

Abstract
The proteasome core particle (CP) is a conserved protease complex that is formed by the stacking of two outer α-rings and two inner β-rings. The α-ring is a heteroheptameric ring of subunits α1 to α7 and acts as a gate that restricts entry of substrate proteins into the catalytic cavity formed by the two abutting β-rings. The 31-kDa proteasome inhibitor (PI31) was originally identified as a protein that binds to the CP and inhibits CP activity in vitro, but accumulating evidence indicates that PI31 is required for physiological proteasome activity. To clarify the in vivo role of PI31, we examined the Saccharomyces cerevisiae PI31 ortholog Fub1. Fub1 was essential in a situation where the CP assembly chaperone Pba4 was deleted. The lethality of Δfub1 Δpba4 was suppressed by deletion of the N terminus of α7 (α7ΔN), which led to the partial activation of the CP. However, deletion of the N terminus of α3, which activates the CP more efficiently than α7ΔN by gate opening, did not suppress Δfub1 Δpba4 lethality. These results suggest that the α7 N terminus has a role in CP activation different from that of the α3 N terminus and that the role of Fub1 antagonizes a specific function of the α7 N terminus.
INTRODUCTION
The 26S proteasome is a multicatalytic protease complex conserved in eukaryotes (1). Its main function is to serve as a selective and regulated mechanism for intracellular protein degradation, mainly in a ubiquitin-dependent manner. The 26S proteasome consists of one 20S core particle (CP) and one or two 19S regulatory particles (RPs) attached to the CP.
The CP exerts proteolytic activity and is made up of four axially stacked heteroheptameric rings: two outer α-rings formed by α1 to α7 and two inner β-rings formed by β1 to β7. Of the seven β-subunits, only β1, β2, and β5 have proteolytic activities. The archaebacterium Thermoplasma acidophilum has a prototype of the CP that consists of a single type of α- and β-subunit, with all the β-subunits being catalytically active. Another difference between archaeal and eukaryotic CPs is the structure of the α-rings. Whereas archaeal CPs have a disordered gate that is permeable to peptide substrates, the eukaryotic α-ring of the CP is primarily in a closed state because the N termini of α1, α2, α3 (Pre9), α6, and α7 project into the opening of the α-ring (2–4). Therefore, the activity of the eukaryotic CP is basically latent. Of the α-subunits, α3 is supposed to be most important because the N terminus of α3 projects directly across the pseudo 7-fold symmetry axis. In addition, its deletion (α3ΔN) causes disorder of the N termini of α1, α5, and α7, leading to an open state of α-rings (5). In vivo, binding of CP activators, such as the RPs, opens the closed α-ring (6, 7).
The CP is assembled with the aid of various proteasome-dedicated chaperones in mammals and Saccharomyces cerevisiae (8–13). The assembly of CPs begins with the formation of the α-ring, assisted by the heterodimeric complexes PAC1-PAC2/Pba1-Pba2 and PAC3-PAC4/Pba3-Pba4 in mammals/yeast. After the formation of the α-ring, β-subunits are recruited to the α-ring in a defined order with the help of another assembly chaperone, Ump1, during which PAC3-PAC4/Pba3-Pba4 detaches from the assembly intermediate, while PAC1-PAC2/Pba1-Pba2 and Ump1 are kept associated with them until the completion of CP assembly. The resultant half-CPs dimerize to form a mature CP. As expected, knockdown of PAC proteins in mammalian cells impairs α-ring formation, resulting in poor maturation of CPs (14, 15). It has also been reported that the amount of CPs decreases in Δpba3 and Δpba4 deletion mutants of yeast (16–19).
The 31-kDa proteasome inhibitor (PI31) was originally identified in mammals as a protein that inhibits the peptidase activity of the CP in vitro (20). PI31 has been shown to compete with PA28 and the RP for binding to the CP in vitro (21, 22). Furthermore, when PI31 was overexpressed in cells, the formation of the immunoproteasome, which is one of the CP subtypes, was attenuated, and thus processing of an immunoproteasome-dependent epitope was impaired (23). In contrast, recent studies have shown that the Drosophila melanogaster PI31 homolog (DmPI31) activated the 26S proteasome in vitro and that knockdown of DmPI31 in flies compromised protein degradation by the proteasome (24, 25), while mammalian PI31 had no effect on the in vitro activity of the 26S proteasome (26). In view of these conflicting findings, the physiological role and the mechanism of action of PI31 in proteasome-mediated protein degradation remain enigmatic.
The PI31 homolog in S. cerevisiae, encoded by YCR076c, has been reported recently as one of the barrier proteins that block the spread of transcriptional gene silencing (27). This protein, named Fub1 (function of boundary), has been shown to associate with the CP subunits. Hatanaka et al. also showed that FUB1/YCR076c exhibited genetic interactions with several proteasome-related genes, suggesting a functional relationship between the PI31 homolog and the proteasome in budding yeast (27). However, whether Fub1 acts on proteasomes positively or negatively has not been fully understood.
In this study, we found a genetic interaction between the FUB1, PBA3, PBA4, and CP α-subunit genes. Deletion of FUB1 was lethal under Pba3- and Pba4-deficient conditions. This lethality was suppressed by Δα3 and α7 with an N-terminal deletion (α7ΔN), both of which lead to the partial activation of the CP. Interestingly, α3ΔN, which activates the CP more efficiently than α7ΔN by gate opening, did not suppress the lethality of the Δfub1 Δpba3 mutant. These findings suggest that Fub1 is not a CP inhibitor in vivo and that it exerts its function via the activity of the CP, which is enhanced by α7ΔN. Our results also suggest that the α7 N terminus has biological significance, a notion that has not received much attention so far.
MATERIALS AND METHODS
Strains and plasmids.
The E. coli strain DH5α was used for propagating plasmids. BL21(DE3) cells were used for expression and purification of recombinant proteins. Yeast strain genotypes are given in Table 1. Yeast knockout strains (catalog number YSC1053) were purchased from Open Biosystems. Media and methods for standard culture, mating, sporulation, tetrad analysis, and transformation were described previously (28, 29). The plasmids used in this study are listed in Table 2.
TABLE 1.
Strains used in the present study
| Strain | Genotype | Strain background |
|---|---|---|
| BY4741 | MATa hisΔ1 leu2Δ0 met15Δ0 ura3Δ0 | |
| BY4742 | MATα hisΔ1 leu2Δ0 lys2Δ0 ura3Δ0 | |
| W303a | MATa his3 leu2 ura3 trp1 ade2 can1 | |
| YT54 | MATa Δpba4::kanMX4 | BY |
| YT116 | MATα Δpba4::HIS3MX6 | BY |
| YT122 | MATa Δpre9::kanMX4 | BY |
| YT124 | MATa Δrpn10::kanMX4 | BY |
| YT126 | MATa Δecm29::kanMX4 | BY |
| YT128 | MATa Δsem1::kanMX4 | BY |
| YT130 | MATa RPN11-3×Flag-kanMX4 | BY |
| YT270 | MATa Δump1::kanMX4 | BY |
| YT385 | MATa α6-3HA-HIS3MX6 | BY |
| YT556 | MATa Δfub1::kanMX4 | BY |
| YT580 | MATa Δrpn4::HIS3 Δfub31::kanMX4 | BY |
| YT595 | MATa GAL1p-PBA3-TRP1α6-3HA-HIS3MX6 | W303 |
| YT678 | MATa/α Δfub31::kanMX4/+ Δpba4::HIS3MX6/+ | BY |
| YT681 | MATa Δfub1::HIS3MX6 | BY |
| YT745 | MATa GAL1p-PBA3-TRP1α6-3HA-HIS3MX6Δfub1::kanMX4 | W303 |
| YT782 | MATa Δblm10::TRP1 Δfub1::kanMX4 | KA |
| YT800 | MATa Δfub1::hphMX6 | BY |
| YT836 | MATα Δrpn10::kanMX4 Δfub31::HIS3MX6 | BY |
| YT837 | MATa Δpre9::kanMX4 Δfub1::HIS3MX6 | BY |
| YT838 | MATα Δrpn10::kanMX4 Δfub1::HIS3MX6 | BY |
| YT839 | MATα Δsem1::kanMX4 Δfub1::HIS3MX6 | BY |
| YT1138 | MATα α3ΔN-URA3 | BY |
| YT1140 | MATα α7ΔN-HIS3 | BY |
| YT1141 | MATα α3ΔN-URA3α7ΔN-HIS3 | BY |
| YT1148 | MATα α7ΔN-HIS3 Δfub1::hphMX6 Δpba4::kanMX4 | BY |
| YT1177 | MATα Δump1::kanMX4 Δfub1::hphMX6 | BY |
| YT1203 | MATα PRE1-3×Flag-kanMX4 Δfub1::hphMX6 | BY |
| YT1209 | MATα PRE1-3×Flag-kanMX4 | BY |
| YT1219 | MATα PRE1-3×Flag-kanMX4α3ΔN-URA3 | BY |
| YT1222 | MATα PRE1-3×Flag-kanMX4α7ΔN-URA3 | BY |
| YT2-1D | MATa Δblm10::TRP1 | KA |
| TD110 | MATa GAL1p-PBA3-TRP1 α6-3HA-HIS3MX6Δfub1::kanMX4 Δprb1::natMX6 | W303 |
| TD132 | GAL1p-PBA3-TRP1 RPN11-3×Flag-natMX6 | W303 |
| TD133 | GAL1p-PBA3-TRP1 Δfub1::kanMX4 RPN11-3×Flag-natMX6 | W303 |
| TD206 | MATa Δfub1::kanMX4 α3G155R-URA3 | BY |
TABLE 2.
