

Highlights
-
•
Expression of recombinant human carboxylesterase I in E. coli is mainly insoluble.
-
•
Refolding using a combination of 1% glycerol and 2 mM β-mercaptoethanol in Tris–HCl, pH 7.5 significantly improved solubility.
-
•
Purified recombinant human CES1 is functionally active and stable.
-
•
We provided efficient method to produce large amount and catalytically active CES1.
Keywords: Carboxylesterases, E. coli, Refolding, Inclusion bodies, Glycerol
Abstract
Human liver carboxylesterase 1 (CES1) plays a critical role in the hydrolysis of various ester- and amide-containing molecules, including active metabolites, drugs and prodrugs. However, it has been problematic to express recombinant CES1 in bacterial expression systems due to low solubility, with the CES1 protein being mainly expressed in inclusion bodies, accompanied by insufficient purity issues. In this study, we report an efficient in vitro method for refolding recombinant CES1 from inclusion bodies. A one-step purification with an immobilized-metal affinity column was utilized to purify His-tagged recombinant CES1. Conveniently, both denaturant and imidazole can be removed while the enzyme is refolded via buffer exchange, a dilution method. We show that the refolding of recombinant CES1 was successful in Tris–HCl at pH 7.5 containing a combination of 1% glycerol and 2 mM β-mercaptoethanol, whereas a mixture of other additives (trehalose, sorbitol and sucrose) and β-mercaptoethanol failed to recover a functional protein. His-tagged recombinant CES1 retains its biological activity after refolding and can be used directly without removing the fusion tag. Altogether, our results provide an alternative method for obtaining a substantial amount of functionally active protein, which is advantageous for further investigations such as structural and functional studies.
Introduction
Human liver carboxylesterase (CES1, EC number: 3.1.1.1, 3.1.1.56), an enzyme responsible for the hydrolysis of ester- and amide-containing molecules, is mainly expressed in the liver where it is crucial for the processing of active metabolites (heroin and cocaine) and is also involved in trans-esterification reactions [1–4]. In addition, CES1 is known to play an important role in the hydrolysis of several classes of drugs and the activation of prodrugs, including angiotensin-converting enzyme (ACE)1 inhibitors and the anti-influenza agent oseltamivir [5–10], also known as Tamiflu, a recommended treatment by the World Health Organization (WHO). Oseltamivir is a prodrug in the form of oseltamivir phosphate, which requires conversion into its active carboxylate form by CES1. Two known CES1 mutations, G143E and p.D260fs, have been shown to impair the function of the enzyme with regard to the activation of oseltamivir, indicating the significant role of CES1 in oseltamivir metabolism [8,9].
Extensive studies have been carried out to investigate the role of CES1 in oseltamivir metabolism in terms of pharmacokinetics, structure and function. Accordingly, different expression systems, purification processes and enzymatic assays have been established, with most previous works employing conventional cell-culture techniques to produce a functional enzyme. Mammalian cell lines, COS7 and Flp-In-293 with the pCDNA5/FRT/V5-His TOPOTA expression system, have frequently been utilized for the production of CES1. Wild-type (WT) as well as mutant proteins have been expressed and investigated with regard to hydrolysis, metabolism and activation of several compounds mediated by CES1, such as p-nitrophenyl acetate (pNPA), oseltamivir, methylphenidate, trandolapril and clopidogrel and its metabolite [6–10]. Insect cells, Sf9 and Sf21, are also preferred for expressing CES1, which is accomplished using a baculovirus expression system [2,3,11,12]. In addition to insect cell culture, there was a report in which Trichoplusia ni whole-insect larvae were used as a production source of CES1 recombinant protein [13]. Finally, yeast expression systems involving Saccharomyces cerevisiae and Pichia pastoris are also used for the expression of CES1 [14]. Although eukaryotic expression systems are suitable for expressing eukaryotic proteins, especially when post-translational modifications are needed, they are relatively complex, expensive and require long time periods to obtain the desired protein compared to bacterial expression systems. Furthermore, eukaryotic expression systems typically yield only small to moderate amounts of protein.
As an alternative to expression in eukaryotic cells, bacteria are among the most extensively hosts used for protein production because expressing proteins in bacterial systems is inexpensive and easy to handle and large amounts of protein are achievable [15,16]. A previous attempt to express CES1 in bacteria failed to produce the functional protein because CES1 was mainly expressed in inclusion bodies [14]. Indeed, this is a major problem with expressing human proteins in bacteria, as the proteins are misfolded and aggregate, preventing the production of a functional eukaryotic protein [17–20]. As a result, various methods have been established to overcome insolubility issues: fusion tags and growth temperature, auto-induction and purification conditions have all been optimized to improve the solubility of proteins expressed in Escherichia coli [21–23]. Moreover, various types of folding aids are employed in bacterial systems. E. coli molecular chaperones, e.g., GroES and GroEL, have shown to promote protein folding when co-expressed with human aromatase and aldehyde dehydrogenase enzymes [24,25]. Additionally, it has been proven that including some chemical chaperones, e.g., polyols, amino acids and polyethylene glycol, in the growth medium is beneficial for producing soluble and functional proteins [26–28]. However, when the aforementioned methods are unsuccessful and the protein is still mainly expressed as inclusion bodies, efficient protocols for refolding proteins is, therefore, necessary to produce biologically active proteins, which will be advantageous for structural and functional studies. Many effective protocols for refolding proteins from inclusion bodies have been established and proven to successfully produce active enzymes. The refolding of proteins can mainly be achieved in two different ways: dialysis and dilution. During the refolding process, chemical chaperons or additives are usually added to assist in protein conformation, and additives such as trehalose, glycerol, sorbitol, arginine and sucrose have been reported to be advantageous for the refolding step [17–19,29–33].
In the present study, we report the heterologous expression of CES1 in E. coli, in which it was mainly expressed in inclusion bodies. Thereafter, different refolding conditions were applied to retain the catalytically active enzyme. In addition to WT CES1, three natural variants (S76N, D204E and A270S) were expressed, purified, refolded and characterized.
Materials and methods
Plasmid constructs and site-directed mutagenesis
In this study, we investigated the expression, purification and refolding of CES1 isoform a (NCBI Reference Sequence: NP_001020366.1), which is one and two amino acids longer than isoforms b and c, respectively. Therefore, the numbering of amino acid residues is herein shifted by one residue compared to previous reports. The CES1 gene was amplified from human liver cDNA (BD Biosciences) using Platinum Taq DNA polymerase (Life Technologies) with a forward primer GGATCCGAATTCCATCCGTCCTCGC and a reverse primer GGTGCTCGAGTCACAGCTCTATGTG. The amplified region was designed to start at amino acid 20, omitting the first 19 amino acids of the signal peptide to prevent complications of post-translational modification. Two restriction enzyme sites were introduced, EcoRI and XhoI, in the forward and reverse primers, respectively (underlined). PCR products were gel purified and digested with EcoRI and XhoI (New England Biolabs). The digested amplicon (encoding 20–568 residues) was cloned into the pET28a expression vector with an N-terminal His-tagged and transformed into the E. coli expression host BL21 (DE3). Three constructs of CES1 natural mutations, S76N, D204E and A270S, were generated using a site-directed mutagenesis kit (Agilent Technologies) with specific primers. All constructs were verified by bidirectional DNA sequencing and restriction digestion to confirm that the desired plasmids were obtained.
