propargylamine, and the resulting amine was acylated with an aminocaproic acid-derived spacer. The resulting adduct (9) was then Fmoc deprotected to afford amine 10, and dimerized using an azido arene template (11, Scheme 2B) to 12. Compound 12 served as a substrate for a second [3+2] cycloaddition, and the resulting intermediate carboxylic acid was then readily converted to SyAM-P2 (2) through a low-yielding sequence involving NHS-ester formation, global deprotection, and acylation with CP33-containing amine 14 (Scheme 2C).
Scheme 2.

With SyAM-P2 (2) in hand, we next evaluated its ability to bind PSMA and FcγRI targets and induce phagocytosis. Although SyAM-P2 was found to bind PSMA somewhat more potently than SyAM-P1 (Kd values of 26 and 40 nM, respectively, SI, Table S1), and also to induce a much greater level of phagocytosis against PSMA-coated bead targets (SI, Figure S4C), it proved no better in cellular phagocytosis assays than its predecessor (1, SI, Figure S4D,E). We therefore developed several additional analogues of SyAM-P2, using chemistry similar to that shown in Scheme 2, to probe the effects of linker length and composition. Thus, two SyAM-P2 analogues containing PEG-based linkers (S.7 and S.8, SI, Supplementary Scheme 2), as well as one additional analogue containing a shorter aminocaproic acid-based linker (S.13, SI, Supplementary Scheme 4), were synthesized and evaluated using both binding and phagocytosis assays described above (SI, Figure S4A,B). However, none of these additional derivatives exhibited greater potency or phagocytic ability compared to SyAM-P2 (2).
We therefore shifted our optimization efforts to the FcγRI-binding domain in SyAM-P constructs. Based on a mathematical model for three-component equilibria recently developed in our laboratory,55 we hypothesized that increasing the number of FcγRI-ligating motifs would enhance both the potency and efficacy of SyAM-P molecules in eliciting targeted immune responses (SI, Figure S5). We therefore designed a bis-bivalent SyAM-P derivative called SyAM-P3 (Figure 1D, 3). Synthesis of 3 was accomplished starting from bis-functionalized azide 12 (Scheme 3). This material was coupled with diamine 16 using a microwave-assisted [3+2] cycloaddition reaction, and then these amines were acylated with CP33-containing NHS-ester 15 (derived as shown in Scheme 2C from intermediate 14) to provide SyAM-P3 in acceptable yield. Once again, linker lengths were chosen in accordance with the coarse-grained modeling studies presented in Figure 1B.
Scheme 3.

Although we were aware that bivalent presentation of CP33 had the potential to elicit target-independent activation of immune cells, we speculated that such effects could be minimized, or completely avoided, as long as the efficacious therapeutic concentration was significantly lower than that needed to induce target-independent activation. In such a scenario, a derivative such as 3 would improve the Kd for the FcγRI–ligand interaction as a consequence of avidity, but require binding to targets displaying a sufficient PSMA density to activate immune effectors cell responses.
In the event, SyAM-P3 (3) was found to be both more potent and more efficacious than earlier-generation molecules in oxidative burst and phagocytosis assays using PSMA-coated beads as targets and varying E:T ratios (Figure 3A,B). Control experiments demonstrated that the oxidative burst responses required the presence of PSMA-labeled beads (SI, Figure S6A), indicating that direct cross-linking of FcγRI by SyAM-P3 (3) was not taking place under these experimental conditions, despite the presence of bivalent CP33 motifs in SyAM-P3 (3). This observation is consistent with prior reports demonstrating that very high concentrations of soluble bivalent-CP33 (10 mM or greater)—approximately 1000-fold higher than what is required for SyAM-P3’s activity (Figure 3A; SI, Figure S6A)—are required to activate effector cells in the absence of targets.46 SyAM-mediated phagocytosis of PSMA-coated beads was significant at varying E:T ratios from the perspective of both the targets phagocytosed (Figure 3B; SI, Figure S7E) and the engagement of effector cells (SI, Figure S7A–D). Phagocytic responses were suppressed in a concentration-dependent manner in the presence of either human IgG or 2-PMPA (SI, Figure S6B,C), confirming that observed levels of phagocytosis require both Fc-receptor- and PSMA-binding interactions with SyAM-P3 (3).