Plasmids used in the present study
| Plasmid | Character | Source |
|---|---|---|
| pRS303 | HIS3 vector for integration | Sikorski and Hieter (45) |
| pRS306 | URA3 vector for integration | Sikorski and Hieter (45) |
| pRS316 | URA3 ARS CEN | Sikorski and Hieter (45) |
| pHY68 | 3×Flag-kanMX6 | Yashiroda and Tanaka (46) |
| pYF2 | GAL1p-Flag URA3 2μ | Gift from Saeki |
| pYF5 | GAL1p-Flag URA3 2μ (cloning sites are different from those in pYF2) | Gift from Saeki |
| pYO326 | URA3 2μ | Qadota et al. (47) |
| pT6 | pYF2-PBA4 | This study |
| pT17 | pRS316-PBA4 | Yashiroda et al. (17) |
| pT168 | pYF2-FUB1 | This study |
| pT169 | pYF2-YDL199C | This study |
| pT170 | pYF2-YGR067C | This study |
| pT171 | pYF2-YGR067C | This study |
| pT172 | pYF2-YOL098C | This study |
| pT173 | pYF2-YOL087C | This study |
| pT174 | pYF2-YBL054W | This study |
| pT175 | pYF2-YDL156W | This study |
| pT176 | pYF2-YOL022C | This study |
| pT177 | pYF2-YHR003C | This study |
| pT261 | pYO326-FUB1 | This study |
| pT271 | pYF5-human PI31 | This study |
| pT314 | pRS303-α7ΔN | This study |
| pT322 | GAL1p-Flag LEU2 2μ | This study |
| pT333 | pT322-FUB1 | This study |
| pT334 | pT322-FUB1 (PI domain) | This study |
| pT335 | pT322-FUB1 (N terminus) | This study |
| pT327 | pT322-FUB1 (N terminus of PI domain) | This study |
| pT328 | pT322-FUB1 (C terminus of PI domain) | This study |
| pMO1 | pYF2-YKL027W | This study |
| pMO5 | pYF2-YNL313C | This study |
| pMO23 | pRS316-FUB1 | This study |
| pMO32 | pET28-FUB1 | This study |
| pYT75 | pRS306-α3ΔN | This study |
Constructions of the α3ΔN and α7ΔN alleles.
For construction of α3ΔN, PCRs were performed using the BY strain genome as a template DNA and the primer sets of T110 (AAGGATCCGGAGAAGCCAGTGATCAAG)/T121 (CTCAGGGGAGAAAATTGTCATGTTTAACACTACTAACTATA) and T122 (TATAGTTAGTAGTGTTAAACATGACAATTTTCTCCCCTGAG)/T123 (AAGTCGACGCCTTCCACCCTGTATAGTT). Then, the resultant PCR fragments and the primers T110 and T123 were used for the second PCR. The obtained fragments were cloned into the EcoRI and SalI sites of pRS306. The constructed plasmid, pYT75, was linearized by EcoRI digestion and integrated into the BY wild-type strain. For α7ΔN, the primer sets for the first PCRs were 797 (ATCTCGAGCATTGACAGCTCAGACGATAG)/798 (CTACCATCGGGGGAAAAAACACTCATTGCTGAAGAGTTATGC) and 799 (GCATAACTCTTCAGCAATGAGTGTTTTTTCCCCCGATGGTAG)/800 (ATGCGGCCGCGCTTCCTGTAGCAGATCGCC). The second PCR was performed using the first PCR fragments and the primer set 797 and 800. The resultant fragments were cloned into pRS303, and then the obtained plasmid, pT314, was linearized by HincII and integrated into the BY wild-type strain.
Protein extraction.
For immunoprecipitation and protein purification, cells were suspended in lysis buffer (50 mM Tris-HCl [pH 7.5], 100 mM NaCl, 10% [vol/vol] glycerol, 1 mM phenylmethylsulfonyl fluoride [PMSF], and 0.25% [vol/vol] Triton X-100). For glycerol gradient analysis, cells were suspended in 25 mM Tris-HCl (pH 7.6), 5 mM MgCl2, 1 mM dithiothreitol (DTT), and 2 mM ATP. Total cell lysates were prepared by vortexing them with glass beads using a Multi-beads shocker cell disruptor (Yasui Kikai), and then the lysates were cleared by centrifugation at 20,000 × g for 10 min at 4°C.
Coimmunoprecipitation.
For Fig. 2D and 9B, cell fractions or appropriate total cell lysates were mixed with anti-Flag M2 agarose beads (Sigma) and incubated for 2 h. Immunoprecipitates were then eluted in lysis buffer containing 200 μg/ml 3×Flag peptides (Sigma). For Fig. 2B, cell fractions were mixed with anti-Fub1 antibodies and incubated for 2 h, after which protein G-Sepharose beads (GE Healthcare) were added and the mixture was incubated for 1 h. The beads were then boiled in 1× sample buffer for SDS-PAGE, and immunoprecipitates were eluted.
FIG 2.

Identification of Fub1 as a proteasome-interacting protein. (A) Suc-LLVY-MCA hydrolyzing activities of wild-type cell lysates fractionated by 8 to 32% glycerol gradient centrifugation and immunoblot analysis with anti-Fub1 antibodies. Fractions 16 to 20 and 26 to 30 were categorized into 20S and 26S subfractions according to their peptidase activities. (B) Coimmunoprecipitation of Fub1 and α5. Fub1 of each subfraction prepared for the experiment in panel A was immunoprecipitated with anti-Fub1 antibodies, followed by immunoblotting for α5. (C) Fractionation of lysates of Rpn11-Flag cells by 4 to 24% glycerol gradient centrifugation. Each fraction was subjected to assay of Suc-LLVY-MCA hydrolyzing activity in the presence of SDS and immunoblot analysis with anti-Flag antibodies. Fractions 20 to 22 and 26 to 30 were categorized into 19S, 20S, and 26S subfractions according to their peptidase activities. The C-terminal Flag tag of Rpn11 did not affect the peptidase activities in all fractions in a comparison with the wild type. (D) Interaction of Fub1 and 26S proteasomes. Rpn11-Flag in 19S, 20S, and 26S subfractions prepared for the experiment represented in panel C was immunoprecipitated with anti-Flag antibodies, and Fub1 in each immunoprecipitate was detected with anti-Fub1 antibodies.
