

Abstract
BACKGROUND AND PURPOSE
We recently found that PKCε was required for spinal analgesic synergy between two GPCRs, δ opioid receptors and α2A adrenoceptors, co-located in the same cellular subpopulation. We sought to determine if co-delivery of μ and δ opioid receptor agonists would similarly result in synergy requiring PKCε.
EXPERIMENTAL APPROACH
Combinations of μ and δ opioid receptor agonists were co-administered intrathecally by direct lumbar puncture to PKCε-wild-type (PKCε-WT) and -knockout (PKCε-KO) mice. Antinociception was assessed using the hot-water tail-flick assay. Drug interactions were evaluated by isobolographic analysis.
KEY RESULTS
All agonists produced comparable antinociception in both PKCε-WT and PKCε-KO mice. Of 19 agonist combinations that produced analgesic synergy, only 3 required PKCε for a synergistic interaction. In these three combinations, one of the agonists was morphine, although not all combinations involving morphine required PKCε. Morphine + deltorphin II and morphine + deltorphin I required PKCε for synergy, whereas a similar combination, morphine + deltorphin, did not. Additionally, morphine + oxymorphindole required PKCε for synergy, whereas a similar combination, morphine + oxycodindole, did not.
CONCLUSIONS AND IMPLICATIONS
We discovered biased agonism for a specific signalling pathway at the level of spinally co-delivered opioid agonists. As the bias is only revealed by an appropriate ligand combination and cannot be accounted for by a single drug, it is likely that the receptors these agonists act on are interacting with each other. Our results support the existence of μ and δ opioid receptor heteromers at the spinal level in vivo.
LINKED ARTICLES
This article is part of a themed section on Opioids: New Pathways to Functional Selectivity. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2015.172.issue-2
Keywords: μ opioid receptor, δ opioid receptor, heteromer, heterodimer, PKCε, intrathecal, spinal, synergy, biased agonism
Introduction
Two analgesic drugs may produce a greater than additive, or synergistic, response when co-delivered at the spinal level (Ossipov et al., 1989; Tallarida et al., 1989; Sutters et al., 1990; Fairbanks and Wilcox, 1999). We have recently shown that PKCε is required for spinal analgesic synergy between agonist pairs acting at α2A-adrenoceptors (receptor nomenclature conforms to BJP's Concise Guide to PHARMACOLOGY, Alexander et al., 2013) and δ-opioid peptide (DOP) receptors (Cox et al., 2015), but not for interactions between α2A-adrenoceptors and μ opioid peptide (MOP) receptors (Schuster et al., 2013). It is thought that the PKCε-dependent interaction between α2A-adrenoceptors and DOP receptors occurs at the level of individual presynaptic terminals in the superficial dorsal horn of the spinal cord because all three proteins (α2A-adrenoceptors, DOP receptors and PKCε) are found to co-localize there (Riedl et al., 2009; Schuster et al., 2013), and the interaction has also been shown to occur in spinal cord slices (Overland et al., 2009) and spinal synaptosomes (Riedl et al., 2009). Thus, it is feasible that other pairs of GPCRs located in the same cell with PKCε could also produce PKCε-dependent synergy. Furthermore, the requirement of PKCε for spinal analgesic synergy between a pair of GPCRs can provide indirect functional evidence that the two receptors in question are expressed in a common cell.
There have been several studies demonstrating interactions between ligands acting at MOP and DOP receptors. Some groups have found enhanced analgesia when MOP and DOP agonists are co-delivered at the spinal level (Sutters et al., 1990; Miaskowski et al., 1992), or locally in the periphery (Joseph and Levine, 2010; Schramm and Honda, 2010), compared with the same agonists delivered alone. Others have shown that both agonists and antagonists of DOP receptors can potentiate analgesic effects of morphine (Gomes et al., 2000; 2004; Abul-Husn et al., 2007). These studies are complemented by work indicating that DOP receptors mediate the development of analgesic tolerance to morphine (Zhu et al., 1999), and that MOP activity regulates cellular trafficking and functional availability of DOP receptors (Cahill et al., 2001; Morinville et al., 2003; Gendron et al., 2006; 2007). Although there is much evidence for functional cross-modulation between MOP and DOP receptors, co-localization of these receptors, specifically within peptidergic primary afferent neurons, has recently been a topic of controversy. Some have suggested that there is very little overlap (Scherrer et al., 2009), whereas others have presented molecular and functional evidence of co-expression and formation of MOP-DOP heteromeric complexes (Daniels et al., 2005; Gupta et al., 2010; Wang et al., 2010; Beaudry et al., 2011; He et al., 2011).
Considering our recent results indicating that PKCε co-localizes with DOP receptors and α2A-adrenoceptors in peptidergic primary afferent neurons where it mediates spinal analgesic synergy between these receptors (Overland et al., 2009; Schuster et al., 2013), we sought to determine if we could provide further functional evidence for co-localization of MOP and DOP receptors in the same population of neurons by evaluating the role of PKCε in analgesic synergy between co-delivered MOP and DOP agonists. In the current study, we tested a battery of MOP and DOP agonist combinations in wild-type (PKCε-WT) and PKCε-knockout mice (PKCε-KO) for analgesic synergy in the hot-water tail-flick assay. We found that while nearly all agonist combinations produced synergistic analgesia, few required PKCε for synergy. Ligand-dependent bias in opioid receptor signalling has been previously proposed to account for pharmacological differences observed between individual opioid agonists of the same class (for review, see Pradhan et al., 2012). The finding that requirement of PKCε for synergy was dependent on the specific ligand combinations used suggests that biased agonism of opioid receptors towards a given signalling pathway can occur at the level of co-delivered agonists simultaneously acting on two different protomers of a heteromeric receptor signalling complex.
Methods
Animals
Adult PKCε-WT and PKCε-KO mice of either sex (20 ± 5 g) were used for all experiments and were bred from pairs of hybrid (50% C57BL/6J, 50%129S4) mice heterozygous for the mutant PKCε gene (Khasar et al., 1999); all animals were maintained on a 12 h light/dark cycle with food and water available ad libitum. The temperature range was 20–24°C. The humidity range was 45 ± 15%. Animals were housed with littermates and were assigned to experimental groups in such a way to distribute age and sex equally across groups. Animals from each category (age/sex) were chosen at random for a given group. The total number of animals used was 454. The authors have consulted the ARRIVE guidelines (Kilkenny et al., 2010) and the associated British Journal of Pharmacology editorial (McGrath et al., 2010) regarding pharmacological studies. All experiments and protocols were as humane as possible and were approved by the Institutional Animal Care and Use Committee of the University of Minnesota.
Behavioural measures
Thermal nociceptive responsiveness was assessed using the warm water (52.5°C) tail immersion assay as previously described (Janssen et al., 1963). Briefly, each animal was gently held wrapped in a cloth, and the tail dipped into a controlled temperature water bath. Withdrawal latency was recorded as the amount of time that passed before a rapid movement of the tail and was not allowed to exceed 12 s. Baseline latency was recorded before drug administration, and subsequent latencies were recorded 7 min after each dose, immediately before the next dose. Each agonist or combination was administered sequentially approximately every 7 min in increasing doses to generate a cumulative dose–response curve, each mouse received no less than three and no more than four doses (Shin and Eisenach, 2003). Most dose–response curves were generated with an n of 5 or 6, or as indicated in each figure or table legend. For morphine and deltorphin II, two dose–response curves were obtained on separate days to confirm the potency and efficacy of these drugs in the PKCε mutant mice. These curves did not differ significantly in potency or efficacy and were combined for a total n of 11 or 12 as indicated in each figure or table legend. These curves were reused in the analysis for each combination tested allowing us to minimize the number of animals necessary to complete this study. Data are presented as % maximum possible effect (MPE) values, which were determined using the following equation: %MPE = 100 × (test – baseline)/(maximum – baseline).