As expected, SyAM-Ps 1, 2, and 3 all exhibit autoinhibitory dose–response curves in bead-based phagocytosis assays (Figure 3B). Fitting these data to a mathematical model for ternary equilibria has allowed us to gain several mechanistic insights into the modes of action and differential activities of SyAM derivatives.55 For example, the high quality of the model fit strongly supports that SyAMs act via a three-component pre-equilibrium, in which formation of the underlying FcγR-SyAM-PSMA complex occurs significantly faster than the induced effector response. The mathematical model also explains why making the FcγRI-targeting side of SyAM-Ps bivalent (2 → 3, blue versus red curves) leads to a greater change in phagocytosis than does the analogous modification to the PSMA-binding side (1 → 2, black versus blue curves). This rationale can be understood in terms of the increases in interaction strength that accompanies bivalency. For systems such as the SyAM-Ps, in which Kds corresponding to each end of the complex differ from each other by more than 10-fold, and are larger in value than the corresponding cell-surface receptor concentrations, our model predicts that improvements in the weaker Kd (here KFcγRI) will enhance both the magnitude (i.e., maximum phagocytosis) and width of dose–response curves. Improving the stronger binding constant (here KPSMA), on the other hand, only affects the width of these curves. Finally, the “zippering” effect between the immune and cancer surfaces leads to positive cooperativity, which also emerges when these curves are fit to our mathematical model. Similar cooperativity trends have previously been observed for antibody-dependent cytotoxicity.56
In the final set of experiments, we monitored SyAM-P3-induced phagocytosis of PSMA-expressing RM1.PGLS cells by FcγRI-expressing U937 monocytic cells (Figure 3C; SI, Figure S8A). As shown, treatment of RM1.PGLS cells with varying concentrations of SyAM-P3 led to significant levels of target cell phagocytosis, over a range of E:T ratios (from 2:1 to 20:1; SI, Figure S11A–C,E). Phagocytosis was dependent on SyAM-P3 concentration, and autoinhibitory at higher concentrations, as predicted by mathematical models. Phagocytosis was suppressed in the presence of either human IgG or 2-PMPA, which are known competitors at FcγRI and PSMA, respectively (Figure 3C; SI, Figures S6E,F and S11D). Furthermore, treatment of the isogenic PSMA-negative RM1 tumor line with SyAM-P3 led to only minimal levels of phagocytosis (SI, Figure S6D). Taken together, these data indicate that SyAM-P3 is inducing phagocytosis in a manner that depends on the presence of PSMA and FcγRI on target and effector cells, respecitvely. Additional control experiments were also performed, which confirmed the viability of phagocytosed targets (SI, Figure S10A–F), as well as that SyAM-P3 is non-cytotoxic in the absence of effector cells (SI, Figure S9). Furthermore, direct head-to-head comparison of SyAM-P3 derivative S.18 and ARM-P8 plus anti-DNP antibody demonstrates comparable levels of phagocytic activity (SI, Figure S6G). Compound S.18 is somewhat more potent than ARM-P8, whereas the latter compound gives rise to slightly higher maximal levels of phagocytosis, perhaps due to the ability of the anti-DNP antibody to engage FcγRs other than FcγRI.
SyAM-induced phagocytosis was then visualized using an Amnis imaging flow cytometer (Figure 3D; SI, Figure S8B). Here effector and target cells were exposed to SyAM-P3, and 30 000 epifluorescence images were obtained for each set of experimental conditions. These experiments allowed us to image double-positive cellular clusters, which fell into three categories—cell–cell attachment (red), phagocytic cup formation (blue), and complete target cell engulfment (green, Figure 3C, right axis)—which reflect early, intermediate, and late stages of the phagocytic process, respectively (calculation of percentages can be found in SI, Table S3). Careful analysis of these high-throughput microscopy data revealed that the percentages of cells in the various phagocytic stages were well-correlated between SyAM-P3 (3) and the positive control of ARM-P8 (SI, Table S3). Also, among cell complexes engaged in phagocytic cup formation, effector cells routinely exhibited surface invagination at the site of cell–cell contact. Such morphological changes were not observed among target cells, consistent with the expectation that double-positive readings result from engulfment of PSMA-expressing cells by FcγRI-positive cells, not vice versa.