FIG 9.
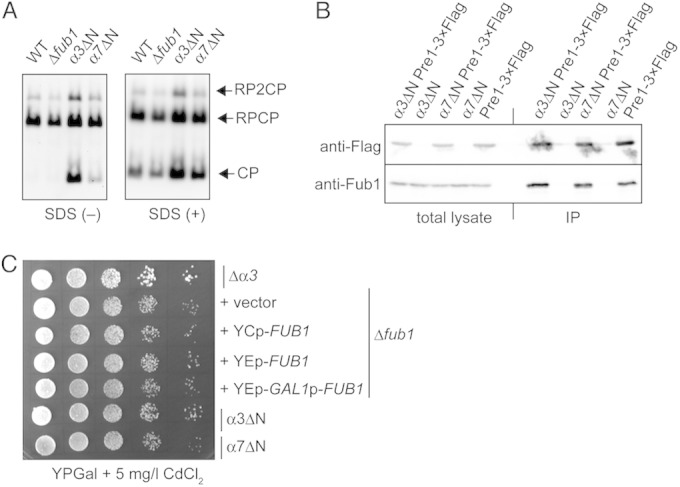
Activities of the CPs lacking the N terminus of α3 or α7. (A) In-gel assay for proteasome activity using native PAGE. Affinity-purified proteasomes using anti-Flag antibodies from each strain bearing Flag-tagged β4 (Pre1) were subjected to native PAGE, followed by in-gel proteasome activity analysis using Suc-LLVY-MCA in the presence or absence of 0.025% SDS. RP2CP, CPs with two RPs; RPCP, CPs with one RP; RP, the 19S regulatory particle. (B) Binding affinity of Fub1 to wild-type, α3ΔN, and α7ΔN CPs. Total lysates and immunoprecipitates (IP) of anti-Flag antibodies were subjected to SDS-PAGE and Western blot analysis using anti-Flag and anti-Fub1 antibodies. (C) Cadmium resistance in the Δα3, Δfub1, FUB1-overexpressed, α3ΔN, and α7ΔN cells. The indicated strains were serially diluted, spotted on YPGal plus 5 mg/liter CdCl2, and cultured at 27°C for 4 days.
Immunological analysis.
SDS-PAGE and Western blotting were performed according to standard protocols. Anti-Fub1, antihemagglutinin (anti-HA; Babco), anti-Flag (Sigma), antiubiquitin (LifeSensor), anti-α5 (our stock), and anti-Pgk1 (Molecular Probes) antibodies were used at various points during the course of this study. Anti-Fub1 was raised in rabbits using recombinant 6×His-Fub1. 6×His-Fub1 was expressed in Escherichia coli using pMO32 (pET28-FUB1) and purified using Ni-nitrilotriacetic acid (Ni-NTA) agarose (Qiagen). The experimental protocols were approved by the Ethics Review Committee for Animal Experimentation of the University of Tokyo.
Proteasome purification.
26S proteasomes and CPs were purified using anti-Flag M2 agarose beads from YT130 (wild-type RPN11-3×Flag), YT1209 (wild-type PRE1-3×Flag), YT1203 (Δfub1 PRE1-3×Flag), YT1219 (α3ΔN PRE1-3×Flag), YT1222 (α7ΔN PRE1-3×Flag), TD132 (GAL1-PBA3 RPN11-3×Flag), and TD133 (GAL1-PBA3 Δfub1 RPN11-3×Flag) cells (30). Peptidase inhibitors were required to inhibit the processing of TD133 proteasome subunits by upregulated Prb1 during the purification procedure (31). For the preparation of CPs used in Fig. 3B, 500 mM NaCl was used for washout of RPs from 26S proteasomes bound to M2 agarose beads during the purification procedure.
FIG 3.

Effects of overexpression and deletion of FUB1 on proteasome activities. (A) Effects of purified recombinant Fub1 on the activities of fractionated CPs and 26S proteasomes. Suc-LLVY-MCA hydrolyzing activities of cell lysates (2 mg) fractionated by 8 to 32% glycerol gradient centrifugation were measured with or without 2 μg/ml of recombinant Fub1 and in the presence (upper panel) or absence (lower panel) of SDS. For 26S proteasomes, we measured fractions 19 to 28. (B) Effects of Fub1 on the activities of CPs and 26S proteasomes in vitro. The indicated concentration of purified Fub1 was mixed with 1 nM purified CPs and 26S proteasomes, followed by measurement of peptidase activities using Suc-LLVY-MCA. Activities of CPs and 26S proteasomes in the absence of Fub1 were set at 100%. (C) Phenotypes of Δfub1 and Fub1-overexpressing cells. Δfub1 cells were transformed with pRS316 (vector), pMO23 (YCp FUB1), or pT261 (YEp FUB1). YCp and YEp denote a single-copy and a multicopy vector, respectively. Resultant transformant cells were streaked onto YPD plates and incubated for 2 days at the indicated temperatures (upper panel). Δump1 cells were used as an AZC-sensitive positive control (lower panel). (D) Detection of polyubiquitinated proteins. Δα3 and Δump1 cells were used as positive controls for the accumulation of polyubiquitinated proteins. Pgk1 (3-phosphoglycerate kinase) was used as a loading control. Protein levels of Fub1 were confirmed by immunoblot analysis using anti-Fub1 antibodies. Ub, ubiquitin. Numbers at the right are molecular masses (in kilodaltons). (E) Suc-LLVY-MCA hydrolyzing activities of FUB1-overexpressed and Δfub1cells. Activities of CPs and 26S proteasomes were measured in the absence (left) or presence (right) of SDS.
Glycerol gradient analysis.
Cell extracts (2 mg of protein) were separated into 32 fractions by centrifugation (22 h, 100,000 × g) in 8 to 32% (vol/vol) glycerol linear gradients as described previously (15).
Assay of proteasome activity.
Peptidase activity was measured using a fluorescent peptide substrate, succinyl-Leu-Leu-Val-Tyr-7-amino-4-methylcoumarin (Suc-LLVY-MCA) (Peptide Institute, Inc.), as described previously (15). A low concentration of 0.025% (wt/vol) SDS was used as an artificial activator of CPs that are usually latent in cells. For the in vitro assay results shown in Fig. 3B, 1 nM affinity-purified CPs or 26S proteasomes and various concentrations of recombinant Fub1 were mixed and preincubated at 37°C for 15 min and then further incubated at 37°C for 1 h in 100 mM Tris-HCl (pH 8.0), 50 μM Suc-LLVY-MCA before peptidase activity was measured.
In-gel peptidase activity assay.
Native-PAGE and in-gel peptidase activity assays were basically performed according to the protocols of Elsasser et al. (32). About 100 μg of total lysates was electrophoresed using 4% native gels at 15 mA for 3 h. The gels were incubated with 100 μM Suc-LLVY-MCA in developing buffer for 5 min. After incubation, gels were exposed to UV light and photographed with a LAS-4000 mini biomolecular imager (Fujifilm).
In vitro ubiquitination and degradation assay.
The experiment for Fig. 5C was performed according to the protocols of Saeki et al. (30). 26S proteasomes were purified using anti-Flag M2 agarose beads from TD132 and TD133 cells, which were cultured not to reach stationary phase in yeast extract-peptone-dextrose (YPD) liquid medium for 2 days. YPD medium was used to shut off the expression of PBA3 under the control of the GAL1 promoter.
FIG 5.