All injections were administered to the intrathecal space between the L5 and L6 vertebrae, which is caudal to the spinal cord and at the level of the cauda equina. No more than four injections were given to each animal for generation of a single dose–response curve. Most mice were used in only one experiment, but some were used in one to two additional experiments; the latter mice were allowed to recover for at least 2 weeks before reuse. Repeat of dose–response curves for morphine and deltorphin II on mice that had been used previously indicated no change in potency or efficacy. There were no signs of tissue damage or pain associated with the injection site.
Data analysis
The ED50, in nmol, and 95% confidence limits (CLs) of all agonists and combinations were calculated using the graded dose–response curve method of Tallarida and Murray (1987). Dose-ratios for drug combinations were estimated based on comparison of ED50 values and/or dose–response curves, and were chosen to approximate equi-effective doses. Isobolographic analyses were performed using the numerical method (Tallarida et al., 1989; Ossipov et al., 1997). All ED50 calculations and statistical comparisons were performed using the JFlashCalc Pharmacological Calculations Program software package generously provided by Dr. Michael Ossipov (Department of Pharmacology, University of Arizona College of Medicine, Tucson, AZ, USA). For all isobolograms, error bars for theoretical additive and observed combination ED50 values represent the vector sum of vertical and horizontal CLs. Interaction index (γ) values were calculated as described by Tallarida (2002) using the equation γ = a/A + b/B, where A and B are the doses of each single drug required for a given effect, and a and b are the relative dose contribution of each drug when given in combination for the same effect. Dose–response curves and isobolograms were produced using GraphPad Prism 4.0 (GraphPad Software Inc., La Jolla, CA, USA).
Drug preparation and administration
Agonists used were dermorphin, deltorphin (both from Bachem, Torrance, CA, USA), deltorphin I (Biotrend, Destin, FL, USA), deltorphin II, SNC80, naltrindole HCl (Tocris Bioscience, Bristol, UK), morphine sulfate, endomorphin II, leu-enkephalin, β-endorphin, codeine HCl, oxycodone HCl, oxymorphone HCl, hydromorphone HCl, DAMGO ([D-Ala2, N-MePhe4, Gly-ol]-enkephalin), fentanyl, DPDPE ([D-Pen2, D-Pen5]-enkephalin) (all from Sigma, St. Louis, MO, USA), α-oxymorphamine HCl, oxymorphindole HCl and oxycodindole HCl (gifts from the lab of Phillip Portoghese, University of Minnesota). Commercially unavailable deltorphin analogue peptides (Tyr-D-Met-Phe-Glu-Leu-Met-Asp, Tyr-D-Met-Phe-Glu-Leu-Met-Asn, Tyr-D-Ala-Phe-Ala-Val-Val-Gly and Tyr-D-Ala-Phe-Ala-Val-Val-Asp) were synthesized by, and purchased from, Biomatik (Wilmington, DE, USA). SNC80 was dissolved with an equimolar amount of tartaric acid in saline. All other drug stocks were prepared in normal saline. All drugs were diluted from stock solution into sterile 0.9% saline and injected intrathecally in a volume of 5 μL in awake mice by the method of Hylden and Wilcox (1980), as modified by Fairbanks (2003).
Results
Mechanisms underlying spinal analgesic synergy between morphine and DOP receptor agonists depend on the specific DOP agonist used
To determine if PKCε is required for spinal analgesic synergy between MOP and DOP receptor agonists, we tested combinations of DOP receptor agonists with morphine. Initially, we tested intrathecal co-delivery of morphine with deltorphin II (DELT-II), SNC80 or DPDPE for antinociceptive synergy in the hot-water tail-flick assay. When delivered individually, each agonist produced comparable antinociception in PKCε-WT and PKCε-KO mice (Table 1). Morphine was delivered intrathecally in approximately equi-effective combination with DELT-II, SNC80 or DPDPE in both PKCε-WT and PKCε-KO mice. All three combinations were synergistic in PKCε-WT mice, but only DELT-II + morphine was additive in PKCε-KO mice (Figure 1, Table 1). Subsequently, we tested several additional DOP receptor agonists in combination with morphine.
Table 1.
Morphine in combination with DOP receptor agonists
| ED50, nmol (95% CL) | |||||
|---|---|---|---|---|---|
| In combination with morphine | |||||
| Genotype | Agonist | Observed combined | Theoretical additive | Outcome (P-value) | Dose ratio with morphine |
| Morphine | |||||
| PKCε-WT | 2.22 (±0.51) | NA | NA | NA | NA |
| PKCε-KO | 1.86 (±0.44) | NA | NA | NA | NA |
| SNC80a | |||||
| PKCε-WT | 49.30 (±6.31) | 1.21 (±0.58) | 27.18 (±3.66) | SYN (2.0 × 10−16) | 25:1 |
| PKCε-KO | 49.50 (±5.42) | 2.00 (±1.12) | 24.97 (±3.57) | SYN (9.5 × 10−15) | |
| DPDPE | |||||
| PKCε-WT | 1.86 (±0.62) | 1.35 (±0.28) | 2.03 (±0.40) | SYN (8.6 × 10−3) | 1:1 |
| PKCε-KO | 1.95 (±0.65) | 1.33 (±0.29) | 1.90 (±0.37) | SYN (0.019) | |
| Deltorphin II | |||||
| PKCε-WT | 2.22 (±0.57) | 1.21 (±0.45) | 2.22 (±0.41) | SYN (1.2 × 10−3) | 1:1 |
| PKCε-KO | 2.16 (±0.53) | 2.32 (±0.92) | 2.00 (±0.36) | ADD (0.50) | |
| Deltorphin I | |||||
| PKCε-WT | 1.39 (±0.44) | 0.49 (±0.19) | 1.71 (±0.37) | SYN (2.3 × 10−7) | 1:1 |
| PKCε-KO | 1.24 (±0.46) | 1.13 (±0.46) | 1.49 (±0.37) | ADD (0.21) | |
| Deltorphin | |||||
| PKCε-WT | 3.11 (±1.28) | 0.22 (±0.07) | 2.59 (±0.57) | SYN (6.3 × 10−11) | 1:1 |
| PKCε-KO | 4.84 (±2.00) | 0.51 (±0.16) | 2.69 (±0.58) | SYN (1.6 × 10−9) | |
| Oxymorphindole | |||||
| PKCε-WT | 1.24 (±0.28) | 0.32 (±0.12) | 1.59 (±0.27) | SYN (1.8 × 10−11) | 1:1 |
| PKCε-KO | 1.28 (±0.36) | 2.04 (±1.22) | 1.52 (±0.30) | ADD (0.39) | |
| Oxycodindole | |||||
| PKCε-W | 10.47 (±2.84) | 3.33 (±1.88) | 6.47 (±1.18) | SYN (5.2 × 10−3) | 5:1 |
| PKCε-KO | 10.44 (±2.90) | 3.45 (±1.96) | 5.91 (±1.11) | SYN (0.028) | |
Single-drug data for SNC80 were previously published (Schuster et al., 2013). Morphine + SNC80 combination data are novel. A P-value < 0.05 indicates a synergistic interaction. For all groups except morphine, deltorphin II and morphine + DPDPE, n = 6. For morphine, n = 12 in PKCε-WT and n = 11 in PKCε-KO. For deltorphin II, n = 11 in PKCε-WT and n = 12 in PKCε-KO. For DPDPE + morphine, n = 10 in PKCε-WT and PKCε-KO. All analyses were performed using JFlashCalc software (M. Ossipov, University of Arizona).
ADD, additive; SYN, synergistic.
Figure 1.