Expression of recombinant CES1
A single colony of BL21 (DE3) harboring the recombinant plasmid was inoculated in 5 ml of LB medium containing 50 μg/ml kanamycin and incubated at 37 °C with 250 rpm shaking overnight. The fresh overnight cultures were inoculated in LB medium in the presence of 50 μg/ml kanamycin at a dilution of 1:100 and grown at 25 °C with 250 rpm shaking until the absorbance at 600 nm (OD600) reached 0.3. Then, the cultures were induced with isopropyl-β-d-thiogalactopyranoside (IPTG, Merck) at a final concentration of 0.5 mM and were further incubated at 25 °C with 200 rpm shaking for 4 h before being harvested by centrifugation at 1000×g for 10 min.
Purification of recombinant CES1
The CES1 protein was purified under denaturing condition due to its main expression in the insoluble fraction. Cell pellets were resuspended in lysis buffer (20 mM sodium phosphate, pH 7.4, 300 mM NaCl, 10 mM imidazole and 2 mM β-mercaptoethanol) and disrupted by sonication. The cell lysate was centrifuged at 20,000×g for 45 min at 4 °C, and the supernatant was removed. To reduce protein interference, the pellet was washed with 2 M urea in lysis buffer by incubating on ice for 15 min and then subjected to centrifugation at 20,000×g for 45 min at 4 °C. The inclusion bodies were solubilized in buffer A (lysis buffer containing 8 M urea) by incubating the solution on ice for 1 h. The solubilized solution was centrifuged for 1 h at 20,000×g and 4 °C. The supernatant was collected and incubated with pre-equilibrated TALON Metal Affinity Resin (BD Biosciences) in buffer A at 4 °C for at least 1 h. The unbound proteins were removed with wash buffer (20 mM sodium phosphate, pH 7.4, 300 mM NaCl, 8 M urea, 20 mM imidazole and 2 mM β-mercaptoethanol; 4 times, 6 ml each). The elution of CES1 was accomplished with elute buffer (20 mM sodium phosphate, pH 7.4, 300 mM NaCl, 8 M urea and 250 mM imidazole, 2 mM β-mercaptoethanol; for 5 times, 4 ml each).
In vitro refolding of recombinant CES1 using the dilution method
In the refolding step, urea (protein denaturant) and imidazole were gradually removed via dilution. This procedure was performed while the protein was refolded by buffer exchange with 50 mM Tris–HCl, pH 7.5, containing different combinations of additives and reducing agents (Table 1) using Amicon Ultra centrifugal filter devices (Millipore). The protein solution was mixed with refolding buffer and subjected to centrifugation at 2465×g (swing bucket) and 4 °C for 10–15 min. The flow-through was discarded; the refolding buffer was added again, and the sample was centrifuged. The step was repeated until the final concentration of both urea and imidazole was less than 0.01 mM. The purity of protein was visualized by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE), and the enzymatic activity was measured as described below. The protein concentration was measured by the Nanodrop method [34] using an extinction coefficient at 280 nm of 86,860 M−1 cm−1 calculated, as with the Protparam server [35].
Table 1.
Refolding conditions for recombinant WT CES1.
| Additive | Reducing agent | Catalytically active CES1 |
|---|---|---|
| 0.2 M trehalose | 2 mM β-mercaptoethanol | No |
| 0.2 M sucrose | 2 mM β-mercaptoethanol | No |
| 0.2 M sorbitol | 2 mM β-mercaptoethanol | No |
| 1% glycerol | 2 mM β-mercaptoethanol | Yes |
| 1% glycerol | 2 mM DTT | No |
Intrinsic fluorescence analysis
Intrinsic fluorescence emission spectra of the refolded proteins (WT and mutants) were collected using a Synergy H1 hybrid reader (Biotek) with a 96-well plate at 25 °C. The excitation wavelength was 280 nm, and the emission spectra were monitored in the range of 300–450 nm. All the spectra were collected after refolding CES1 in 50 mM Tris–HCl, pH 7.5, containing 1% glycerol and 2 mM β-mercaptoethanol.
Circular dichroism (CD) spectrum analysis
Far UV-CD spectra were recorded using a Jasco spectrometer, model J-815, with a 1 mm pathlength. The measurements were carried out at room temperature. The spectra were collected over a wavelength range of 190–260 nm. CD spectra for WT and the three mutants were collected after refolding in 50 mM Tris–HCl, pH 7.5, containing 1% glycerol and 2 mM β-mercaptoethanol. Three scans were averaged for each protein sample, and the buffer was also subtracted.
Enzyme activity assay
CES1 activity was measured spectrophotometrically by following the conversion of pNPA to p-nitrophenol (pNP) at 405 nm using a UV-2700 UV–VIS spectrophotometer (Shimadzu). The substrate was dissolved in acetonitrile. The enzyme activity assay was performed in a cuvette with a final volume of 1 ml. In brief, the reaction mixture containing 930 μl of 50 mM Tris–HCl, pH 7.5, 40 μl absolute ethanol and 10 μl of various concentrations of pNPA was incubated at 37 °C for 5 min. After that, 20 μl of purified enzyme was added, and the reaction was further incubated at the same temperature for 10 min. The absorbance 405 nm was monitored for 10 min. Each reaction was conducted in four replicates, and the initial linear measurement was used for determining the slope of the initial velocity. Data from the spectrophotometer were exported to Excel to calculate the rate of product formation and were expressed as micromole of pNP produced per minute per milligram protein (μmol/min/mg) as calculated using the extinction coefficient for pNP at 405 nm (18,000 M−1 cm−1). Steady-state kinetic parameters, KM, kcat and Vmax, were obtained by fitting the collected data to the Michaelis–Menten equation using GraphPad Prism software (GraphPad Software).