Fub1 is unlikely to be involved in proteasome assembly. (A) Δpba4 cells were transformed with pYF5 (YEp GAL1p), pT17 (YCp PBA4), pT261 (YEp FUB1), and pT168 (YEp GAL1p-FUB1), and transformed cells were cultured on yeast extract-peptone-galactose (YPGal) for 2 days at 37°C and on YPGal plus 5 mM AZC for 3 days at 27°C. (B) Total lysates of the indicated cells were prepared after the cells were cultured in YPD medium for 2 days to shut off the expression of PBA3 and fractionated by 8 to 32% glycerol gradient centrifugation. Suc-LLVY-MCA hydrolyzing activity was measured in the presence of SDS, and each fraction was subjected to immunoblotting for α5. (C) In vitro degradation of ubiquitinated T7-Sic1PY-6×His. 26S proteasomes were purified from TD132 (GAL1p-PBA3 RPN11-3×Flag) and TD133 (GAL1p-PBA3 Δfub1 RPN11-3×Flag) strains cultured as described for panel B and were subjected to SDS-PAGE and Coomassie blue staining (left). Ubiquitinated T7-Sic1PY-6×His was incubated with the purified 26S proteasomes for the indicated times. Reaction mixtures were subjected to immunoblot analysis using anti-T7 antibodies (right). (D) The Suc-LLVY-MCA hydrolyzing activity of fractionated lysates of the indicated cells was measured in the presence of SDS. (E) Effect of Δprb1 on cell viability. One thousand cells of each indicated genotype were spread on YPGal plates to count the viable cells after PBA3 shutoff for 2 days in YPD medium. (F) Detection of polyubiquitinated proteins in wild-type, YT54 (Δpba4), YT595 (GAL1p-PBA3), and YT745 (GAL1p-PBA3Δfub1) cells cultured as described for panel B. Total lysates of the indicated strains were subjected to SDS-PAGE, followed by immunoblot analysis using antiubiquitin antibodies.
Detection of polyubiquitinated proteins.
We followed a method to detect polyubiquitinated proteins described previously (33). Cells were suspended in 200 ml cold ethanol containing 2 mM PMSF. Cells were lysed by agitation with 200 ml glass beads for 10 min. Cells lysates were dried and suspended in sample buffer for Western blotting. The primary antibody was anti-Ub antibody (LifeSensors), and the secondary antibody was anti-mouse IgG horseradish peroxidase (Jackson ImmunoResearch).
Identification of Prb1.
YT745 (GAL1p-PBA3 Δfub1::kanMX4) cells were cultured in 1 liter of YPD for 32 h at 2.5 × 107 cells/ml. After fractionation by glycerol gradient centrifugation, fractions containing nonproteasomal peptidase activity were collected and subjected to a Resource Q anion-exchange column. The resultant positive fractions were further applied to the Resource S cation-exchange column. Purified proteins were concentrated with acetone precipitation, digested with trypsin, and identified using mass spectrometry (AB Sciex TOF/TOF 5800 system).
Screening for suppressor mutations of Δfub1 Δpba4 cells.
Δfub1 Δpba4 cells bearing pMO23 (pRS316-FUB1) were mutagenized according to the protocol of Burke et al. (29). Mutagenized cells were spread onto YPD plates and incubated for 3 days at 27°C. Colonies were replicated on SD plates containing 0.5 μg/ml 5-fluoroorotic acid (5-FOA). Among 105 strains resistant to 5-FOA, one strain had a temperature-sensitive and recessive mutation. We screened a genomic library and isolated a plasmid which suppressed this temperature sensitivity. The responsible gene was identified as PRE9 by subcloning, and the mutation site was determined by sequence analysis of PRE9 in the revertant.
Crystallization and data collection.
S. cerevisiae CPs with the α7ΔN mutation were affinity purified from the YT1222 strain using anti-Flag M2 agarose beads (Sigma) and concentrated to 7.2 mg/ml by ultrafiltration in a buffer containing 25 mM Tris-HCl (pH 7.5) and 1 mM DTT. Crystals were grown by the hanging-drop vapor diffusion method at 293 K in drops containing a mixture of 1 μl of protein solution and 1 μl of reservoir solution, which consisted of 0.1 M MES (morpholineethanesulfonic acid; pH 6.5), 50 mM magnesium acetate, and 14% (vol/vol) 2-methyl-2,4-pentanediol (MPD). Diffraction data were collected under cryogenic conditions on the BL44XU beamline at Spring8 (Harima, Japan). Data processing and reduction were conducted with HKL-2000 software (34).
Structure determination and refinement.
The structure of the CP with α7ΔN was solved by molecular replacement with the wild-type yeast CP structure (Protein Data Bank [PDB] accession number 1RYP) (2) as a search model. Molecular replacement was performed with the MOLREP program (35). The models were subsequently improved through alternate cycles of manual rebuilding using Coot (36) and refinement with the program REFMAC5 (37). Data collection, phasing, and refinement statistics are summarized in Table 3. Structure figures were generated using PyMOL (38).
TABLE 3.
Data collection and refinement statistics
| Parametera | Value(s)b or group |
|---|---|
| Data collection | |
| Wavelength (Å) | 0.9 |
| Unit cell a, b, c (Å) | 137.2, 298.6, 145.4 |
| Unit cell α, β, γ (°) | 90, 113.1, 90 |
| Space group | P21 |
| Resolution range (Å) | 50.0–3.15 (3.20–3.15) |
| Total no. of reflections | 536,846 |
| No. of unique reflections | 183,783 |
| Completeness (%) | 87.0 (82.2) |
| Redundancy | 3.4 (3.2) |
| Rmerge (%) | 13.5 (42.2) |
| 〈I/σ(I)〉 | 13.7 (3.5) |
| Refinement statistics | |
| Resolution range (Å) | 48.2–3.15 |
| No. of reflections used | 151,713 |
| Free R reflections (%) | 8,012 |
| Rwork/Rfree (%) | 17.3/24.2 |
| RMSD bond length (Å) | 0.011 |
| RMSD bond angle (°) | 1.75 |
| No. of protein atoms | 49,372 |
| B factor (Å2) for protein | 52.1 |
RMSD, root mean square difference.
The values for the highest-resolution shell are in parentheses.
Protein structure accession number.
The atomic coordinate for the α7ΔN CP has been deposited under PDB accession number 3WXR.
RESULTS
Fub1 is associated mainly with the 20S CP.
In an attempt to identify proteasome-interacting proteins with a novel function, we selected 12 uncharacterized S. cerevisiae proteins that have been reported to physically interact with the proteasome from the BioGRID interaction database (39). These interactions were detected by large-scale analyses using the affinity capture-mass spectrometry or yeast two-hybrid system. Therefore, to validate the interaction of these proteins with the proteasome, each of the 12 proteins with N-terminal Flag tags was ectopically overexpressed in yeast cells and immunoprecipitated, followed by immunoblot analysis with antibodies to the endogenously HA-tagged CP subunit α6. Of the 12 proteins, Ycr076c most reliably coprecipitated with α6 (Fig. 1). Ycr076c has homology with human PI31 (hPI31) and was recently identified as Fub1 (27).
FIG 1.
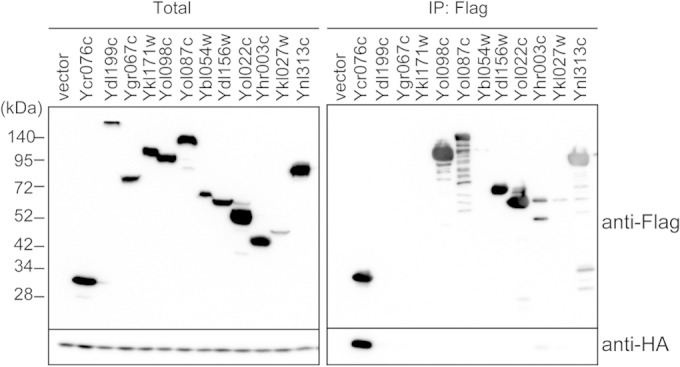
Identification of uncharacterized proteasome-interacting proteins. Uncharacterized candidates for proteasome-interacting proteins were N-terminally tagged with a Flag tag. Flag-tagged proteins were overexpressed by the GAL1 promoter in the strain bearing an α6-3HA allele, and then these proteins were immunoprecipitated (IP) using anti-Flag antibodies. Anti-HA antibodies were used to examine whether Flag-tagged proteins were coimmunoprecipitated with proteasomes.