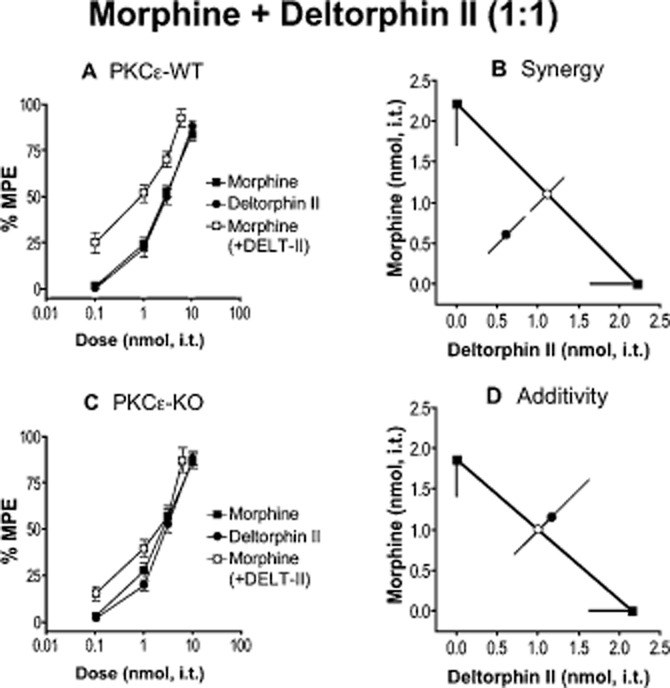
Morphine and deltorphin II (DELT-II) require PKCε for spinal analgesic synergy. (A, C) Dose–response curves for morphine, DELT-II and a 1:1 combination of the two in PKCε-WT (A) and PKCε-KO (C) mice. Error bars for each data point represent SEM. (B, D) Isobolograms showing DELT-II dose and ED50 (square) on the x-axis, morphine dose and ED50 (square) on the y-axis, theoretical additive ED50 for a 1:1 combination (open circle on line of theoretical additivity), and observed combined ED50 (filled circle). Error bars represent 95% confidence limits. (B) Morphine and DELT-II produce analgesic synergy at the spinal level in PKCε-WT mice. (D)The combination of morphine and DELT-II is additive in PKCε-KO mice. For morphine, n = 12 in PKCε-WT and n = 11 in PKCε-KO. For DELT-II, n = 11 in PKCε-WT and N = 12 in PKCε-KO. For morphine + DELT-II, n = 6 for PKCε-WT and PKCε-KO. Numerical details including ED50 value for each dose–response curve, and P-value for each interaction can be found in Table 1.
Other deltorphin peptides, deltorphin (DELT) and deltorphin I (DELT-I), were chosen for their similarity for DELT-II. Each peptide produced comparable spinal antinociception in PKCε-WT and PKCε-KO mice when delivered alone (Table 1), and was subsequently delivered in approximately equi-effective combination with morphine. We found that DELT-I, which is nearly structurally identical to DELT-II, yielded a PKCε-dependent interaction with morphine like DELT-II (Table 1); however, DELT in combination with morphine resulted in synergy that did not require PKCε (Figure 2, Table 1).
Figure 2.
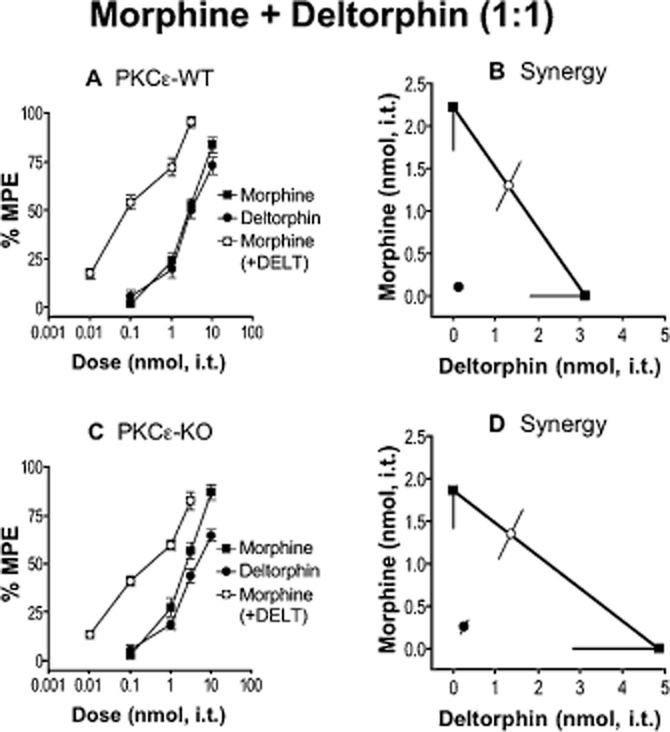
Morphine and deltorphin (DELT) do not require PKCε for spinal analgesic synergy. (A, C) Dose–response curves for morphine, DELT and a 1:1 combination of the two in PKCε-WT (A) and -KO (C) mice. Error bars for each data point represent SEM. (B, D) Isobolograms showing DELT dose and ED50 (square) on the x-axis, morphine dose and ED50 (square) on the y-axis, theoretical additive ED50 for a 1:1 combination (open white circle on line of theoretical additivity), and observed combined ED50 (filled circle). Error bars represent 95% confidence limits. (B) Morphine and DELT produce analgesic synergy at the spinal level in PKCε-WT mice. (D) The combination of morphine and DELT remains synergistic in PKCε-KO mice. For morphine, n = 12 in PKCε-WT and n = 11 in PKCε-KO. For DELT and morphine + DELT, n = 6 in PKCε-WT and PKCε-KO. Numerical details including ED50 value for each dose–response curve, and P-value for each interaction can be found in Table 1.
In addition to SNC80, we tested another non-peptide DOP agonist, oxymorphindole (OMI), for synergy with morphine, and dependence on PKCε. Oxymorphindole produced comparable antinociception in PKCε-WT and PKCε-KO mice when delivered intrathecally alone (Table 1). The combination of OMI + morphine was synergistic in PKCε-WT mice, but was additive in PKCε-KO mice (Figure 3, Table 1). We then tested an additional small-molecule DOP receptor agonist, oxycodindole (OCI), which has a very similar structure to OMI, for spinal analgesic synergy with morphine. Oxycodindole produced comparable antinociception in PKCε-WT and PKCε-KO mice when delivered intrathecally alone (Table 1). Unlike OMI, spinal co-delivery of OCI with morphine in approximately equi-effective combination resulted in synergy that did not require PKCε (Figure 4, Table 1). All numerical details including statistical analyses are reported in Table 1.
Figure 3.
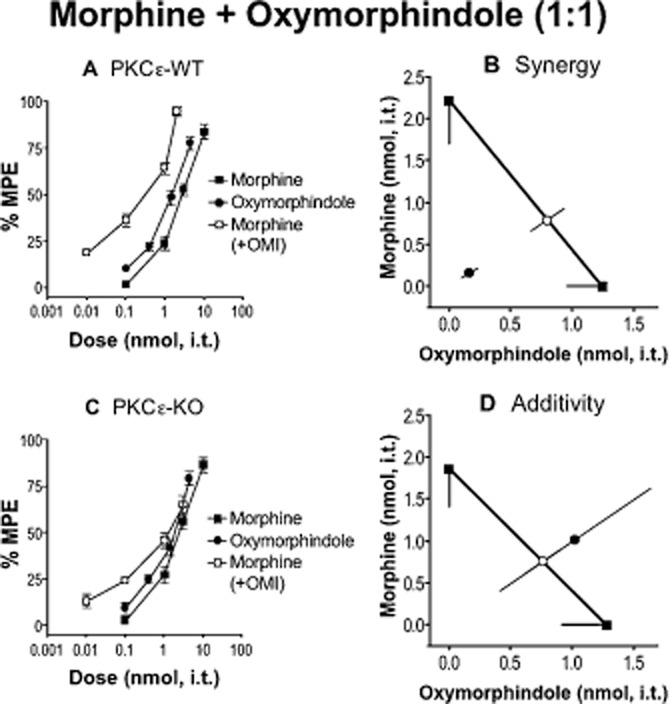
Morphine and oxymorphindole (OMI) require PKCε for spinal analgesic synergy. (A, C) Dose–response curves for morphine, OMI and a 1:1 combination of the two in PKCε-WT (A) and -KO (C) mice. Error bars for each data point represent SEM. (B, D) Isobolograms showing OMI dose and ED50 (square) on the x-axis, morphine dose and ED50 (square) on the y-axis, theoretical additive ED50 for a 1:1 combination (open circle on line of theoretical additivity), and observed combined ED50 (filled circle). Error bars represent 95% confidence limits. (B) Morphine and OMI produce analgesic synergy at the spinal level in PKCε-WT mice. (D) The combination of morphine and OMI is additive in PKCε-KO mice. For morphine, n = 12 in PKCε-WT and n = 11 in PKCε-KO. For OMI and morphine + OMI, n = 6 for PKCε-WT and PKCε-KO. Numerical details including ED50 value for each dose–response curve, and P-value for each interaction can be found in Table 1.