Western blot analysis
A Western blot analysis was performed to confirm the expression of recombinant CES1. Purified proteins were loaded onto a 12% SDS-PAGE, separated by electrophoresis and transferred onto a polyvinylidene difluoride (PVDF) membrane. The membrane was blocked with phosphate-buffered saline (PBS) containing 5% skimmed milk at room temperature for 1 h and then incubated at 4 °C overnight with an anti-his antibody (Pierce), at a dilution of 1:3000 in the same solution. After washing 4 times with PBS containing 0.05% Tween 20 (PBST) for 15 min each, the membrane was incubated with a horseradish peroxidase (HRP)-conjugated anti-mouse secondary antibody (Pierce) diluted 1:2500 in PBST. Prior to development, the membrane was washed 4 times with PBST for 15 min each. Finally, the proteins were detected using the ECL system (Merck Millipore), and the membrane was visualized using an ImageQuant LAS4000 mini system (GE Healthcare Life Sciences).
Statistical analysis
All enzyme assays were conducted in four replicates, and an unpaired t-test of independent experiments was performed using GraphPad Prism software (GraphPad Software). The results were considered significant at a p-value ⩽ 0.05.
Results
Expression, purification and refolding of recombinant CES1
The cDNA coding region of CES1 isoform a (amino acid residues 20–568; NCBI Reference Sequence: NP_001020366.1) was amplified by PCR and cloned into the EcoRI and XhoI sites of the pET28a expression vector (Fig. 1) for recombinant expression in E. coli BL21 (DE3). It is worth noting that CES1 isoform a is one amino acid longer than the previously reported isoform b. Isoform a has a longer signal peptide, of 19 amino acids, whereas isoform b has a signal peptide of only 18. As a consequence, the numbering of amino acid residues in this study is shifted by one amino acid compared to the previous reports for isoform b. As shown in Fig. 2, recombinant CES1 protein was expressed differently when grown at different temperatures (37 °C and 25 °C before induction at 25 °C). It was notable that the expression of CES1 grown at 25 °C was much higher than in cells grown at 37 °C; however, the lower temperature resulted in significantly low solubility because of the large quantity of protein expressed. As a result, the protein was found in inclusion bodies; SDS–PAGE analysis revealed a band at around 62 kDa corresponding to human CES1. While CES1 grown at 37 °C produced comparable amount of protein in both soluble and insoluble fractions. Unfortunately, we were unable to obtain CES1 from the soluble fraction grown at 37 °C due to low purity. Therefore, attempts were made to improve the solubility of CES1, with additives into growth medium. However, neither glycerol nor sorbitol had an effect on CES1 solubility (Fig. 3), and the protein was purified under denaturing condition using 8 M urea to solubilize the misfolded protein. Purification of the His-tagged recombinant protein was accomplished in one step using affinity column chromatography. After purification, urea and imidazole were removed, as they interfere with the activity assay. The details of the purification of CES1s, both WT and mutants, are shown in Table 2. The purity of the purified protein was greater than 95%, as shown by SDS–PAGE (Fig. 4).
Fig. 1.

Amino acid sequence of CES1 isoform a. The first 19-amino acids, highlighted in gray, comprise the signal peptide sequence, which was omitted during cloning. Amino acids known to be associated with the biological function are boxed. Natural variants created in our experiment are underlined. The N-glycosylation site is circled, and disulfide bonds are shown by linkage of two cysteine residues.
Fig. 2.

SDS–PAGE analysis of recombinant WT CES1 expressed at different temperatures. (A) Cells were grown at 37 °C, induced with 0.5 mM IPTG and incubated at 25 °C for 4 h before harvest. Lane M, molecular mass marker proteins; lane 1, uninduced cells; lane 2, induced cells; lane 3, inclusion bodies and lane 4, soluble fraction. (B) Cells were grown at 25 °C and induced with 0.5 mM IPTG; lane 1, uninduced cells; lane 2, induced cells; lane 3, inclusion bodies and lane 4, soluble fraction. The arrow indicates the expected size of recombinant CES1.
Fig. 3.

SDS–PAGE analysis of recombinant WT CES1 expressed at 25 °C in the presence of additives in the growth medium. (A) Final concentration of 1% glycerol at the time of induction; Lane M, molecular mass marker proteins; lane 1, uninduced cells; lane 2, induced cells; lane 3, inclusion bodies and lane 4, soluble fraction. (B) A final concentration of 0.2 M sorbitol was added to growth medium at the time of induction; lane 1, uninduced cells; lane 2, induced cells; lane 3, inclusion bodies and lane 4, soluble fraction. The arrow indicates the expected size of recombinant CES1.
Table 2.
Purification of recombinant human CES1 from E. coli.
| Construct | Purification step | Total protein (mg) | Specific activity (μmol/min/mg) | Yield (%) | Refolding yield (%) |
|---|---|---|---|---|---|
| WT | Solubilized in 8 M urea | 27.1 | – | 100 | 9.0 |
| Cobalt resin column | 13.4 | – | 49.4 | ||
| Refolding | 1.2 | 73 | 4.4 | ||
| S76N | Solubilized in 8 M urea | 31.2 | – | 100 | 10.2 |
| Cobalt resin column | 16.6 | – | 53.2 | ||
| Refolding | 1.7 | 128 | 5.4 | ||
| D204E | Solubilized in 8 M urea | 25.7 | – | 100 | 19.1 |
| Cobalt resin column | 11.6 | – | 45.1 | ||
| Refolding | 2.21 | 193 | 8.6 | ||
| A270S | Solubilized in 8 M urea | 29.3 | – | 100 | 11.9 |
| Cobalt resin column | 12.6 | – | 43.0 | ||
| Refolding | 1.5 | 109 | 5.1 | ||
Fig. 4.
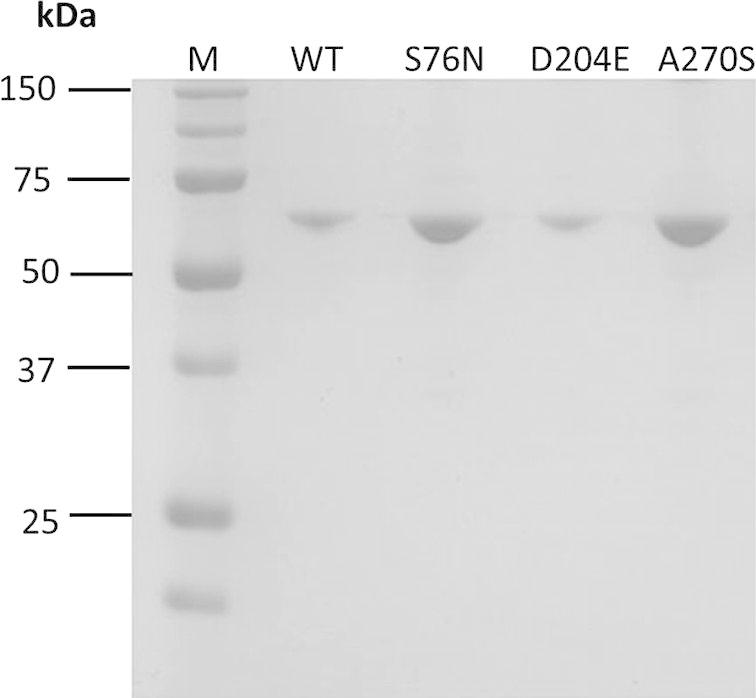
SDS–PAGE analysis of purified recombinant CES1s, WT, S76N, D204E and A270S, after refolding in 50 mM Tris–HCl, pH 7.5, containing 1% glycerol and 2 mM β-mercaptoethanol.