To examine how Fub1 interacts with the proteasome in yeast, cell lysates were fractionated by glycerol gradient centrifugation, followed by measurement of peptidase activity of each fraction in the presence of 0.025% SDS, which artificially activates the CP (Fig. 2A). Fub1 was sedimented mainly in fractions 2 to 12, where no assembled CP existed, as revealed by the absence of any peptidase activity (Fig. 2A). However, a small but significant amount of Fub1 was detected in fractions 16 to 30, which correspond to the fractions containing the 20S CP and the 26S proteasome. Fub1 was relatively enriched in the 20S fractions compared to the 26S fractions. The 20S fractions (fractions 16 to 20) and 26S fractions (fractions 26 to 30) were subjected to immunoprecipitation using anti-Fub1 antibodies. Immunoblot analysis revealed that Fub1 was coprecipitated with CP subunit α5 in the 20S and the 26S fractions, indicating that Fub1 was associated with both the 20S CP and the 26S proteasome (Fig. 2B). To further confirm this, lysates of Rpn11-Flag-expressing cells were fractionated, followed by immunoprecipitation of fractions containing free 19S RPs (fractions 20 to 22) as well as the 26S fractions (fractions 26 to 30) with anti-Flag antibodies (Fig. 2C). Fub1 was coprecipitated with Rpn11-Flag in the 26S fractions but not with Rpn11-Flag in the free 19S RP fractions (Fig. 2D). These results demonstrate that Fub1 is associated mainly with the 20S CP and only to a lesser extent with the 26S proteasome and that the association of Fub1 with the proteasome is mediated through the CP.
Fub1 does not inhibit proteasome activity in vivo.
There are conflicting reports as to the effect of PI31 on proteasome function. The original paper showed that mammalian PI31 inhibits 20S CP activity in vitro, but other reports indicate that PI31 cooperates with the proteasome in protein degradation in fruit flies (20, 24–26).
To test whether Fub1 exhibits an inhibitory effect on the CP in vitro, purified recombinant Fub1 was added to each fraction of yeast lysates separated by glycerol gradient centrifugation (Fig. 3A). In the presence of 0.025% SDS, which not only acts as an artificial activator of CPs but also abolishes the interaction between the CP and the RP, Fub1 completely abrogated the activation of the CP (Fig. 3A, upper panel). However, in the absence of SDS, Fub1 had no effect on the peptidase activity of the 26S proteasome (Fig. 3A, lower panel). We also examined the effect of recombinant Fub1 on the activities of purified CPs and 26S proteasomes. Consistently with the results in Fig. 3A, Fub1 inhibited the CP activity in a dose-dependent manner, but no effect on the 26S proteasome was observed (Fig. 3B). These results suggest that Fub1 can inhibit the free CP but does not inhibit the 26S proteasome in vitro.
Next, to investigate the in vivo function of Fub1, phenotypes of Δfub1 and Fub1-overexpressing cells were examined. Δfub1 cells were viable and did not show temperature sensitivity at 37°C, just like Δfub1 cells expressing Fub1 from a single-copy plasmid (YCp) (Fig. 3C, upper panel). Furthermore, growth rates of Δfub1 and Fub1-overexpressing cells, either from a single-copy plasmid or a multicopy plasmid (YEp), were indistinguishable from Δfub1 cells on a plate containing the proline analog l-azetidine-2-carboxylic acid (AZC), while Δump1 cells showed severe sensitivity to AZC (Fig. 3C, lower panel).
We then observed whether ubiquitinated proteins accumulated by overexpression or deletion of Fub1. While Δα3 and Δump1 caused the accumulation of ubiquitinated proteins, neither overproduction nor deletion of Fub1 did (Fig. 3D). Furthermore, any obvious differences in proteasome peptidase activities were not observed between Δfub1 and Fub1-overexpressing cells (Fig. 3E). These results suggest that Fub1 does not work as a CP inhibitor in vivo. However, the functional relevance of Fub1 to proteasome-mediated protein degradation is still unclear since deletion and overexpression of Fub1 did not result in any obvious defect in cell growth and proteasome activity.
Double deletion of FUB1 and PBA4 causes synthetic lethality.
To get insights into the function of Fub1, we searched for genetic interactions by crossing Δfub1 cells with various proteasome-related-gene disruptants, such as Δpba4, Δump1, Δrpn4, Δα3, Δrpn10, Δsem1, Δecm29, and Δblm10 mutants. Of these crosses, Δfub1 Δpba4 double disruptions were lethal, as revealed by tetrad analysis of Δfub1/+ Δpba4/+ cell-derived asci (Fig. 4A, dashed circles). The other double mutants were viable and did not show a growth defect even at 35°C (Fig. 4B).
FIG 4.

Synthetic lethality of Δfub1 Δpba4. (A) Dissection of diploid cells bearing Δfub1 and Δpba4 alleles. Spores in the dashed circles were expected to have Δfub1 and Δpba4 mutations. (B) Comparison of growth levels between single and double mutant strains lacking Fub1 and proteasome-related proteins. Double mutants lacking Fub1 and proteasome-related proteins were obtained by crossing Δfub1 cells with various proteasome-related disruptant cells, followed by tetrad analysis. The resultant strains were streaked onto YPD plates and incubated for 2 days at 27°C or 35°C.
The Pba3-Pba4 heterodimer acts as a chaperone for CP assembly. Deletion of either Pba3 or Pba4 causes inefficient CP assembly and thus decreases assembled CPs (16–19). The synthetic lethality of Δpba4 Δfub1 raises the possibility that Fub1 is also involved in proteasome assembly. However, this possibility seems unlikely because Δfub1 cells produced normal amounts of CPs and 26S proteasomes (Fig. 3E). The inability of FUB1 overexpression to suppress the sensitivity of Δpba4 cells to high temperature and AZC also supports the view that Fub1 and Pba3/Pba4 have different roles (Fig. 5A). It is still possible that Fub1 works as an assembly chaperone only when Pba3-Pba4 is absent. To test this possibility, we constructed a Δfub1 strain in which the promoter of PBA3 was replaced with a GAL1 promoter, repressing the expression of PBA3 with the addition of glucose. This strain stopped growing 2 days after incubation in glucose-containing media. The proteasomal peptidase activity of the cells at this time point was measured and compared with that of cells in which only Pba3 was depleted (Fig. 5B). Activities of 20S CPs were barely detectable in both strains because of PBA3 shutoff (Fig. 5B) (17). If Fub1 were involved in the CP assembly like Pba3-Pba4, a further decrease in the 26S proteasome would be expected with the deletion of Fub1. However, there were no significant differences between the peptidase activities and the α5 protein levels in the fractions corresponding to the 26S proteasome (fractions 22 to 28), irrespective of the presence or absence of Fub1 (Fig. 5B). These results suggest that Fub1 is not involved in CP assembly and that Δfub1 Δpba4 lethality is not due to a further decrease in the amount of proteasomes.
To know whether proteasomes assembled in Δpba3 Δfub1 cells have defects in the degradation of ubiquitinated proteins, we also tested in vitro degradation of ubiquitinated T7-Sic1PY-6×His using 26S proteasomes purified from GAL1p-PBA3 and GAL1p-PBA3 Δfub1 cells cultured in the presence of glucose (30). Coomassie blue staining of purified 26S proteasomes showed that there was no apparent difference in the SDS-PAGE migration patterns of the two strains, which supports the idea that Fub1 is not involved in proteasome assembly (Fig. 5C, left panel). These proteasomes deubiquitinated and degraded ubiquitinated T7-Sic1PY-6×His proteins similarly (Fig. 5C, right panel). These results suggest that the activity of 26S proteasomes was not impaired by the deletion of Fub1, at least with respect to degradation of ubiquitinated T7-Sic1PY-6×His.