Figure 4.
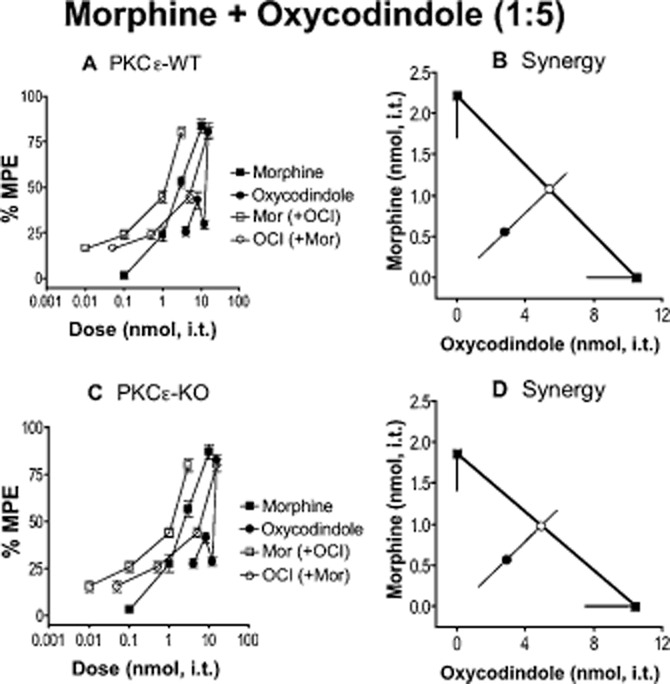
Morphine and oxycodindole (OCI) do not require PKCε for spinal analgesic synergy. (A, C) Dose–response curves for morphine, OCI and a 1:5 (morphine : OCI) combination of the two (open squares for relative contribution of morphine, and open circles for relative combination of OCI) in PKCε-WT (A) and -KO (C) mice. Error bars for each data point represent SEM. (B, D) Isobolograms showing OCI dose and ED50 (square) on the x-axis, morphine dose and ED50 (square) on the y-axis, theoretical additive ED50 for a 1:5 combination (open white circle on line of theoretical additivity), and observed combined ED50 (filled circle). Error bars represent 95% confidence limits. (B) Morphine and OCI produce analgesic synergy at the spinal level in PKCε-WT mice. (D) The combination of morphine and OCI remains synergistic in PKCε-KO mice. For morphine, n = 12 in PKCε-WT and n = 11 in PKCε-KO. For OCI and morphine + OCI, n = 6 for PKCε-WT and PKCε-KO. Numerical details including ED50 value for each dose–response curve, and P-value for each interaction can be found in Table 1.
Analgesic synergy between deltorphin II and MOP receptor agonists other than morphine does not require PKCε
To determine if other MOP receptor agonists would synergize with DELT-II in a PKCε-dependent manner similar to morphine, we tested several MOP receptor agonists delivered intrathecally in approximately equi-effective doses with DELT-II. Each of the MOP receptor agonists, endomorphin II, DAMGO, fentanyl, codeine, hydromorphone, oxymorphone, oxycodone and oxymorphamine, produced comparable spinal antinociception in both PKCε-WT and PKCε-KO mice when delivered singly (Table 2). Each agonist that produced antinociceptive synergy with DELT-II did so in both PKCε-WT and PKCε-KO mice, suggesting that PKCε-dependent synergy of a MOP receptor agonist with DELT-II may be unique to morphine. Numerical details including statistical analyses are reported in Table 2.
Table 2.
Deltorphin II in combination with MOP receptor agonists
| ED50, nmol (95% CL) | |||||
|---|---|---|---|---|---|
| In combination with deltorphin II | |||||
| Genotype | Agonist | Observed combined | Theoretical additive | Outcome (P-value) | Dose ratio with deltorphin II |
| Deltorphin II | |||||
| PKCε-WT | 2.22 (±0.57) | NA | NA | NA | NA |
| PKCε-KO | 2.16 (±0.53) | NA | NA | NA | NA |
| Endomorphin IIa | |||||
| PKCε-WT | 1.24 (±0.58) | 1.71 (±0.43) | 1.85 (±0.46) | SYN (0.029) | 1:3 |
| PKCε-KO | 1.16 (±0.55) | 1.21 (±0.34) | 1.78 (±0.43) | SYN (0.037) | |
| DAMGO | |||||
| PKCε-WT | 8.7 × 10−4 (±2.0 × 10−4) | 0.10 (±0.03) | 0.63 (±0.11) | SYN (3.6 × 10−12) | 1:1000 |
| PKCε-KO | 7.8 × 10−4 (±1.5 × 10−4) | 0.13 (±0.06) | 0.57 (±0.09) | SYN (7.6 × 10−11) | |
| Fentanyl | |||||
| PKCε-WT | 0.98 (±0.28) | 0.20 (±0.06) | 1.36 (±0.29) | SYN (1.6 × 10−10) | 1:1 |
| PKCε-KO | 1.23 (±0.41) | 0.26 (±0.07) | 1.61 (±0.36) | SYN (1.2 × 10−9) | |
| Codeine | |||||
| PKCε-WT | 1.80 (±0.56) | 2.19 (±0.55) | 1.99 (±0.42) | ADD (0.55) | 1:1 |
| PKCε-KO | 1.90 (±0.46) | 2.52 (±0.65) | 2.02 (±0.36) | ADD (0.17) | |
| Hydromorphone | |||||
| PKCε-WT | 0.05 (±0.01) | 0.14 (±0.03) | 0.45 (±0.09) | SYN (3.7 × 10−8) | 1:10 |
| PKCε-KO | 0.05 (±0.01) | 0.15 (±0.03) | 0.44 (±0.08) | SYN (6.5 × 10−9) | |
| Oxymorphone | |||||
| PKCε-WT | 0.06 (±0.02) | 0.14 (±0.02) | 0.52 (±0.11) | SYN (6.4 × 10−9) | 1:10 |
| PKCε-KO | 0.06 (±0.01) | 0.14 (±0.02) | 0.49 (±0.09) | SYN (2.0 × 10−9) | |
| α-Oxymorphamine | |||||
| PKCε-WT | 1.12 (±0.30) | 0.75 (±0.12) | 1.67 (±0.32) | SYN (1.3 × 10−6) | 1:2 |
| PKCε-KO | 1.21 (±0.31) | 0.65 (±0.09) | 1.71 (±0.32) | SYN (2.6 × 10−8) | |
| Oxycodone | |||||
| PKCε-WT | 1.21 (±0.34) | 0.32 (±0.09) | 1.57 (±0.32) | SYN (8.1 × 10−10) | 1:1 |
| PKCε-KO | 1.30 (±0.34) | 0.31 (±0.10) | 1.62 (±0.31) | SYN (5.9 × 10−11) | |
Single-drug data for endomorphin II were previously published (Schuster et al., 2013). Deltorphin II + endomorphin II combination data are novel. A P-value < 0.05 indicates a synergistic interaction. For all groups except deltorphin II, n = 6. For deltorphin II, n = 11 in PKCε-WT and n = 12 in PKCε-KO. All analyses were performed using JFlashCalc software (M. Ossipov, University of Arizona).
ADD, additive; SYN, synergistic.