In this study, we used different combinations of various additives and reducing agents for refolding recombinant CES1 (Table 1). CES1 has four cysteine residues and two disulfide bonds (Fig. 1); therefore, the influence of reducing agents during the refolding process was investigated. Refolding buffer Tris–HCl, pH 7.5, containing a combination of sorbitol, trehalose, sucrose and β-mercaptoethanol failed to enhance solubility, resulting in non-functional enzyme (Table 1). Interestingly, only the combination of 1% glycerol and 2 mM β-mercaptoethanol was successful for refolding CES1, resulting in a functionally active enzyme. When β-mercaptoethanol was replaced with DTT, which is known to be an effective reducing agent, no enzymatic activity was observed. The proteins obtained after refolding were subjected to spectroscopic characterization, intrinsic fluorescence and CD analyses. The intrinsic fluorescence and CD spectra are shown in Figs. 5 and 6, respectively. The recombinant WT and mutant proteins exhibited similar patterns of intrinsic fluorescence and CD spectra, indicating that both WT and mutant CES1s share similar secondary structure, with α-helices as the main ordered structure.
Fig. 5.
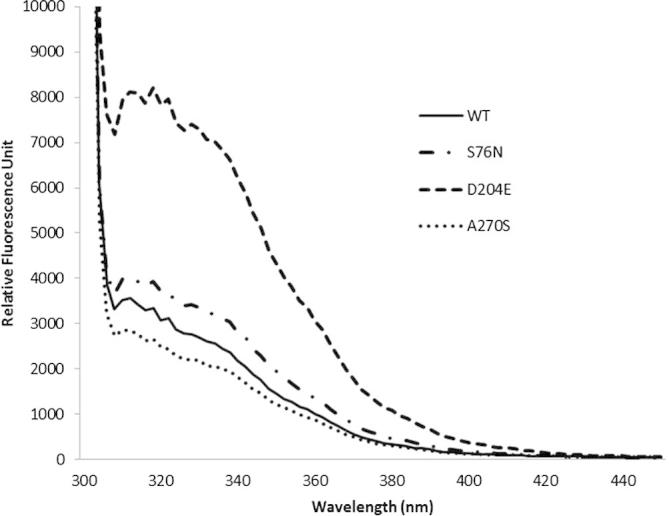
Intrinsic fluorescence emission spectra of refolded CES1s, WT, S76N, D204E and A270S. Protein refolding was performed in 50 mM Tris–HCl, pH 7.5, containing 1% glycerol and 2 mM β-mercaptoethanol.
Fig. 6.
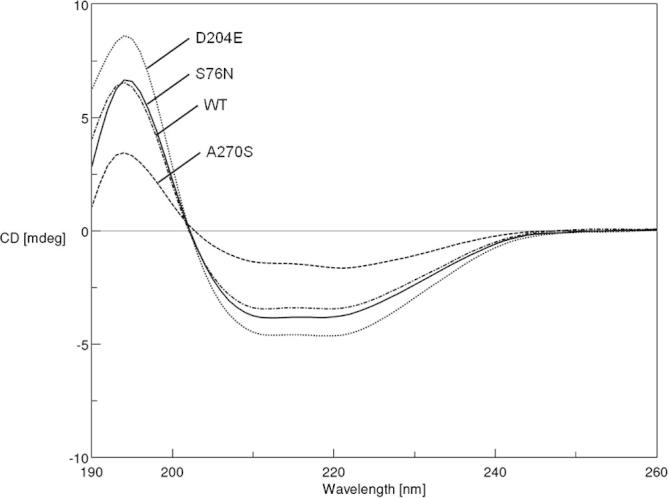
Circular dichroism (CD) spectra of refolded CES1s, WT, S76N, D204E and A270S. Protein refolding was performed in 50 mM Tris–HCl, pH 7.5, containing 1% glycerol and 2 mM β-mercaptoethanol.
Enzymatic activity assay
Enzymatic assays were performed in Tris–HCl, pH 7.5. The recombinant CES1 enzymes were catalytically active after refolding in buffer containing 1% glycerol and 2 mM β-mercaptoethanol (Fig. 7). The kinetic parameters (KM, kcat and Vmax) of WT and non-synonymous variant CES1s were determined for the pNPA substrate by measuring the rate of enzymatic formation of pNP at different substrate concentrations and fitting the obtained data to the Michaelis–Menten equation with a nonlinear regression analysis. Typical Michaelis–Menten kinetics was observed for recombinant CES1 (Fig. 8), and the kinetic parameters are shown in Table 3. WT CES1 demonstrated promising hydrolysis of pNPA, with KM, kcat and Vmax values of 579 ± 79 μM, 7.6 ± 0.4 s−1 and 73 ± 4 μmol/min/mg protein, respectively. When compared to other expression systems, the purified WT CES1 from the present study exhibited significant specific activity toward pNPA (Table 4). Each of the natural mutants exhibited different enzymatic activities when compared to the WT enzyme (p-value ⩽ 0.05). The Vmax values of S76N, D204E and A270S were 128 ± 10, 193 ± 15 and 109 ± 6 μmol/min/mg protein, respectively, higher than that of WT. The KM values of the mutants were 389 ± 81, 417 ± 98 and 263 ± 53 μM for S76N, D204E and A270S, respectively. Though the mutants showed smaller KM values than WT, they were still in a comparable range. This result suggested that the binding affinity of the enzyme toward pNPA is not greatly altered by mutation at residues 76 and 204, whereas the mutation at position 270 results in a binding affinity that is ∼2-fold greater than that of WT. The kcat values of S76N, D204E and A270S were 13 ± 1, 20 ± 2, 11 ± 0.6 s−1, respectively, higher than that of the WT enzyme.
Fig. 7.