Conclusions
Here we report the first homogeneous, synthetic molecules capable of mimicking the ability of antibodies to target FcγR-dependent immune effector responses selectively to cancer cells. These molecules are completely synthetic, and possess much lower molecular weights than protein-containing agents. Our findings offer insight into the structural parameters required for the design of functional synthetic antibodies, and indicate that the Fc receptor and target proteins can be brought into close enough proximity to enable immune response activation, even when effector and targeting domains are much closer together than in Fc-antibody-target complexes. Furthermore, the improvement in efficacy seen in compound 3 establishes the importance of avidity in mediating immune effector responses. Although a bivalent targeting domain (as seen in 2) mimics the natural F(ab)2 structure, the linking of a pair of FcγRI binding motifs in 3 affords an even larger improvement in efficacy without inducing target-independent immune cell activation.
Although not explicitly addressed in this report, we believe that the SyAM strategy has the potential to combine many of the advantageous properties of small-molecule and biologic therapies while overcoming some of the drawbacks seen with protein-based agents. For example, even the largest of the proposed SyAMs is only approximately 5% of the molecular weight of an antibody, and thus SyAMs have the potential to penetrate into solid tumors more efficiently than antibodies. Although SyAMs contain a peptidic component, due to its relatively small size, along with its cyclic nature and N-terminal acetylation, this motif is highly unlikely to be processed and presented on MHC proteins, and to induce unwanted “anti-SyAM” immune responses. Because of the modular nature of SyAMs, one can imagine incorporating entirely non-peptidic functionality for activating immune cells in next-generation SyAMs. SyAMs also offer a potential advantage over our previously reported antibody-recruiting molecules, in that they are capable of eliciting immune responses by direct binding to immune cell receptors, removing the need for endogenous antibodies, whose concentrations and/or affinities can vary among individuals.45,57 Like antibodies, SyAMs are expected to enhance trafficking of cancer-specific antigens through immune cells, and thus have the potential to give rise to long-lasting immunity.4,58−61 Therefore, SyAMs have the potential to serve simultaneously as treatment and vaccine.
Unlike many conventional small molecules, SyAM constructs will require neither cell permeability nor the ability to interfere with protein–ligand interactions in order to recruit immune cells and function effectively as cytotoxic agents. In analogy to antibodies,62 SyAMs can function through a mechanism that affords multiple levels of regulation: synthetic constructs are expected only to induce cell killing upon binding cancer cells in sufficiently large amounts to initiate FcγRI cross-linking and activate immune effector responses. Because both human prostate cancer cells and tumor neovasculature express PSMA at extremely high levels,63−67 SyAMs are expected to exhibit excellent selectivity for a range of cancer-relevant targets. This behavior stands in contrast to many traditional chemotherapeutics or toxin conjugates, which can kill “off-target” cells due to either non-specific uptake of lethal toxins or low levels of target antigen expression.68 Unlike antibodies, which bind both activating and inhibitory FcγRs,69,70 SyAMs selectively bind to only FcγRI, whose primary role is to activate immune-mediated cytotoxicity.20,21 This property would also avoid the deposition of complement, which has been shown to impair Fc responses in vivo.71 In theory, the convergent synthesis reported herein will enable rapid modification, and therefore allow for application of this strategy against a wide variety of disease-associated cells, viruses,57 or proteins, thus broadening the scope and the utility of our approach. These molecules provide new and exciting avenues for development of next-generation, customizable immunotherapeutics.
Acknowledgments
We dedicate this work to the memory of Prof. Carlos F. Barbas III. This work was supported by funding from the NIH (GM100283-01) and Bristol-Myers-Squibb (OCR4997.11). We thank Chad Miller for the generation of Figure S1, as well as discussions of computational and modeling techniques. We also thank Dr. Jan Konvalinka and Dr. Cyril Barinka for kindly providing avi-tagged recombinant human PSMA protein, and Dr. Thihan Padukkavidana for his careful reading of this manuscript.