Lack of requirement for PKCε in opioid–opioid interactions involving endogenous agonists
Two endogenous opioid ligands, β-endorphin and leu-enkephalin, were co-administered intrathecally in approximately equi-effective doses in both PKCε-WT and PKCε-KO mice, and tested using the hot-water tail-flick assay. There was no difference in efficacy or potency of either agonist between PKCε-WT and PKCε-KO mice, and co-delivery resulted in antinociceptive synergy that did not require PKCε (Table 3). We also tested each of these endogenous ligands in combination with either morphine or DPDPE. All four combinations produced antinociceptive synergy in PKCε-WT and PKCε-KO mice. Numerical details including statistical analyses are reported in Table 3.
Table 3.
Interactions involving endogenous opioid agonists
| ED50, nmol (95% CL) | |||||
|---|---|---|---|---|---|
| Genotype | Agonist(s) | Observed combined | Theoretical additive | Outcome (P-value) | Dose ratio |
| β-Endorphin | |||||
| PKCε-WT | 0.76 (±0.25) | NA | NA | NA | NA |
| PKCε-KO | 0.74 (±0.29) | NA | NA | NA | NA |
| leu-Enkephalin | |||||
| PKCε-WT | 1.42 (±0.49) | NA | NA | NA | NA |
| PKCε-KO | 1.49 (±0.47) | NA | NA | NA | NA |
| β-Endorphin: leu-Enkephalin | |||||
| PKCε-WT | NA | 0.11 (±0.03) | 0.99 (±0.25) | SYN (2.6 × 10−9) | 1:1 |
| PKCε-KO | NA | 0.32 (±0.13) | 0.99 (±0.28) | SYN (4.2 × 10−5) | |
| β-Endorphin: Morphine | |||||
| PKCε-WT | NA | 0.10 (±0.02) | 1.36 (±0.30) | SYN (3.2 × 10−11) | 1:2 |
| PKCε-KO | NA | 0.19 (±0.05) | 1.24 (±0.30) | SYN (6.2 × 10−9) | |
| leu-Enkephalin: Morphine | |||||
| PKCε-WT | NA | 0.09 (±0.02) | 1.73 (±0.40) | SYN (4.7 × 10−11) | 1:1 |
| PKCε-KO | NA | 0.14 (±0.04) | 1.66 (±0.34) | SYN (8.4 × 10−12) | |
| β-Endorphin: DPDPE | |||||
| PKCε-WT | NA | 0.16 (±0.03) | 1.37 (±0.32) | SYN (5.9 × 10−10) | 1:3 |
| PKCε-KO | NA | 0.33 (±0.10) | 1.38 (±0.35) | SYN (2.5 × 10−7) | |
| leu-Enkephalin: DPDPE | |||||
| PKCε-WT | NA | 0.19 (±0.05) | 1.72 (±0.43) | SYN (3.2 × 10−9) | 1:3 |
| PKCε-KO | NA | 0.32 (±0.09) | 1.81 (±0.45) | SYN (2.0 × 10−8) | |
A P-value < 0.05 indicates a synergistic interaction. For all groups, n = 6. All analyses were performed using JFlashCalc software (M. Ossipov, University of Arizona).
SYN, synergistic.
Discussion and conclusions
The results presented here show a clear difference in signalling pathways when different combinations of agonists are co-administered. Instead of a biased agonist effect presenting itself at the level of the individual ligand, these results suggest that biased agonism of a receptor complex for a given signalling pathway can occur at the level of co-delivered agonists. For example, morphine co-delivered with DELT-II, DELT-I or OMI resulted in a synergistic interaction that differed in mechanism from the combination of morphine with any other agonist tested in that it required PKCε. Furthermore, synergy resulting from co-delivery of DELT-II with any of the other tested agonists also differed in mechanism from the morphine + DELT-II combination. Thus, the bias in signalling did not lie with a single agonist, but required a specific combination of two agonists. These results are in agreement with the previously proposed concepts that MOP and DOP receptors form heteromers at the spinal level in vivo, and that these heteromers are capable of multiple signalling mechanisms dependent on the ligand, or ligands, bound to the receptor complex (Daniels et al., 2005; Gupta et al., 2010; Wang et al., 2010; Costantino et al., 2012; Yekkirala et al., 2013).
In recent years, a growing body of evidence has indicated that MOP and DOP receptors co-localize at the spinal level where they have regulatory effects on one another, and that these two receptors are able to physically interact and form heteromers. Studies performed in vitro have indicated that co-expression of MOP and DOP receptors enables interactions by co-administered agonists (Gomes et al., 2000; 2004), and that binding of single agonists as well as agonist-induced activity are different in cells expressing MOP or DOP receptors alone compared with cells expressing MOP and DOP receptors together (Yekkirala et al., 2010; Metcalf et al., 2012). Additional in vitro work has shown that dimerization of MOP and DOP receptors can lead to changes in downstream signalling (George et al., 2000; Rozenfeld and Devi, 2007), and that these changes appear to be in part due to interactions involving the carboxy termini of receptors (Fan et al., 2005). Collectively, these studies suggest that heteromer-induced signalling is a plausible and reliable mechanism for ligand bias. It is possible that binding of the appropriate agonist combination, such as morphine + DELT-II, allows an interaction between the intracellular portions of each receptor to enable otherwise unavailable signalling mechanisms leading to involvement of PKCε in the observed interaction.
There is also strong pharmacological evidence that MOP and DOP receptors form heteromers in vivo, which are differentially activated by ligands with subtle variations. Daniels et al. (2005) demonstrated that central delivery of a series of bivalent ligands consisting of two opioid pharmacophores, specifically a MOP receptor agonist and a DOP receptor antagonist connected by a chemical spacer of variable length, yielded distinct pharmacological profiles dependent specifically on the length of the spacer. These results were attributed to variations in the ability of the connected pharmacophores to simultaneously bind their respective receptors and stabilize a form of heteromer that resulted in altered signalling pathways (Yekkirala et al., 2013). Given that a change in spacer length of roughly 5 Å led to a 10-fold shift in analgesic potency, altered the development of tolerance and promoted dramatic differences in downstream signalling (Daniels et al., 2005; Yekkirala et al., 2013), it appears that MOP-DOP heteromers are highly sensitive to variations in ligand binding of each protomer in the overall heteromer complex. The finding that co-delivery of the two monovalent analogues (one pharmacophore with a spacer) of a bivalent ligand (two pharmacophores connected by a chemical spacer) did not produce the same effects as the bivalent ligand suggests that stabilization of a highly specific conformation of a heteromeric receptor complex must be achieved for the observed changes in signalling to occur (Daniels et al., 2005; Yekkirala et al., 2013). Our observation that modest differences in the structure of the DOP receptor agonist injected with morphine can determine which signalling pathway is utilized could be explained by the induction of different heteromeric receptor complex conformations.
The fact that morphine + DELT-I yielded the same result as morphine + DELT-II is not surprising given that these two deltorphin analogues share a nearly identical structure. The difference in result when morphine was combined with DELT rather than DELT-I/DELT-II suggested that the specific structure of the peptide agonist used might determine the signalling bias of the combination. We explored this possibility by synthesizing and testing four deltorphin analogue peptides. While the results of these experiments confirmed that changes to the peptide structure were crucial to determining the agonism produced, the results failed to illuminate the key characteristics distinguishing DELT-I/DELT-II from DELT (see Supporting Information).
The fact that we achieved comparable results with morphine + DELT-II/DELT-I and morphine + OMI suggests that this phenomenon of biased agonism, which only occurs when certain DOP receptor agonists are delivered in combination with morphine, is not unique to opioid peptides. Interestingly, OCI delivered in combination with morphine yielded PKCε-independent synergy (analogous to the morphine SNC-80 combination), unlike morphine-OMI synergy, which was PKCε-dependent (analogous to the morphine DELT-I or DELT-II combinations). Oxymorphindole and oxycodindole are small-molecule DOP receptor agonists. They differ structurally by the 3-phenol, which is masked as a methyl ether in OCI (Figure 5). It is unclear how this difference can account for the difference in requirement of PKCε for spinal antinociceptive synergy with morphine. However, it is apparent that OMI and DELT-I/DELT-II interact functionally with morphine at the spinal level in a very similar way despite large structural differences.