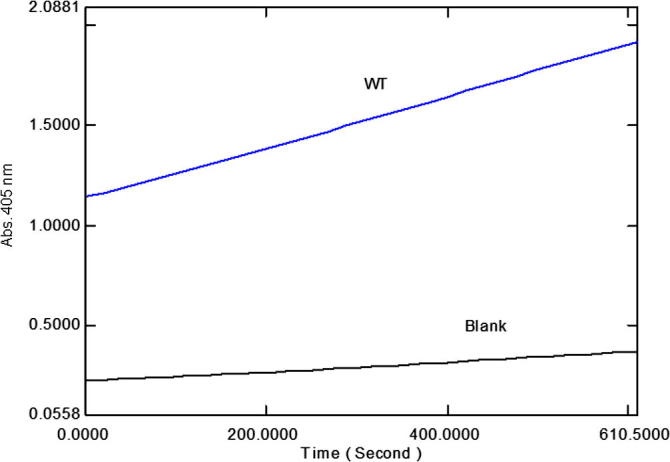
A representative time course of pNPA hydrolysis by WT CES1. The reaction was performed in a 1 ml volume containing Tris–HCl, pH 7.5, absolute ethanol, 100 mM pNPA and 20 μl purified WT CES1 at 37 °C. Hydrolysis was determined after incubating the reaction at 37 °C for 10 min, and the formation of pNP was followed at 405 nm for 10 min. A blank reaction was performed by replacing the enzyme with buffer.
Fig. 8.
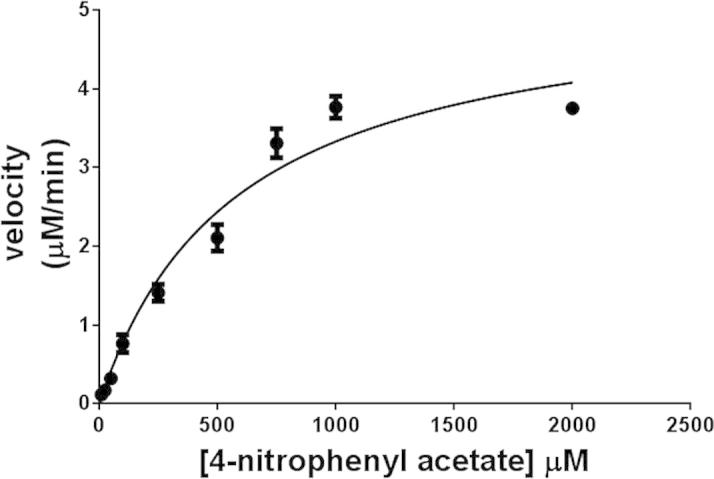
A representative Michaelis–Menten plot of WT CES1. Enzyme kinetics of pNPA hydrolysis catalyzed by WT CES1. The initial velocity was obtained by subtracting the blank hydrolysis from the enzymatic hydrolysis at each substrate concentration.
Table 3.
Kinetic parameters of CES1, WT and mutants. Data are expressed as the mean ± standard deviation (S.D.).
| Construct | KM (μM) | Vmax (μmol/min/mg) | kcat (s−1) |
|---|---|---|---|
| Wild type | 579 ± 79 | 73 ± 4 | 7.6 ± 0.4 |
| S76N | 389 ± 81 | 128 ± 10 | 13 ± 1 |
| D204E | 417 ± 98 | 193 ± 15 | 20 ± 2 |
| A270S | 263 ± 53 | 109 ± 6 | 11 ± 0.6 |
Table 4.
Comparison of specific activity of human CES1 toward pNPA.
Western blot analysis
A Western blot analysis was performed to confirm the expression of recombinant CES1. Fig. 9 shows the Western blot analysis of the WT and mutant proteins. All the recombinant proteins were expressed at similar levels, except for D204E, which was expressed at a lower level than the other constructs.
Fig. 9.
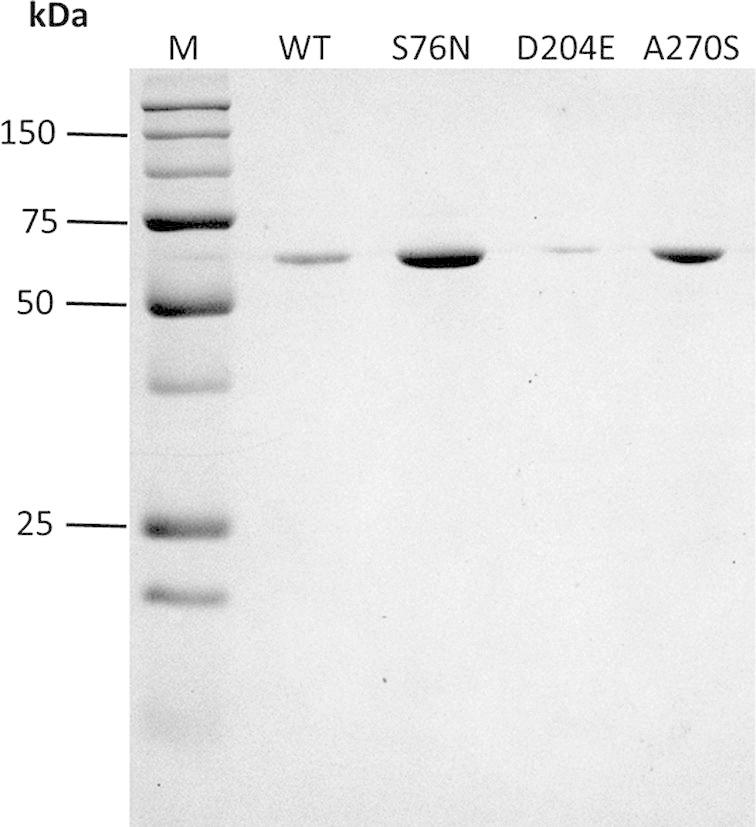
Western blot analysis of purified 6×His recombinant CES1, WT, S76N, D204E and A270S after refolding in 50 mM Tris–HCl, pH 7.5, containing 1% glycerol and 2 mM β-mercaptoethanol. Proteins were immunodetected using an anti-His (N-term) antibody.
Discussion
CES1 plays an important role in metabolism of drugs, such as cocaine and heroin, including oseltamivir, as well as the activation of various xenobiotics [2,4,5,9]. In addition, this enzyme also catalyzes the hydrolysis of long-chain fatty acids and thioesters. Owing to its significance, extensive work has been performed to investigate the role of CES1 using recombinant protein and characterization employing a variety of substrates, including heroin, cocaine, oseltamivir, aspirin, clopidogrel, methyl-4-nitrobenzoate and pNPA [2,4,8,11]. However, such studies have been carried out mainly in eukaryotic cells, mostly insect and mammalian cells [2,5,9,11]. Although eukaryotic expression systems are commonly used for producing recombinant proteins, they are complex and usually produce small to moderate amounts of protein. In contrast, bacterial expression systems are inexpensive, easy to handle and produce large amounts of protein and are therefore widely used in the production of numerous proteins. Morton and Potter [14] attempted to express human CES1 in bacteria and to increase the solubility of the protein by using different bacterial strains (Top10 and BL21 (DE3)), reducing the growth temperature and renaturing the protein with guanidine and other detergents but failed to obtain the functional enzyme, as the protein was largely expressed as inclusion bodies.