Supporting Information Available
All chemical compound characterization, supplementary synthetic schemes, and additional biological controls and validation of the SyAMs. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
∥ P.J.M.: Chemistry Department, The Scripps Research Institute, Jupiter, FL 33458
Author Present Address
# A.X.Z.: AstraZeneca R&D, Waltham, MA 02451
Author Present Address
⊗ E.F.D.: NYU Langone Medical Center, New York, NY 10016
Author Present Address
∇ M.D.K.: Axela Inc., Toronto, ON, Canada M9W 1B3
Author Contributions
⊥ P.J.M. and K.J.F. contributed equally to this work and are listed by seniority.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Beck A.; Wurch T.; Bailly C.; Corvaia N. Nat. Rev. Immunol. 2010, 10, 345. [DOI] [PubMed] [Google Scholar]
- Scott A. M.; Wolchok J. D.; Old L. J. Nat. Rev. Cancer 2012, 12, 278. [DOI] [PubMed] [Google Scholar]
- Nelson A. L.; Dhimolea E.; Reichert J. M. Nat. Rev. Drug Discovery 2010, 9, 767. [DOI] [PubMed] [Google Scholar]
- Lu Y.; You F.; Vlahov I.; Westrick E.; Fan M.; Low P. S.; Leamon C. P. Mol. Pharmaceutics 2007, 4, 695. [DOI] [PubMed] [Google Scholar]
- McEnaney P. J.; Parker C. G.; Zhang A. X.; Spiegel D. A. ACS Chem. Biol. 2012, 7, 1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuesta A. M.; Sainz-Pastor N.; Bonet J.; Oliva B.; Alvarez-Vallina L. Trends Biotechnol. 2010, 28, 355. [DOI] [PubMed] [Google Scholar]
- James N. D.; Atherton P. J.; Jones J.; Howie A. J.; Tchekmedyian S.; Curnow R. T. Br. J. Cancer 2001, 85, 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honeychurch J.; Tutt A. L.; Valerius T.; Heijnen I. A. F. M.; Van de Winkel J. G. J.; Glennie M. J. Blood 2000, 96, 3544. [PubMed] [Google Scholar]
- Shekhar C. Chem. Biol. 2008, 15, 877. [DOI] [PubMed] [Google Scholar]
- Kufer P. Trends Biotechnol. 2004, 22, 238. [DOI] [PubMed] [Google Scholar]
- Hansel T. T.; Kropshofer H.; Singer T.; Mitchell J. A.; George A. J. T. Nat. Rev. Drug Discovery 2010, 9, 325. [DOI] [PubMed] [Google Scholar]
- Allen T. M. Nat. Rev. Cancer 2002, 2, 750. [DOI] [PubMed] [Google Scholar]
- Weber C. A.; Mehta P. J.; Ardito M.; Moise L.; Martin B.; De Groot A. S. Adv. Drug Delivery Rev. 2009, 61, 965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran N. Nat. Biotechnol. 2011, 29, 5. [DOI] [PubMed] [Google Scholar]
- Slusher B. S.; Vornov J. J.; Thomas A. G.; Hurn P. D.; Harukuni I.; Bhardwaj A.; Traystman R. J.; Robinson M. B.; Britton P.; Lu X. C. M.; Tortella F. C.; Wozniak K. M.; Yudkoff M.; Potter B. M.; Jackson P. F. Nat. Med. 1999, 5, 1396. [DOI] [PubMed] [Google Scholar]
- Kularatne S. A.; Wang K.; Santhapuram H.-K. R.; Low P. S. Mol. Pharmaceutics 2009, 6, 780. [DOI] [PubMed] [Google Scholar]
- Ghosh A.; Heston W. D. J. Cell. Biochem. 2004, 91, 528. [DOI] [PubMed] [Google Scholar]
- Tasch J.; Gong M.; Sadelain M.; Heston W. D. Crit. Rev. Immunol. 2001, 21, 249. [PubMed] [Google Scholar]
- Holmes E. H.; Greene T. G.; Tino W. T.; Boynton A. L.; Aldape H. C.; Misrock S. L.; Murphy G. P. Prostate Supp. 1996, 7, 25. [PubMed] [Google Scholar]
- Nimmerjahn F.; Ravetch J. Nat. Rev. Immunol. 2008, 8, 34. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn F. Springer Semin. Immun. 2006, 28, 305. [DOI] [PubMed] [Google Scholar]
- Bruhns P.; Iannascoli B.; England P.; Mancardi D. A.; Fernandez N.; Jorieux S.; Daeron M. Blood 2009, 113, 3716. [DOI] [PubMed] [Google Scholar]