In GAL1p-PBA3 Δfub1 cells, an unusual peptidase peak emerged in fractions 4 to 6, where no CP existed (Fig. 5B). We purified the responsible peptidase and identified it as a vacuolar protease, Prb1, by using mass spectrometry and introducing the Δprb1 mutation into GAL1p-PBA3 Δfub1 cells (Fig. 5D). However, the emergence or strong induction of Prb1 does not seem to be the cause of lethality of Δfub1 Δpba4 cells because deletion of PRB1 in GAL1p-PBA3 Δfub1 cells did not suppress the lethal phenotype (Fig. 5E).
We then further searched the phenotypic difference between GAL1p-PBA3 Δfub1 and GAL1p-PBA3 cells and found that the deletion of Fub1 enhanced the accumulation of polyubiquitinated proteins in the Pba3-depleted background (Fig. 5F). Although we could not observe any significant defects in degradation of ubiquitinated T7-Sic1PY-6×His using purified proteasomes from GAL1p-Pba3 Δfub1 cells (Fig. 5C), this result indicates that Fub1 is required for efficient turnover of polyubiquitinated proteins in vivo, at least in the absence of Pba3-Pba4.
hPI31 complements the loss of Fub1.
PI31 proteins are conserved among eukaryotes from yeast to mammals. Human PI31 (hPI31) shares approximately 20% amino acid identity with Fub1 (27). Taking advantage of the lethality of Δfub1 Δpba4, we examined whether hPI31 complements Δfub1.
hPI31 cDNA was introduced into a Δfub1/+ Δpba4/+ diploid strain, followed by spore dissection of this strain. This analysis showed that G418-resistant (Δfub1) His+ (Δpba4) cells are viable, which indicates that hPI31 complements the lethality of Δfub1 Δpba4 (Fig. 6A, left and middle [clone a]). hPI31-dependent growth of Δfub1 Δpba4 cells was further confirmed by the loss of viability of Δfub1 Δpba4 plus hPI31 cells on 5-fluoroorotic acid (5-FOA)-containing medium (Fig. 6A, right [clone a]). 5-FOA is toxic to URA3+ cells and causes the loss of an hPI31 plasmid that has URA3 as a marker. These results demonstrate that hPI31 can substitute for Fub1 and suggest that the functions of PI31 proteins are conserved among eukaryotes.
FIG 6.
Complementation of Δfub1 Δpba4 lethality by human PI31 and deletion variants of Fub1. (A) Human PI31-dependent growth of Δfub1 Δpba4 cells. The heterozygous Δfub1/+ Δpba4/+ diploid cells bearing pT271 (YEp GAL1p-human PI31 URA3) were sporulated and subjected to tetrad analysis. Resultant strains were streaked on a plate containing 5-fluoroorotic acid (5-FOA), which prevents the growth of URA3+ cells. (B) Complementing abilities of Fub1 deletion mutants. Full-length or mutant Fub1 was expressed as an N-terminally Flag-tagged protein using the pT322 (YEp GAL1p LEU2) vector in Δfub1 Δpba4 cells bearing pMO23 (YCp FUB1 URA3). Transformant cells were diluted serially and spotted on a plate containing galactose and 5-FOA. PRR denotes the proline-rich region. (C) Detection of Fub1 deletion variants by immunoblot analysis. Cell lysates of strains used for the experiment shown in panel B were subjected to SDS-PAGE and immunoblot analysis using anti-Fub1 antibodies. Pgk1 was used as a loading control. Numbers at the right are molecular masses (in kilodaltons).
The C-terminal proline-rich region is essential for Fub1 function.
Two well-characterized domains exist in PI31 proteins (24, 40). The N-terminal FP domain of human and fly PI31 has been shown to bind to the F-box proteins Fbxo7 and Nutcracker (fly homolog of Fbxo7), respectively. The C-terminal proline-rich region (PRR) has been shown to be responsible for the inhibitory effect on the CP in vitro (21). Fub1 also has the PRR, but the FP domain does not seem to be well conserved in Fub1.
We constructed Fub1 deletion mutants, each of which lacks either the N-terminal region (ΔN) or the PRR (ΔPRR) (Fig. 6B). Truncated open reading frames (ORFs) with Flag tags were introduced into Δfub1 Δpba4 cells containing the FUB1-URA3 plasmid. A URA3 counterselection assay using 5-FOA revealed that the PRR was sufficient to complement the loss of FUB1 and that the N-terminal domain was dispensable for viability. The PRR was further divided into two halves (Fig. 6B, PRR-N, PRR-C), but neither complemented Δfub1 Δpba4 lethality.
Furthermore, the N-terminal domain is likely important for the stability of Fub1 protein, since Fub1ΔN was hardly detectable, although this construct complemented Δfub1 Δpba4 lethality (Fig. 6C).
Δα3 suppresses Δfub1 Δpba4 lethality.
To obtain clues as to the reason why simultaneous deletion of Δfub1 and Δpba4 was lethal, we screened suppressor mutations of Δfub1 Δpba4 lethality and identified a mutation in the α3 (PRE9) ORF that resulted in the replacement of Gly155 with Arg (determined to be responsible). Indeed, the α3-G155R mutation suppressed the lethality of Δfub1 Δpba4 (Fig. 7A, circled cells).
FIG 7.

Suppression of Δfub1 Δpba4 synthetic lethality by the mutations in α3. (A) Dissection of diploid cells bearing Δfub1::kan, Δpba4::HIS3, and α3-G155R-URA3. All circled G418-resistant and His+ cells were Ura+. (B) Dissection of diploid cells bearing Δfub1::hph, Δpba4::HIS3, and Δα3::kan. The circled cell with all three mutations was viable. +hyg, with hygromycin.
It is likely that the G155R mutation in α3 disturbs the interaction between α3 and α4 because the Gly155 of α3 is located on the surface in contact with α4, suggesting that deletion of α3 might be equivalent to α3-G155R (2). We examined whether Δα3 suppressed Δfub1 Δpba4 lethality. Δα3 Δpba4 cells were crossed with Δfub1 cells. This revealed that the Δfub1 Δpba4 Δα3 triple mutant was viable (Fig. 7B, circle). This result confirms that G155R of α3 and deletion of α3 have the same effect on the CP and demonstrates that deletion of α3 suppresses Δfub1 Δpba4 lethality.
N-terminal deletion of α7, but not that of α3, suppresses Δfub1 Δpba4 lethality.
α3 is the only CP subunit whose deletion is not lethal in S. cerevisiae. In Δα3 cells, another copy of α4 is incorporated in place of α3, forming an α4-α4-type CP (41). The α-ring of the CP is usually closed owing to the N terminus of α3, which serves as a gatekeeper of the CP (5). Thus, the α4-α4-type CP is supposed to be in a constitutively active form without association of RPs or activators. We speculated that an open conformation of the CP might suppress the lethality of Δfub1 Δpba4.
To test this possibility, we constructed a strain in which the 9 N-terminal residues of α3 were deleted (α3ΔN). However, in contrast to our speculation, α3ΔN failed to suppress Δfub1 Δpba4 lethality (Fig. 8A, both of the dashed circles and either of the dashed triangles). Then, we further made a strain lacking the 11 N-terminal residues of α7 (α7ΔN), because simultaneous deletion of the N termini of α3 and α7 has been shown to be more efficient for activation of the CP than the single deletion of the N terminus of either α3 or α7 (42). To our surprise, this experiment revealed that a single deletion of the α7 N terminus was enough to suppress Δfub1 Δpba4 lethality (Fig. 8B, circles). α7ΔN cells were able to grow almost normally in the presence of AZC and at high temperature (Fig. 8C). To exclude the possibility that α7ΔN merely suppresses the phenotype of Δpba4 cells, we observed the phenotypes of the α7ΔN Δpba4 strain. α7ΔN failed to suppress the AZC sensitivity and temperature sensitivity of Δpba4 cells (Fig. 8C). Thus, this observation indicates that α7ΔN is not just a suppressor mutation of Δpba4 but that α7ΔN is a specific suppressor of Δfub1 Δpba4.