Figure 5.

Variation in DOP receptor agonist structure corresponds to outcome of intrathecal co-delivery with morphine. Table illustrates outcomes of morphine delivered in combination with each DOP receptor agonist in PKCε-WT and PKCε-KO. ‘Syn’ or ‘Add’ indicates a synergistic or additive interaction respectively as determined by isobolographic analysis. Interaction index values are given in parentheses as a secondary measure of synergism (or lack thereof). A value of 1 indicates pure additivity. Increasing distance of values from 1, either above or below, indicates departure from additivity, towards either antagonism or synergism respectively. Morphine structure is illustrated at the top left. Structures for each DOP receptor agonist are shown on the right with coloured highlights indicating specific structural differences between similar agonists yielding opposing outcomes with morphine. For morphine, n = 12 in PKCε-WT and n = 11 in PKCε-KO. For DELT-II, n = 11 in PKCε-WT and n = 12 in PKCε-KO. For all other groups, n = 6 for PKCε-WT and PKCε-KO.
Comparisons of the ligands based solely on DOP receptor affinity or efficacy (Clark et al., 1997) (in vitro individual cloned receptors or homomers) do not display any consistent pattern relating to the current results. This lack of a cohesive pattern with receptor homomer data implies that the agonists are acting at MOP-DOP heteromers, which, in turn, implies that the receptors are co-localized in the same neurons. For example, if MOP and DOP receptors were activated in different cells, it would be expected that previous data using receptor homomers would be better able to predict requirement of PKCε for synergy as receptors acting independently should produce consistent responses for a given agonist. Furthermore, several studies have shown that MOP and DOP receptor agonists interact at the level of single cells to modulate spinal pain signalling (Cahill et al., 1996; Daniels et al., 2005; Gupta et al., 2010; Wang et al., 2010; Beaudry et al., 2011), and it has recently been shown that other agonist combinations requiring PKCε for synergy act on receptors co-located within the same cells (Overland et al., 2009; Schuster et al., 2013). The preceding points along with the evidence presented for involvement of receptor heteromers suggest that the differential effects of co-delivered ligands occur due to simultaneous binding of both protomers of a MOP-DOP heteromer. These results serve as another example of the high sensitivity of GPCRs to ligand structure and binding, and further illustrate the increased complexity in opioid receptor signalling made possible by co-delivery of two agonists.
Specific morphine signalling mechanisms at MOP-DOP heteromers may be relevant to our current results; Rozenfeld and Devi (2007) found that formation of MOP-DOP heteromers results in constitutive interaction with β-arrestin2 rather than the interaction with Gαi observed with homomeric receptors. Interestingly, it has been shown that knockout of β-arrestin2 results in enhanced morphine analgesia (Bohn et al., 1999; 2002), and that this effect is specific to morphine, as it was not observed with the MOP receptor agonists fentanyl or oxycodone (Raehal and Bohn, 2011). This morphine-specific effect is pertinent to our current results considering that morphine + DELT-II resulted in a different signalling interaction than fentanyl + DELT-II and oxycodone + DELT-II. Interestingly, Zheng et al. (2011) showed another difference between morphine and other MOP receptor agonists (including DAMGO and fentanyl) in that under basal conditions morphine resulted in significantly increased activation of PKCε compared to the other agonists. Several recent studies have indicated that morphine-induced signalling differs from that of other MOP receptor agonists (Borgland et al., 2003; Raehal and Bohn, 2011; Zheng et al., 2011; Pradhan et al., 2012). Our results that no other MOP receptor agonist besides morphine produced PKCε-dependent synergy with DELT-II mirror the recent indication that morphine is an agonist with functional properties highly distinct from other MOP receptor agonists.
It is thought that morphine alone can lead to activation of PKCε (Zheng et al., 2011), but it is unclear how PKCε is involved in synergistic interactions between morphine and some DOP receptor agonists. Mittal et al. (2012) found that inhibition of PKC attenuated the enhanced morphine analgesia observed in β-arrestin2 knockout animals, although a specific PKC isoform was not identified. This mechanism provides one possible explanation of our current results in that simultaneous binding of morphine and an appropriate DOP receptor ligand with the individual protomers in a heteromer may cause dissociation from β-arrestin2 and enhanced analgesia through a PKCε-dependent mechanism. Another possible mechanism for involvement of PKCε in analgesic synergy stems from involvement of PKCε in receptor trafficking. In a basal state, DOP receptors are thought to be located primarily in intracellular locations, and trafficked to the surface via large dense-core vesicles (LDCVs) upon various stimuli such as exposure to a DOP receptor agonist (Bao et al., 2003; Zhao et al., 2011) or treatment with morphine (Cahill et al., 2001; Morinville et al., 2003; Gendron et al., 2006). Interestingly, Sweitzer et al. (2004) found that inhibition of PKCε reduced stimulus-evoked release of calcitonin gene-related peptide, which is transported in LDCVs similar to DOP receptors (Zhao et al., 2011). In line with these observations, it has been shown that inhibition of PKC prevents stimulus-induced increases in functional competence of DOP receptors (Patwardhan et al., 2005; Rowan et al., 2009). There is also evidence that PKCε is involved in vesicular trafficking and regulation of surface availability of other receptors (Csukai et al., 1997; Chou et al., 2010). Although there is clearly a link between PKCε, DOP receptors and MOP receptors, a straightforward increase in functional competence of DOP receptors via activation of PKCε by MOP receptors cannot explain why so few combinations of a DOP agonist with morphine result in PKCε-dependent synergy.
Bias in signalling of GPCRs upon binding of a defined agonist has become an increasingly well-accepted concept. The current report is the first to demonstrate this phenomenon at the level of interactions between two opioid agonists that are co-delivered at the spinal level in vivo. These results expand the potential utility of opioid agonist co-delivery to include activation of specific signalling mechanisms that cannot be achieved with delivery of a single agonist.
Acknowledgments
These studies were made possible by the generous support of the National Institutes of Health through an R01 DA015438, NIDA (G. L. W.); P01 NS053709, NINDS (R. O. M.); T32 NS048944, NINDS (Timothy J. Ebner) and an F31 NS063634, NINDS (D. J. S.). We thank Phillip S. Portoghese for providing α-oxymorphamine, oxymorphindole and oxycodindole.
Glossary
Abbreviations
- CL
confidence limit
- DELT
deltorphin
- DELT-I
deltorphin I
- DELT-II
deltorphin II
- DOP
δ opioid peptide
- MOP
μ opioid peptide
- OCI
oxycodindole
- OMI
oxymorphindole
- MPE
maximum possible effect
Author contributions
D. J. S. designed experiments, analysed data, interpreted data, prepared figures, and wrote and edited the manuscript. M. D. M. designed experiments, interpreted data, and wrote and edited the manuscript. K. F. K. performed all experiments and collected the data. R. O. M. provided the breeding stock for the PKCε-KO mice, and edited the manuscript. C. A. F. edited the manuscript. G. L. W. designed experiments, interpreted the data, wrote and edited the manuscript, and provided funding.
Conflicts of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
http://dx.doi.org/10.1111/bph.12774
Figure S1 Illustration of differences between various deltorphin peptides tested for PKCε-dependent synergy with morphine. (A) Deltorphin structure is shown on the left. Synthesized peptides are shown on the right with differences from deltorphin highlighted by coloured boxes. (B) Deltorphin II structure is shown on the left. Synthesized peptides are shown on the right with differences from deltorphin II highlighted by coloured boxes.
Table S1 Morphine in combination with deltorphin analogue peptides.