- Doppalapudi V. R.; Huang J.; Liu D.; Jin P.; Liu B.; Li L.; Desharnais J.; Hagen C.; Levin N. J.; Shields M. J.; Parish M.; Murphy R. E.; Del Rosario J.; Oates B. D.; Lai J.-Y.; Matin M. J.; Ainekulu Z.; Bhat A.; Bradshaw C. W.; Woodnutt G.; Lerner R. A.; Lappe R. W. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 22611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui H.; Thomas J. D.; Burke T. R.; Rader C. J. Biol. Chem. 2012, 287, 28206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C. H.; Axup J. Y.; Lawson B. R.; Yun H.; Tardif V.; Choi S. H.; Zhou Q.; Dubrovska A.; Biroc S. L.; Marsden R.; Pinstaff J.; Smider V. V.; Schultz P. G. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 17796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agnew H. D.; Rohde R. D.; Millward S. W.; Nag A.; Yeo W. S.; Hein J. E.; Pitram S. M.; Tariq A. A.; Burns V. M.; Krom R. J.; Fokin V. V.; Sharpless K. B.; Heath J. R. Angew. Chem., Int. Ed. 2009, 48, 4944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodadek T. Nat. Chem. 2009, 1, 183. [DOI] [PubMed] [Google Scholar]
- Kodadek T. Curr. Opin. Chem. Biol. 2010, 14, 721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park B. W.; Zhang H. T.; Wu C.; Berezov A.; Zhang X.; Dua R.; Wang Q.; Kao G.; Oapos; Rourke D. M.; Greene M. I.; Murali R. Nat. Biotechnol. 2000, 18, 194. [DOI] [PubMed] [Google Scholar]
- Murali R.; Greene M. I. Pharma 2012, 5, 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X.-Q.; Wang H.; Cai B.; Wang L.-L.; Yue S.-T. Nat. Biotechnol. 2007, 25, 921. [DOI] [PubMed] [Google Scholar]
- Bird R.; Hardman K.; Jacobson J.; Johnson S.; Kaufman B.; Lee S.; Lee T.; Pope S.; Riordan G.; Whitlow M. Science 1988, 242, 423. [DOI] [PubMed] [Google Scholar]
- Holliger P.; Prospero T.; Winter G. Proc. Natl. Acad. Sci. U.S.A. 1993, 90, 6444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmsen M. M.; De Haard H. J. Appl. Microbiol. Biotechnol. 2007, 77, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel D. A. Nat. Chem. Biol. 2010, 6, 871. [DOI] [PubMed] [Google Scholar]
- Spiegel D. A. Exp. Rev. Clin. Pharmacol. 2013, 6, 223. [DOI] [PubMed] [Google Scholar]
- Ide K.; Wang H.; Tahara H.; Liu J.; Wang X.; Asahara T.; Sykes M.; Yang Y.-G.; Ohdan H. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackerman M. E.; Moldt B.; Wyatt R. T.; Dugast A. S.; McAndrew E.; Tsoukas S.; Jost S.; Berger C. T.; Sciaranghella G.; Liu Q.; Irvine D. J.; Burton D. R.; Alter G. J. Immunol. Methods 2011, 366, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S. T.; Kelton W.; Kang T. H.; Ng D. T.; Andersen J. T.; Sandlie I.; Sarkar C. A.; Georgiou G. ACS Chem. Biol. 2012, 8, 368. [DOI] [PubMed] [Google Scholar]
- Ashraf S. Q.; Umana P.; Mossner E.; Ntouroupi T.; Brunker P.; Schmidt C.; Wilding J. L.; Mortensen N. J.; Bodmer W. F. Br. J. Cancer 2009, 101, 1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards J. O.; Karki S.; Lazar G. A.; Chen H.; Dang W.; Desjarlais J. R. Mol. Cancer Ther. 2008, 7, 2517. [DOI] [PubMed] [Google Scholar]
- Barinka C.; Rojas C.; Slusher B.; Pomper M. Curr. Med. Chem. 2012, 19, 856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillier S. M.; Maresca K. P.; Lu G.; Merkin R. D.; Marquis J. C.; Zimmerman C. N.; Eckelman W. C.; Joyal J. L.; Babich J. W. J. Nucl. Med. 2013, 54, 1369. [DOI] [PubMed] [Google Scholar]
- Bacich D. J.; Pinto J. T.; Tong W. P.; Heston W. D. Mamm. Genome 2001, 12, 117. [DOI] [PubMed] [Google Scholar]