FIG 8.

N-terminal deletion of α7, but not that of α3, suppresses Δfub1 Δpba4 lethality. (A) Dissection of diploid cells bearing Δfub1::hph, Δpba4::kan, and α3ΔN-URA3. In α3ΔN, 9 residues (GSRRYDSRT) were deleted from the N terminus of α3. Dashed circles represent the locations of the spores expected to have Δfub1 Δpba3 α3ΔN triple mutations. Either one of the two spores located in dashed triangles is also expected to have Δfub1 Δpba3 α3ΔN triple mutations. (B) Dissection of diploid cells bearing Δfub1::hph, Δpba4::kan, α3ΔN-URA3, and α7ΔN-HIS3. In α7ΔN, 11 residues (TSIGTGYDLSN) were deleted from the N terminus of α7. The circled viable colonies have α7ΔN mutations (His+) along with Δfub1 Δpba3 but do not have α3ΔN (Ura−). (C) Inability of α7ΔN to suppress Δpba4 phenotypes. Wild-type (WT), Δpba4, α7ΔN, and α7ΔNΔpba4 cells were streaked onto a plate containing YPD plus 5 mM AZC and incubated for 2 days at 27°C (left). The same cells were also streaked onto a YPD plate and incubated for 1 day at 37°C (right).
To understand the property of CPs in α7ΔN cells, purified proteasomes from the lysates of wild-type, Δfub1, α7ΔN, and α3ΔN cells were resolved by native-PAGE, followed by an in-gel peptidase assay in the presence or absence of 0.025% SDS (Fig. 9A). Wild-type cells have free CPs, RPCPs (CPs associated with one RP), and RP2CPs (CPs associated with two RPs). RPCPs and RP2CPs exhibited peptidase activity in the absence of SDS, whereas free, latent CPs were activated only when SDS was added. Δfub1 cells showed essentially the same profile as wild-type cells, consistent with the observation that Δfub1 did not apparently affect overall proteasome activity (Fig. 3E). As expected, free CPs of α3ΔN cells were active even in the absence of SDS. α7ΔN cells showed essentially the same phenotype as α3ΔN cells with respect to the peptidase activity of the CP, although its activity was weaker than that of α3ΔN. These results indicate that the CPs lacking the N terminus of either α3 or α7 allow small peptides to enter the catalytic chamber.
It is possible that Fub1 binds to the N terminus of α7 and change its structural arrangement. Thus, we then examined whether α7ΔN affects the binding of Fub1 to CPs. Wild-type, α3ΔN, and α7ΔN proteasomes bearing Pre1-3×Flag were immunoprecipitated with anti-Flag antibodies, and immunoprecipitates were subjected to Western blot analysis using anti-Fub1 antibodies. This experiment revealed that both α7ΔN and α3ΔN did not affect binding of Fub1 to CPs (Fig. 9B). Thus, although the physiological role of Fub1 is closely related to that of the α7 N terminus, binding of Fub1 to the CP does not depend on the N terminus of α7.
We then focused on the unique phenotype of Δα3 cells. Δα3 cells were shown to be resistant to cadmium (16). We examined whether α7ΔN cells also show cadmium resistance. Consistently with the previous finding, Δα3 cells grew better than Δfub1 cells bearing YCp-FUB1, which were equivalent to wild-type cells. However, neither α7ΔN nor α3ΔN cells were resistant to cadmium (Fig. 9C). Δfub1 and Fub1-overexpressed cells also did not show cadmium resistance. These results suggest that cadmium resistance is not involved in the suppression of Δfub1 Δpba4 lethality.
Crystal structure of the α7ΔN CP.
Finally, we determined the crystal structure of the α7ΔN CP and compared it with the structures of wild-type and α3ΔN CPs (Table 3 and Fig. 10). From the top, whereas the α3ΔN α-ring had a clear open gate, α7ΔN did not seem to cause any significant structural changes in the α-ring (Fig. 10A). However, we noticed that the N terminus of α1 protruded from the α-ring of α7ΔN CP when viewed from the side, while the N terminus of α7 protruded in the wild-type CP (Fig. 10B). Protrusion of the N terminus of α1 causes steric hindrance with the N terminus of α7, and therefore the N terminus of α1 locates at the molecular surface of the α-ring in the wild-type (Fig. 10C and D). Unfortunately, we cannot see the conformation of the N terminus of α1 in the α3ΔN α-ring because the structure of this region is not reported in the PDB (Fig. 10D). If protrusion of the N terminus of α1 is specific for the α-ring of α7ΔN CP, this feature might be responsible for recovery from Δfub1 Δpba3 lethality by the α7ΔN mutation.
FIG 10.

Crystal structure of the α7ΔN α-ring. (A) Top views of the structures of the wild-type (PDB accession number 1RYP), α3ΔN (PDB accession number 1G0U), and α7ΔN α-rings (PDB accession number 3WXR). A C-α diagram (upper left) and surface plots of the α-ring are shown. N termini of α3 and α7 are colored magenta and red in a C-α diagram, respectively. The same colors are used for α3 and α7 subunits in surface plots as in the C-α diagram. (B) Side views of the α-rings of wild-type and α7ΔN CPs. Note that the 3 N-terminal residues of α6 are not visible in α7ΔN CP. (C) Closeup view of the N-terminal region of the α7 (wild-type)-α1 (α7ΔN) complex model. The dashed circle shows a collision between Thr5 of α7 and Ala10 of α1. (D) Closeup view around the N-terminal regions of α1 and α7 in the α-rings of wild-type, α3ΔN, and α7ΔN CPs. The sequence numbering follows that of the wild-type structural data (1RYP).
DISCUSSION
Mammalian PI31 was originally identified as an inhibitor of CP activity in vitro (20). We showed that Fub1, a yeast S. cerevisiae PI31 ortholog, also bound to the proteasome and exhibited an inhibitory effect on the in vitro activity of the CP. However, our overall results do not indicate that Fub1 acts on the proteasome's function negatively, as we did not detect any deficiencies in proteasomal protein degradation by overexpressing Fub1 in yeast cells. Instead, our new finding of the synthetic lethality of Δfub1 with Δpba4 suggests that Fub1 positively regulates proteasome function in yeast, since Pba3-Pba4 is an assembly chaperone complex for CPs and Δpba4 causes a CP assembly defect.
Recently, Steller's group found that fruit fly PI31, DmPI31, works in cooperation with 26S proteasomes (24, 25). They showed that ectopic expression of DmPI31 suppressed an eye phenotype caused by proteasome dysfunction, whereas reducing DmPI31 function enhanced proteasome defects. They also showed that ADP ribosylation of DmPI31 by tankyrase was important for promoting assembly of the 26S proteasome. However, there are no tankyrase homologs in S. cerevisiae. It remains an open question whether alternative modifications exist for the regulation of Fub1 function in yeast.
Although there might be some differences between the regulatory systems of Fub1 and other PI31 homologs, their main function seems to be conserved among eukaryotes. At least, Fub1 and hPI31 have overlapping functions because hPI31 complemented Δfub1 Δpba4 lethality. Fub1 was detected mainly in fractions where no assembled CP existed by our glycerol gradient centrifugation analysis. However, we further showed that the only C-terminal proline-rich region (PRR) is essential for suppression of Δfub1 Δpba4 lethality, and the PRR is responsible for the in vitro inhibition of the CP's activity. Thus, our results suggest that Fub1 exerts its main function via the interaction with the CP, although we cannot exclude the possibility that Fub1 has some other functions irrespective of the CP.