Molecular chaperones such as GroES/GroEL are known to facilitate the folding of many proteins expressed in bacteria and have been applied and proven to be successful in the expression of human aromatase and aldehyde dehydrogenase [24,25]. Initially, we co-expressed GroES/GroEL and pET32a–CES1 in Origami (DE3) cells grown at 37 °C and induced with IPTG when the absorbance at 600 nm reached 0.6, harvesting the cultures after 16 h of induction. Unfortunately, the protein was primarily expressed in the insoluble fraction, and purification of soluble CES1 was not attainable because of inadequate purity (data not shown). Because using pET32a as an expression vector and co-expressing with GroES/GroEL failed to yield a practical amount of soluble protein, we continued our expression experiments in the absence of these molecular chaperones and sub-cloned CES1 into the pET28a vector. Subsequently, we attempted to increase the solubility of the protein by lowering the temperature before and after IPTG induction. As shown in Fig. 2A, a small band at approximately 62 kDa was observed when the cells were grown at 37 °C and induced at 25 °C, indicating the improved solubility of WT CES1. The small soluble CES1 protein was then subjected to purification using an immobilized-metal affinity column; however, this purification was unsuccessful, as a large amount of contaminating proteins was present (data not shown). Expressing and inducing CES1 at 25 °C had no influence on solubility (Fig. 2B). Although decreasing the growth temperature to 25 °C resulted in a higher yield of protein, this was accompanied by low solubility, as a large amount of protein was expressed in inclusion bodies (Fig. 2B). Thereafter, we performed expression at 25 °C because of the greater protein yield.
Chemical chaperones are known to help correct the conformation of proteins via different mechanisms. Improved solubility has been observed for the cytokinin biosynthetic enzyme dimethylallyl pyrophosphate: 5′-AMP transferase (DMAPP: AMP transferase), green fluorescent protein (GFP) and single-chain Fv when sorbitol was added to the growth medium [26–28]. Moreover, the addition of 0.4% glycerol to the culture medium enhanced the solubility of a human phenylalanine hydroxylase mutant enzyme [36], resulting in both higher solubility and activity. Accordingly, at the time of induction, we included chemical chaperones, glycerol and sorbitol in the growth medium at final concentrations of 1% and 0.2 M, respectively but found that these additives were not beneficial for expressing CES1 (Fig. 3).
Recombinant CES1 was significantly expressed in the insoluble fraction, and we purified under denaturing conditions in which 8 M urea was used to solubilize the insoluble protein. Additives such as trehalose, sorbitol, glycerol, arginine and sucrose may enhance the solubility of numerous proteins when included in the refolding buffer [17,19,29,31–33]. In addition, it has been shown that the presence of reducing agents also improves protein solubility [19,29]. Therefore, the misfolded protein obtained was subjected to refolding in the presence of a combination of different chemical chaperones, trehalose, sucrose, glycerol and sorbitol, and reducing agents, β-mercaptoethanol and dithiothreitol (DTT) (Table 1). Fortunately, refolding in Tris–HCl, pH 7.5 containing 1% glycerol and 2 mM β-mercaptoethanol was successful, producing functional CES1. After refolding, intrinsic fluorescence and CD spectra for the recombinant WT and mutant proteins were analyzed. Each CES1 protein showed the same pattern of intrinsic fluorescence and CD spectra. The spectroscopic characterization results indicate that both WT and mutant CES1s share similar secondary structure, with α-helices as the main ordered structure. The presence of mutations at residues 76, 204 and 270 had no effect on the secondary structure of human CES1 (Figs. 5 and 6). According to the procedures presented here, it was evident that the refolding method was efficient for both WT and mutant CES1: all of the recombinants were catalytically active, with high specific activity (Table 3). The yield in the first purification step ranged from 43% to 53%, and the refolding yield ranged between 9% and 19%, resulting in 1–2 mg protein, which is practical for functional characterization.
To verify that our recombinant CES1 was functionally active after refolding, we investigated the catalytic activity of the WT enzyme. pNPA was chosen as the substrate to study CES1 activity because this compound is a sensitive and established model substrate. As shown in Fig. 7, WT CES1 was hydrolytically active toward pNPA. In comparison with recombinant human CES1 expressed in eukaryotic systems, our refolded CES1 exhibited a significant specific activity comparable to that of CES1 expressed in mammalian COS7 cells. In fact, our refolded CES1 showed greater specific activity than the enzyme expressed in insect and mammalian (Flp-In-293) cells (Table 4). This finding indicates that recombinant CES1 expressed in E. coli and refolded in Tris–HCl, pH 7.5, containing 1% glycerol and 2 mM β-mercaptoethanol is catalytically active, showing similar activity to the native enzyme. As the WT enzyme was successfully purified and refolded, producing a catalytically active enzyme, we then sought to examine whether this refolding system is applicable to the mutant proteins. Additionally, the effect of these mutations on the catalytic activity of CES1 would provide information regarding the significance of polymorphisms to metabolism. Therefore, we created three natural mutants, which are reported in the literature and a database of single-nucleotide polymorphism (SNP) from NCBI (NCBI: dbSNP), S76N, D204E and A270 [10,37]. These three variants were included in our study because the frequency of these mutations is relatively high, as reported by NCBI: dbSNP [37]. Our results illustrated that the recombinant mutant proteins were catalytically active, with kinetic properties similar to that of the WT enzyme (Table 3). This result indicates that these mutations, S76N, D204E and A270S, are not critical for the catalytic activity of the enzyme, as they had little effect on activity. Apparently, these mutant enzymes are moderately more active than the WT enzyme. This is in agreement with an in vitro functional study on the enzymatic activity of nonsynonymous variants (G18V, S82L and A269S; numbering according to CES1 isoform b) that reported no significant difference from that of the WT enzyme with regard to the hydrolysis of clopidogrel, 2-oxo-clopidogrel and methylphenidate [10]. Other natural mutations (V21I, I32V, S58N, C70F, G156D, R182H, D186E; numbering according to CES1 isoform b) also exhibited activity comparable to the WT enzyme regarding the hydrolysis of oseltamivir and clopidogrel, with the exception that C70F was catalytically inactive against both substrates, whereas R182H showed markedly reduced activity toward oseltamivir [4,5].
Conclusions
In present study, we, for the first time, developed an efficient method for the refolding of denaturant-solubilized recombinant human CES1 while retaining the biological activity of the enzyme. We found that the combination of 1% glycerol and 2 mM β-mercaptoethanol in Tris–HCl at pH 7.5 significantly improved the solubility of denaturant-solubilized human CES1 during the refolding process. This refolding condition was applicable to the WT enzyme as well as three natural variants. The biological function of CES1 was retained after refolding, and this purified His-tagged CES1 can be used directly without removing the fusion tag. To the best of our knowledge, no details of CES1 refolding have been reported before. The methods presented in this study allow the production of a significant amount as well as a functionally active human CES1 in a bacterial expression system for further functional studies on the biological role of this enzyme.