References
- Abul-Husn NS, Sutak M, Milne B, Jhamandas K. Augmentation of spinal morphine analgesia and inhibition of tolerance by low doses of mu- and delta-opioid receptor antagonists. Br J Pharmacol. 2007;151:877–887. doi: 10.1038/sj.bjp.0707277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. CGTP Collaborators. The Concise Guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013;170:1459–1562. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao L, Jin SX, Zhang C, Wang LH, Xu ZZ, Zhang FX, et al. Activation of delta opioid receptors induces receptor insertion and neuropeptide secretion. Neuron. 2003;37:121–133. doi: 10.1016/s0896-6273(02)01103-0. [DOI] [PubMed] [Google Scholar]
- Beaudry H, Dubois D, Gendron L. Activation of spinal mu- and delta-opioid receptors potently inhibits substance P release induced by peripheral noxious stimuli. J Neurosci. 2011;31:13068–13077. doi: 10.1523/JNEUROSCI.1817-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT. Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science. 1999;286:2495–2498. doi: 10.1126/science.286.5449.2495. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Lefkowitz RJ, Caron MG. Differential mechanisms of morphine antinociceptive tolerance revealed in (beta)arrestin-2 knock-out mice. J Neurosci. 2002;22:10494–10500. doi: 10.1523/JNEUROSCI.22-23-10494.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgland SL, Connor M, Osborne PB, Furness JB, Christie MJ. Opioid agonists have different efficacy profiles for G protein activation, rapid desensitization, and endocytosis of mu-opioid receptors. J Biol Chem. 2003;278:18776–18784. doi: 10.1074/jbc.M300525200. [DOI] [PubMed] [Google Scholar]
- Cahill CM, White TD, Sawynok J. Synergy between mu/delta-opioid receptors mediates adenosine release from spinal cord synaptosomes. Eur J Pharmacol. 1996;298:45–49. doi: 10.1016/0014-2999(95)00775-x. [DOI] [PubMed] [Google Scholar]
- Cahill CM, Morinville A, Lee MC, Vincent JP, Collier B, Beaudet A. Prolonged morphine treatment targets delta opioid receptors to neuronal plasma membranes and enhances delta-mediated antinociception. J Neurosci. 2001;21:7598–7607. doi: 10.1523/JNEUROSCI.21-19-07598.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou WH, Wang D, McMahon T, Qi ZH, Song M, Zhang C, et al. GABAA receptor trafficking is regulated by protein kinase C(epsilon) and the N-ethylmaleimide-sensitive factor. J Neurosci. 2010;30:13955–13965. doi: 10.1523/JNEUROSCI.0270-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark MJ, Emmerson PJ, Mansour A, Akil H, Woods JH, Portoghese PS, et al. Opioid efficacy in a C6 glioma cell line stably expressing the delta opioid receptor. J Pharmacol Exp Ther. 1997;283:501–510. [PubMed] [Google Scholar]
- Costantino CM, Gomes I, Stockton SD, Lim MP, Devi LA. Opioid receptor heteromers in analgesia. Expert Rev Mol Med. 2012;14:e9. doi: 10.1017/erm.2012.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox BM, Christie MJ, Devi L, Toll L, Traynor JR. Challenges for opioid receptor nomenclature: IUPHAR Review 9. Br J Pharmacol. 2015;172:317–323. doi: 10.1111/bph.12612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csukai M, Chen CH, De Matteis MA, Mochly-Rosen D. The coatomer protein beta'-COP, a selective binding protein (RACK) for protein kinase Cepsilon. J Biol Chem. 1997;272:29200–29206. doi: 10.1074/jbc.272.46.29200. [DOI] [PubMed] [Google Scholar]
- Daniels DJ, Lenard NR, Etienne CL, Law PY, Roerig SC, Portoghese PS. Opioid-induced tolerance and dependence in mice is modulated by the distance between pharmacophores in a bivalent ligand series. Proc Natl Acad Sci U S A. 2005;102:19208–19213. doi: 10.1073/pnas.0506627102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairbanks CA. Spinal delivery of analgesics in experimental models of pain and analgesia. Adv Drug Deliv Rev. 2003;55:1007–1041. doi: 10.1016/s0169-409x(03)00101-7. [DOI] [PubMed] [Google Scholar]
- Fairbanks CA, Wilcox GL. Moxonidine, a selective alpha2-adrenergic and imidazoline receptor agonist, produces spinal antinociception in mice. J Pharmacol Exp Ther. 1999;290:403–412. [PubMed] [Google Scholar]
- Fan T, Varghese G, Nguyen T, Tse R, O'Dowd BF, George SR. A role for the distal carboxyl tails in generating the novel pharmacology and G protein activation profile of mu and delta opioid receptor hetero-oligomers. J Biol Chem. 2005;280:38478–38488. doi: 10.1074/jbc.M505644200. [DOI] [PubMed] [Google Scholar]
- Gendron L, Lucido AL, Mennicken F, O'Donnell D, Vincent JP, Stroh T, et al. Morphine and pain-related stimuli enhance cell surface availability of somatic delta-opioid receptors in rat dorsal root ganglia. J Neurosci. 2006;26:953–962. doi: 10.1523/JNEUROSCI.3598-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendron L, Esdaile MJ, Mennicken F, Pan H, O'Donnell D, Vincent JP, et al. Morphine priming in rats with chronic inflammation reveals a dichotomy between antihyperalgesic and antinociceptive properties of deltorphin. Neuroscience. 2007;144:263–274. doi: 10.1016/j.neuroscience.2006.08.077. [DOI] [PubMed] [Google Scholar]
- George SR, Fan T, Xie Z, Tse R, Tam V, Varghese G, et al. Oligomerization of mu- and delta-opioid receptors. Generation of novel functional properties. J Biol Chem. 2000;275:26128–26135. doi: 10.1074/jbc.M000345200. [DOI] [PubMed] [Google Scholar]
- Gomes I, Jordan BA, Gupta A, Trapaidze N, Nagy V, Devi LA. Heterodimerization of mu and delta opioid receptors: a role in opiate synergy. J Neurosci. 2000;20:RC110. doi: 10.1523/JNEUROSCI.20-22-j0007.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes I, Gupta A, Filipovska J, Szeto HH, Pintar JE, Devi LA. A role for heterodimerization of mu and delta opiate receptors in enhancing morphine analgesia. Proc Natl Acad Sci U S A. 2004;101:5135–5139. doi: 10.1073/pnas.0307601101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A, Mulder J, Gomes I, Rozenfeld R, Bushlin I, Ong E, et al. Increased abundance of opioid receptor heteromers after chronic morphine administration. Sci Signal. 2010;3:ra54. doi: 10.1126/scisignal.2000807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He SQ, Zhang ZN, Guan JS, Liu HR, Zhao B, Wang HB, et al. Facilitation of mu-opioid receptor activity by preventing delta-opioid receptor-mediated codegradation. Neuron. 2011;69:120–131. doi: 10.1016/j.neuron.2010.12.001. [DOI] [PubMed] [Google Scholar]
- Hylden JL, Wilcox GL. Intrathecal morphine in mice: a new technique. Eur J Pharmacol. 1980;67:313–316. doi: 10.1016/0014-2999(80)90515-4. [DOI] [PubMed] [Google Scholar]
- Janssen PA, Niemegeers CJ, Dony JG. The inhibitory effect of fentanyl and other morphine-like analgesics on the warm water induced tail withdrawl reflex in rats. Arzneimittelforschung. 1963;13:502–507. [PubMed] [Google Scholar]
- Joseph EK, Levine JD. Mu and delta opioid receptors on nociceptors attenuate mechanical hyperalgesia in rat. Neuroscience. 2010;171:344–350. doi: 10.1016/j.neuroscience.2010.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khasar SG, Lin YH, Martin A, Dadgar J, McMahon T, Wang D, et al. A novel nociceptor signaling pathway revealed in protein kinase C epsilon mutant mice. Neuron. 1999;24:253–260. doi: 10.1016/s0896-6273(00)80837-5. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalf MD, Yekkirala AS, Powers MD, Kitto KF, Fairbanks CA, Wilcox GL, et al. The delta opioid receptor agonist SNC80 selectively activates heteromeric mu-delta opioid receptors. ACS Chem Neurosci. 2012;3:505–509. doi: 10.1021/cn3000394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miaskowski C, Sutters KA, Taiwo YO, Levine JD. Antinociceptive and motor effects of delta/mu and kappa/mu combinations of intrathecal opioid agonists. Pain. 1992;49:137–144. doi: 10.1016/0304-3959(92)90200-U. [DOI] [PubMed] [Google Scholar]