- Murelli R. P.; Zhang A. X.; Michel J.; Jorgensen W. L.; Spiegel D. A. J. Am. Chem. Soc. 2009, 131, 17090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonetto S.; Spadola L.; Buchanan A. G.; Jermutus L.; Lund J. FASEB J. 2009, 23, 575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang A. X.; Murelli R. P.; Barinka C.; Michel J.; Cocleaza A.; Jorgensen W. L.; Lubkowski J.; Spiegel D. A. J. Am. Chem. Soc. 2010, 132, 12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J.; Ellsworth J. L.; Hamacher N.; Oak S. W.; Sun P. D. J. Biol. Chem. 2011, 286, 40608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb H. C.; Finn M. G.; Sharpless K. B. Angew. Chem., Int. Ed. 2001, 40, 2004. [DOI] [PubMed] [Google Scholar]
- Rostovtsev V. V.; Green L. G.; Fokin V. V.; Sharpless K. B. Angew. Chem., Int. Ed. 2002, 41, 2596. [DOI] [PubMed] [Google Scholar]
- Tornøe C. W.; Christensen C.; Meldal M. J. Org. Chem. 2002, 67, 3057. [DOI] [PubMed] [Google Scholar]
- Kårehed K.; Dimberg A.; Dahl S.; Nilsson K.; Öberg F. Mol. Immunol. 2007, 44, 615. [DOI] [PubMed] [Google Scholar]
- Yamazaki T.; Kawai C.; Yamauchi A.; Kuribayashi F. Trop. Med. Int. Health 2011, 39, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsche C. E.; McEnaney P. J.; Zhang A. X.; Spiegel D. A. ACS Chem. Biol. 2011, 7, 316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglass E. F.; Miller C. J.; Sparer G.; Shapiro H.; Spiegel D. A. J. Am. Chem. Soc. 2013, 135, 6092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underhill D. M.; Goodridge H. S. Nat. Rev. Immunol. 2012, 12, 492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker C. G.; Domaoal R. A.; Anderson K. S.; Spiegel D. A. J. Am. Chem. Soc. 2009, 131, 16392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Vugt M. J.; Kleijmeer M. J.; Keler T.; Zeelenberg I.; van Dijk M. A.; Leusen J. H. W.; Geuze H. J.; van de Winkel J. G. J. Blood 1999, 94, 808. [PubMed] [Google Scholar]
- Rawool D. B.; Bitsaktsis C.; Li Y.; Gosselin D. R.; Lin Y.; Kurkure N. V.; Metzger D. W.; Gosselin E. J. J. Immunol. 2008, 180, 5548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas-Rudolph D.; Du Clos T. W.; Snapper C. M.; Mold C. J. Immunol. 2007, 178, 7283. [DOI] [PubMed] [Google Scholar]
- Dhodapkar M. V.; Dhodapkar K. M.; Li Z. Curr. Opin. Immunol. 2008, 20, 512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Garcia E.; Rosales C. J. Leukocyte Biol. 2002, 72, 1092. [PubMed] [Google Scholar]
- Speno H.; Luthi-Carter R.; Macias W.; Valentine S.; Joshi A.; Coyle J. Mol. Pharmacol. 1999, 55, 179. [DOI] [PubMed] [Google Scholar]
- Bostwick D. G.; Pacelli A.; Blute M.; Roche P.; Murphy G. P. Cancer 1998, 82, 2256. [DOI] [PubMed] [Google Scholar]
- Wright G. L.; Haley C.; Beckett M. L.; Schellhammer P. F. Urol. Oncol. 1995, 1, 18. [DOI] [PubMed] [Google Scholar]
- Chang S. S.; Reuter V. E.; Heston W. D. W.; Bander N. H.; Grauer L. S.; Gaudin P. B. Cancer Res. 1999, 59, 3192. [PubMed] [Google Scholar]
- Adams G.; Weiner L. Nat. Biotechnol. 2005, 23, 1147. [DOI] [PubMed] [Google Scholar]
- Carlson C. B.; Mowery P.; Owen R. M.; Dykhuizen E. C.; Kiessling L. L. ACS Chem. Biol. 2007, 2, 119. [DOI] [PubMed] [Google Scholar]
- Stavenhagen J. B.; Gorlatov S.; Tuaillon N.; Rankin C. T.; Li H.; Burke S.; Huang L.; Johnson S.; Bonvini E.; Koenig S. Cancer Res. 2007, 67, 8882. [DOI] [PubMed] [Google Scholar]
- Clynes R.; Towers T.; Presta L.; Ravetch J. Nat. Med. 2000, 6, 443. [DOI] [PubMed] [Google Scholar]
- Wang S.-Y.; Veeramani S.; Racila E.; Cagley J.; Fritzinger D. C.; Vogel C.-W.; St John W.; Weiner G. J. Blood 2009, 114, 5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.