Conflict of interest statement
The authors state that there is no conflict of interest.
Author contributions
Conceived and designed the experiments: UB, KP, ND, MI. Performed the experiments: UB, KP, SJ. Contributed reagents/materials/analysis tools: UB, MI, ND. Wrote the paper: UB, ND, MI.
Acknowledgements
This study was supported by Faculty of Tropical Medicine, Mahidol University and Wellcome Trust Mahidol University Oxford Tropical Medicine Research Programme, Wellcome Trust of Great Britain.
Footnotes
Abbreviations used: CES1, carboxylesterase 1; ACE, angiotensin-converting enzyme; WHO, World Health Organization; WT, wild-type; pNPA, p-nitrophenyl acetate; SDS–PAGE, sodium dodecyl sulfate–polyacrylamide gel electrophoresis; CD, circular dichroism; pNP, p-nitrophenol; PVDF, polyvinylidene difluoride; PBS, phosphate-buffered saline; HRP, horseradish peroxidase; GFP, green fluorescent protein; DTT, dithiothreitol; SNP, single-nucleotide polymorphism; IPTG, isopropyl-β-d-thiogalactopyranoside.
Contributor Information
Usa Boonyuen, Email: usa.boo@mahidol.ac.th.
Kamoltip Promnares, Email: kamoltip.p@psu.ac.th, kpromnares@gmail.com.
Suwapat Junkree, Email: suwapat.jun@mahidol.ac.th.
Nichloas P.J. Day, Email: nickd@tropmedres.ac.
Mallika Imwong, Email: noi@tropmedres.ac.
References
- 1.Laizure S.C., Mandrell T., Gades N.M., Parker R.B. Cocaethylene metabolism and interaction with cocaine and ethanol: role of carboxylesterases. Drug Metab. Dispos. 2003;31:16–20. doi: 10.1124/dmd.31.1.16. [DOI] [PubMed] [Google Scholar]
- 2.Hatfield M.J., Tsurkan L., Hyatt J.L., Yu X., Edwards C.C., Hicks L.D., Wadkins R.M., Potter P.M. Biochemical and molecular analysis of carboxylesterase-mediated hydrolysis of cocaine and heroin. Br. J. Pharmacol. 2010;160:1916–1928. doi: 10.1111/j.1476-5381.2010.00700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nishi K., Huang H., Kamita S.G., Kim I.H., Morisseau C., Hammock B.D. Characterization of pyrethroid hydrolysis by the human liver carboxylesterase hCE-1 and hCE-2. Arch. Biochem. Biophys. 2006;445:115–123. doi: 10.1016/j.abb.2005.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tang M., Mukundan M., Yang J., Charpentier N., LeCluyse E.L., Black C., Yang D., Shi D., Yan B. Antiplatelet agents aspirin and clopidogrel are hydrolyzed by distinct carboxylesterases, and clopidogrel is transesterificated in the presence of ethyl alcohol. J. Pharmacol. Exp. Ther. 2006;319:1467–1476. doi: 10.1124/jpet.106.110577. [DOI] [PubMed] [Google Scholar]
- 5.Shi D., Yang J., Yang D., LeCluyse E.L., Black C., You L., Akhlaghi F., Yan B. Anti-influenza prodrug oseltamivir is activated by carboxylesterase human carboxylesterase 1, and the activation is inhibited by antiplatelet agent clopidogrel. J. Pharmacol. Exp. Ther. 2006;319:1477–1484. doi: 10.1124/jpet.106.111807. [DOI] [PubMed] [Google Scholar]
- 6.Wadkins R.M., Morton C.L., Weeks J.K., Oliver L., Wierdl M., Danks M.K., Potter P.M. Structural constraints affect the metabolism of 7-ethyl-10-[4-(1-piperidino)-1-piperidino]carbonyloxycamptothecin (CPT-11) by carboxylesterases. Mol. Pharmacol. 2001;60:355–362. doi: 10.1124/mol.60.2.355. [DOI] [PubMed] [Google Scholar]
- 7.Zhu H.J., Appel D.I., Johnson J.A., Chavin K.D., Markowitz J.S. Role of carboxylesterase 1 and impact of natural genetic variants on the hydrolysis of trandolapril. Biochem. Pharmacol. 2009;77:1266–1272. doi: 10.1016/j.bcp.2008.12.017. [DOI] [PubMed] [Google Scholar]
- 8.Zhu H.J., Markowitz J.S. Activation of the antiviral prodrug oseltamivir is impaired by two newly identified carboxylesterase 1 variants. Drug Metab. Dispos. 2009;37:264–267. doi: 10.1124/dmd.108.024943. [DOI] [PubMed] [Google Scholar]
- 9.Zhu H.J., Patrick K.S., Yuan H.J., Wang J.S., Donovan J.L., DeVane C.L., Malcolm R., Johnson J.A., Youngblood G.L., Sweet D.H., Langaee T.Y., Markowitz J.S. Two CES1 gene mutations lead to dysfunctional carboxylesterase 1 activity in man: clinical significance and molecular basis. Am. J. Hum. Genet. 2008;82:1241–1248. doi: 10.1016/j.ajhg.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhu H.J., Wang X., Gawronski B.E., Brinda B.J., Angiolillo D.J., Markowitz J.S. Carboxylesterase 1 as a determinant of clopidogrel metabolism and activation. J. Pharmacol. Exp. Ther. 2013;344:665–672. doi: 10.1124/jpet.112.201640. [DOI] [PubMed] [Google Scholar]
- 11.Williams E.T., Bacon J.A., Bender D.M., Lowinger J.J., Guo W.K., Ehsani M.E., Wang X., Wang H., Qian Y.W., Ruterbories K.J., Wrighton S.A., Perkins E.J. Characterization of the expression and activity of carboxylesterases 1 and 2 from the beagle dog, cynomolgus monkey, and human. Drug Metab. Dispos. 2011;39:2305–2313. doi: 10.1124/dmd.111.041335. [DOI] [PubMed] [Google Scholar]
- 12.Williams E.T., Ehsani M.E., Wang X., Wang H., Qian Y.W., Wrighton S.A., Perkins E.J. Effect of buffer components and carrier solvents on in vitro activity of recombinant human carboxylesterases. J. Pharmacol. Toxicol. Methods. 2008;57:138–144. doi: 10.1016/j.vascn.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 13.Greenblatt H.M., Otto T.C., Kirkpatrick M.G., Kovaleva E., Brown S., Buchman G., Cerasoli D.M., Sussman J.L. Structure of recombinant human carboxylesterase 1 isolated from whole cabbage looper larvae. Acta Crystallogr. F Struct. Biol. Cryst. Commun. 2012;68:269–272. doi: 10.1107/S1744309112003326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morton C.L., Potter P.M. Comparison of Escherichia coli, Saccharomyces cerevisiae, Pichia pastoris, Spodoptera frugiperda, and COS7 cells for recombinant gene expression: application to a rabbit liver carboxylesterase. Mol. Biotechnol. 2000;16:193–202. doi: 10.1385/MB:16:3:193. [DOI] [PubMed] [Google Scholar]