- Mittal N, Tan M, Egbuta O, Desai N, Crawford C, Xie CW, et al. Evidence that behavioral phenotypes of morphine in beta-arr2-/- mice are due to the unmasking of JNK signaling. Neuropsychopharmacology. 2012;37:1953–1962. doi: 10.1038/npp.2012.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morinville A, Cahill CM, Esdaile MJ, Aibak H, Collier B, Kieffer BL, et al. Regulation of delta-opioid receptor trafficking via mu-opioid receptor stimulation: evidence from mu-opioid receptor knock-out mice. J Neurosci. 2003;23:4888–4898. doi: 10.1523/JNEUROSCI.23-12-04888.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossipov MH, Lopez Y, Bian D, Nichols ML, Porreca F. Synergistic antinociceptive interactions of morphine and clonidine in rats with nerve-ligation injury. Anesthesiology. 1997;86:196–204. doi: 10.1097/00000542-199701000-00024. [DOI] [PubMed] [Google Scholar]
- Ossipov MH, Suarez LJ, Spaulding TC. Antinociceptive interactions between alpha2-adrenergic and opiate agonists at the spinal level in rodents. Anesth Analg. 1989;68:194–200. [PubMed] [Google Scholar]
- Overland AC, Kitto KF, Chabot-Dore AJ, Rothwell PE, Fairbanks CA, Stone LS, et al. Protein kinase C mediates the synergistic interaction between agonists acting at alpha2-adrenergic and delta-opioid receptors in spinal cord. J Neurosci. 2009;29:13264–13273. doi: 10.1523/JNEUROSCI.1907-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patwardhan AM, Berg KA, Akopain AN, Jeske NA, Gamper N, Clarke WP, et al. Bradykinin-induced functional competence and trafficking of the delta-opioid receptor in trigeminal nociceptors. J Neurosci. 2005;25:8825–8832. doi: 10.1523/JNEUROSCI.0160-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan AA, Smith ML, Kieffer BL, Evans CJ. Ligand-directed signalling within the opioid receptor family. Br J Pharmacol. 2012;167:960–969. doi: 10.1111/j.1476-5381.2012.02075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raehal KM, Bohn LM. The role of beta-arrestin2 in the severity of antinociceptive tolerance and physical dependence induced by different opioid pain therapeutics. Neuropharmacology. 2011;60:58–65. doi: 10.1016/j.neuropharm.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl MS, Schnell SA, Overland AC, Chabot-Dore AJ, Taylor AM, Ribeiro-da-Silva A, et al. Coexpression of alpha 2A-adrenergic and delta-opioid receptors in substance P-containing terminals in rat dorsal horn. J Comp Neurol. 2009;513:385–398. doi: 10.1002/cne.21982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan MP, Ruparel NB, Patwardhan AM, Berg KA, Clarke WP, Hargreaves KM. Peripheral delta opioid receptors require priming for functional competence in vivo. Eur J Pharmacol. 2009;602:283–287. doi: 10.1016/j.ejphar.2008.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozenfeld R, Devi LA. Receptor heterodimerization leads to a switch in signaling: beta-arrestin2-mediated ERK activation by mu-delta opioid receptor heterodimers. FASEB J. 2007;21:2455–2465. doi: 10.1096/fj.06-7793com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherrer G, Imamachi N, Cao YQ, Contet C, Mennicken F, O'Donnell D, et al. Dissociation of the opioid receptor mechanisms that control mechanical and heat pain. Cell. 2009;137:1148–1159. doi: 10.1016/j.cell.2009.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramm CL, Honda CN. Co-administration of delta- and mu-opioid receptor agonists promotes peripheral opioid receptor function. Pain. 2010;151:763–770. doi: 10.1016/j.pain.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster DJ, Kitto KF, Overland AC, Messing RO, Stone LS, Fairbanks CA, et al. Protein kinase C{epsilon} is required for spinal analgesic synergy between delta opioid and alpha-2a adrenergic receptor agonist pairs. J Neurosci. 2013;33:13538–13546. doi: 10.1523/JNEUROSCI.4013-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin SW, Eisenach JC. Intrathecal morphine reduces the visceromotor response to acute uterine cervical distension in an estrogen-independent manner. Anesthesiology. 2003;98:1467–1471. doi: 10.1097/00000542-200306000-00025. discussion 1466A. [DOI] [PubMed] [Google Scholar]
- Sutters KA, Miaskowski C, Taiwo YO, Levine JD. Analgesic synergy and improved motor function produced by combinations of mu-delta- and mu-kappa-opioids. Brain Res. 1990;530:290–294. doi: 10.1016/0006-8993(90)91297-t. [DOI] [PubMed] [Google Scholar]
- Sweitzer SM, Wong SM, Peters MC, Mochly-Rosen D, Yeomans DC, Kendig JJ. Protein kinase C epsilon and gamma: involvement in formalin-induced nociception in neonatal rats. J Pharmacol Exp Ther. 2004;309:616–625. doi: 10.1124/jpet.103.060350. [DOI] [PubMed] [Google Scholar]
- Tallarida R, Murray R. Manual of Pharmacological Calculations with Computer Programs. New York: Springer; 1987. [Google Scholar]
- Tallarida RJ. The interaction index: a measure of drug synergism. Pain. 2002;98:163–168. doi: 10.1016/s0304-3959(02)00041-6. [DOI] [PubMed] [Google Scholar]
- Tallarida RJ, Porreca F, Cowan A. Statistical analysis of drug-drug and site-site interactions with isobolograms. Life Sci. 1989;45:947–961. doi: 10.1016/0024-3205(89)90148-3. [DOI] [PubMed] [Google Scholar]
- Wang HB, Zhao B, Zhong YQ, Li KC, Li ZY, Wang Q, et al. Coexpression of delta- and mu-opioid receptors in nociceptive sensory neurons. Proc Natl Acad Sci U S A. 2010;107:13117–13122. doi: 10.1073/pnas.1008382107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yekkirala AS, Kalyuzhny AE, Portoghese PS. Standard opioid agonists activate heteromeric opioid receptors: evidence for morphine and [d-Ala(2)-MePhe(4)-Glyol(5)]enkephalin as selective mu-delta agonists. ACS Chem Neurosci. 2010;1:146–154. doi: 10.1021/cn9000236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yekkirala AS, Kalyuzhny AE, Portoghese PS. An immunocytochemical-derived correlate for evaluating the bridging of heteromeric mu-delta opioid protomers by bivalent ligands. ACS Chem Biol. 2013;8:1412–1416. doi: 10.1021/cb400113d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Wang HB, Lu YJ, Hu JW, Bao L, Zhang X. Transport of receptors, receptor signaling complexes and ion channels via neuropeptide-secretory vesicles. Cell Res. 2011;21:741–753. doi: 10.1038/cr.2011.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Chu J, Zhang Y, Loh HH, Law PY. Modulating micro-opioid receptor phosphorylation switches agonist-dependent signaling as reflected in PKCepsilon activation and dendritic spine stability. J Biol Chem. 2011;286:12724–12733. doi: 10.1074/jbc.M110.177089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, King MA, Schuller AG, Nitsche JF, Reidl M, Elde RP, et al. Retention of supraspinal delta-like analgesia and loss of morphine tolerance in delta opioid receptor knockout mice. Neuron. 1999;24:243–252. doi: 10.1016/s0896-6273(00)80836-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Illustration of differences between various deltorphin peptides tested for PKCε-dependent synergy with morphine. (A) Deltorphin structure is shown on the left. Synthesized peptides are shown on the right with differences from deltorphin highlighted by coloured boxes. (B) Deltorphin II structure is shown on the left. Synthesized peptides are shown on the right with differences from deltorphin II highlighted by coloured boxes.
Table S1 Morphine in combination with deltorphin analogue peptides.