- 15.Chou C.P. Engineering cell physiology to enhance recombinant protein production in Escherichia coli. Appl. Microbiol. Biotechnol. 2007;76:521–532. doi: 10.1007/s00253-007-1039-0. [DOI] [PubMed] [Google Scholar]
- 16.Sorensen H.P., Mortensen K.K. Advanced genetic strategies for recombinant protein expression in Escherichia coli. J. Biotechnol. 2005;115:113–128. doi: 10.1016/j.jbiotec.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 17.Dong X.Y., Huang Y., Sun Y. Refolding kinetics of denatured-reduced lysozyme in the presence of folding aids. J. Biotechnol. 2004;114:135–142. doi: 10.1016/j.jbiotec.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 18.Fan Y.Q., Lee J., Oh S., Liu H.J., Li C., Luan Y.S., Yang J.M., Zhou H.M., Lü Z.R., Wang Y.L. Effects of osmolytes on human brain-type creatine kinase folding in dilute solutions and crowding systems. Int. J. Biol. Macromol. 2012;51:845–858. doi: 10.1016/j.ijbiomac.2012.07.026. [DOI] [PubMed] [Google Scholar]
- 19.Mollania N., Khajeh K., Ranjbar B., Rashno F., Akbari N., Fathi-Roudsari M. An efficient in vitro refolding of recombinant bacterial laccase in Escherichia coli. Enzyme Microb. Technol. 2013;52:325–330. doi: 10.1016/j.enzmictec.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 20.Sahdev S., Khattar S.K., Saini K.S. Production of active eukaryotic proteins through bacterial expression systems: a review of the existing biotechnology strategies. Mol. Cell. Biochem. 2008;307:249–264. doi: 10.1007/s11010-007-9603-6. [DOI] [PubMed] [Google Scholar]
- 21.Guo W., Cao L., Jia Z., Wu G., Li T., Lu F., Lu Z. High level soluble production of functional ribonuclease inhibitor in Escherichia coli by fusing it to soluble partners. Protein Expr. Purif. 2011;77:185–192. doi: 10.1016/j.pep.2011.01.015. [DOI] [PubMed] [Google Scholar]
- 22.Semba H., Ichige E., Imanaka T., Atomi H., Aoyagi H. Efficient production of active form recombinant cassava hydroxynitrile lyase using Escherichia coli in low-temperature culture. Methods Mol. Biol. 2010;643:133–144. doi: 10.1007/978-1-60761-723-5_10. [DOI] [PubMed] [Google Scholar]
- 23.Studier F.W. Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 24.Kagawa N. Efficient expression of human aromatase (CYP19) in E. coli. Methods Mol. Biol. 2011;705:109–122. doi: 10.1007/978-1-61737-967-3_7. [DOI] [PubMed] [Google Scholar]
- 25.Voulgaridou G.P., Mantso T., Chlichlia K., Panayiotidis M.I., Pappa A. Efficient E. coli expression strategies for production of soluble human crystallin ALDH3A1. PLoS ONE. 2013;8:e56582. doi: 10.1371/journal.pone.0056582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blackwell J.R., Horgan R. A novel strategy for production of a highly expressed recombinant protein in an active form. FEBS Lett. 1991;295:10–12. doi: 10.1016/0014-5793(91)81372-f. [DOI] [PubMed] [Google Scholar]
- 27.Prasad S., Khadatare P.B., Roy I. Effect of chemical chaperones in improving the solubility of recombinant proteins in Escherichia coli. Appl. Environ. Microbiol. 2011;77:4603–4609. doi: 10.1128/AEM.05259-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sandee D., Tungpradabkul S., Kurokawa Y., Fukui K., Takagi M. Combination of Dsb coexpression and an addition of sorbitol markedly enhanced soluble expression of single-chain Fv in Escherichia coli. Biotechnol. Bioeng. 2005;91:418–424. doi: 10.1002/bit.20524. [DOI] [PubMed] [Google Scholar]
- 29.Asad S., Dabirmanesh B., Ghaemi N., Etezad S.M., Khajeh K. Studies on the refolding process of recombinant horseradish peroxidase. Mol. Biotechnol. 2013;54:484–492. doi: 10.1007/s12033-012-9588-6. [DOI] [PubMed] [Google Scholar]
- 30.Feng S., Yan Y.B. Effects of glycerol on the compaction and stability of the wild type and mutated rabbit muscle creatine kinase. Proteins. 2008;71:844–854. doi: 10.1002/prot.21744. [DOI] [PubMed] [Google Scholar]
- 31.Rariy R.V., Klibanov A.M. Correct protein folding in glycerol. Proc. Natl. Acad. Sci. U.S.A. 1997;94:13520–13523. doi: 10.1073/pnas.94.25.13520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tiwari A., Bhat R. Stabilization of yeast hexokinase A by polyol osmolytes: correlation with the physicochemical properties of aqueous solutions. Biophys. Chem. 2006;124:90–99. doi: 10.1016/j.bpc.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 33.Vagenende V., Yap M.G., Trout B.L. Mechanisms of protein stabilization and prevention of protein aggregation by glycerol. Biochemistry. 2009;48:11084–11096. doi: 10.1021/bi900649t. [DOI] [PubMed] [Google Scholar]
- 34.Desjardins P., Hansen J.B., Allen M. Microvolume protein concentration determination using the NanoDrop 2000c spectrophotometer. J. Vis. Exp. 2009;33:e1610. doi: 10.3791/1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gasteiger E., Hoogland C., Gattiker A., Duvaud S., Wilkins M.R., Appel R.D., Bairoch A. Humana Press; New Jersey: 2005. Protein Identification and Analysis Tools on the ExPASy Server: The Proteomics Protocols Handbook. [Google Scholar]
- 36.Leandro P., Lechner M.C., Tavares de Almeida I., Konecki D. Glycerol increases the yield and activity of human phenylalanine hydroxylase mutant enzymes produced in a prokaryotic expression system. Mol. Genet. Metab. 2001;73:173–178. doi: 10.1006/mgme.2001.3172. [DOI] [PubMed] [Google Scholar]
- 37.<http://www.ncbi.nlm.nih.gov/snp/?term=CES1&SITE=NcbiHome&submit=Go> National Center for Biotechnology Information (2011, October 13), retrieved 15.11.2011.