The most striking finding in this paper is that α7ΔN, but not α3ΔN, suppressed Δfub1 Δpba4 synthetic lethality. It has been reported that α7ΔN has little effect on the activation of CP peptidase activities (42). However, Bajorek et al. also showed that the degradation of casein by CPs was drastically enhanced by α7ΔN when combined with α3ΔN, suggesting a role for the N terminus of α7 in protein degradation by the CP (42). Furthermore, in contrast to the observation by Bajorek et al., we showed that α7ΔN resulted in the production of the activated CP, although its activation was much weaker than α3ΔN′s. Our α7ΔN has a 1-amino-acid-shorter N terminus than Bajorek's one. This difference might account for the observation that our α7ΔN conferred partial activation on CPs in our in-gel peptidase assay.
To further understand the property of the α7ΔN CP, we determined the crystal structure of the α7ΔN CP. The α7ΔN CP looked almost similar to the wild-type CP, but the N terminus of α1 protruded and moved distally from the α-ring. Our findings suggest that the N terminus of α7 might be one of the key factors that regulate CPs by modifying the conformation of α-rings or blocking the degradation of some specific proteins and that Fub1 might play some roles in protein degradation, which would be inhibited by the N terminus of α7 in the absence of Fub1. Alternatively, it is also possible that the protruding N terminus of α1 strengthens the subunit's association with some other proteins, and its association consequently may promote some kind of protein degradation, for which Fub1 is required.
Recombinant Fub1 and in vivo overexpression of Fub1 did not activate the peptidase activities of latent CPs. These results suggest that Fub1 is not likely to act as a simple opener of the α-ring gate but rather that Fub1 might be required for degradation of some specific substrates and be functional only when it is associated with its substrates. It has been shown that the CP can degrade intrinsically unstructured proteins (IUPs) directly without ubiquitination (43). The PRR might be important for capturing IUPs, analogously to recognition of unfolded proteins by San1 (44), and direct them to degradation by the CPs. The observation that the PRR of Fub1, which was enough to suppress the loss of Fub1, was extremely unstable may support this idea.
On the other hand, the N-terminal region of Fub1 is important for the stability of Fub1, because the PRR alone was extremely unstable. N termini of hPI31 and DmPI31 are known to interact with an F-box protein, Fbxo7, and its fly homolog Nutcracker, respectively. Interestingly, binding of DmPI31 with Nutcracker stabilizes DmPI31. Although the biological significance of the interaction between these PI31 proteins and their partners still remains to be solved, exploring such proteins in yeast would be helpful to understand the functions of Fub1 and PI31 more precisely.
ACKNOWLEDGMENTS
We thank Y. Saeki for providing the materials for in vitro degradation assay. We also thank M. Ozaki and T. Kameyama for their assistance with this work and Y. Sakurai and L. Kogleck for critically reading the manuscript.
Funding for this work was provided by the Japan Society for the Promotion of Science (25221102 and 26000014).
REFERENCES
- 1.Tanaka K. 2009. The proteasome: overview of structure and functions. Proc Jpn Acad Ser B Phys Biol Sci 85:12–36. doi: 10.2183/pjab.85.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Groll M, Ditzel L, Lowe J, Stock D, Bochtler M, Bartunik HD, Huber R. 1997. Structure of 20S proteasome from yeast at 2.4 A resolution. Nature 386:463–471. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- 3.Forster A, Masters EI, Whitby FG, Robinson H, Hill CP. 2005. The 1.9 A structure of a proteasome-11S activator complex and implications for proteasome-PAN/PA700 interactions. Mol Cell 18:589–599. doi: 10.1016/j.molcel.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 4.Lowe J, Stock D, Jap B, Zwickl P, Baumeister W, Huber R. 1995. Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 A resolution. Science 268:533–539. doi: 10.1126/science.7725097. [DOI] [PubMed] [Google Scholar]
- 5.Groll M, Bajorek M, Kohler A, Moroder L, Rubin DM, Huber R, Glickman MH, Finley D. 2000. A gated channel into the proteasome core particle. Nat Struct Biol 7:1062–1067. doi: 10.1038/80992. [DOI] [PubMed] [Google Scholar]
- 6.Stadtmueller BM, Ferrell K, Whitby FG, Heroux A, Robinson H, Myszka DG, Hill CP. 2010. Structural models for interactions between the 20S proteasome and its PAN/19S activators. J Biol Chem 285:13–17. doi: 10.1074/jbc.C109.070425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu Y, Smith DM, Kim HM, Rodriguez V, Goldberg AL, Cheng Y. 2010. Interactions of PAN′s C-termini with archaeal 20S proteasome and implications for the eukaryotic proteasome-ATPase interactions. EMBO J 29:692–702. doi: 10.1038/emboj.2009.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bedford L, Paine S, Sheppard PW, Mayer RJ, Roelofs J. 2010. Assembly, structure, and function of the 26S proteasome. Trends Cell Biol. 20:391–401. doi: 10.1016/j.tcb.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kusmierczyk AR, Hochstrasser M. 2008. Some assembly required: dedicated chaperones in eukaryotic proteasome biogenesis. Biol Chem 389:1143–1151. doi: 10.1515/BC.2008.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramos PC, Dohmen RJ. 2008. PACemakers of proteasome core particle assembly. Structure 16:1296–1304. doi: 10.1016/j.str.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 11.Rosenzweig R, Glickman MH. 2008. Chaperone-driven proteasome assembly. Biochem Soc Trans 36:807–812. doi: 10.1042/BST0360807. [DOI] [PubMed] [Google Scholar]
- 12.Murata S, Yashiroda H, Tanaka K. 2009. Molecular mechanisms of proteasome assembly. Nat Rev Mol Cell Biol 10:104–115. doi: 10.1038/nrm2630. [DOI] [PubMed] [Google Scholar]
- 13.Matias AC, Ramos PC, Dohmen RJ. 2010. Chaperone-assisted assembly of the proteasome core particle. Biochem Soc Trans 38:29–33. doi: 10.1042/BST0380029. [DOI] [PubMed] [Google Scholar]
- 14.Hirano Y, Hayashi H, Iemura S, Hendil KB, Niwa S, Kishimoto T, Kasahara M, Natsume T, Tanaka K, Murata S. 2006. Cooperation of multiple chaperones required for the assembly of mammalian 20S proteasomes. Mol Cell 24:977–984. doi: 10.1016/j.molcel.2006.11.015. [DOI] [PubMed] [Google Scholar]
- 15.Hirano Y, Hendil KB, Yashiroda H, Iemura S, Nagane R, Hioki Y, Natsume T, Tanaka K, Murata S. 2005. A heterodimeric complex that promotes the assembly of mammalian 20S proteasomes. Nature 437:1381–1385. doi: 10.1038/nature04106. [DOI] [PubMed] [Google Scholar]
- 16.Kusmierczyk AR, Kunjappu MJ, Funakoshi M, Hochstrasser M. 2008. A multimeric assembly factor controls the formation of alternative 20S proteasomes. Nat Struct Mol Biol 15:237–244. doi: 10.1038/nsmb.1389. [DOI] [PubMed] [Google Scholar]
- 17.Yashiroda H, Mizushima T, Okamoto K, Kameyama T, Hayashi H, Kishimoto T, Niwa S, Kasahara M, Kurimoto E, Sakata E, Takagi K, Suzuki A, Hirano Y, Murata S, Kato K, Yamane T, Tanaka K. 2008. Crystal structure of a chaperone complex that contributes to the assembly of yeast 20S proteasomes. Nat Struct Mol Biol 15:228–236. doi: 10.1038/nsmb.1386. [DOI] [PubMed] [Google Scholar]
- 18.Hoyt MA, McDonough S, Pimpl SA, Scheel H, Hofmann K, Coffino P. 2008. A genetic screen for Saccharomyces cerevisiae mutants affecting proteasome function, using a ubiquitin-independent substrate. Yeast 25:199–217. doi: 10.1002/yea.1579. [DOI] [PubMed] [Google Scholar]
- 19.Le Tallec B, Barrault MB, Courbeyrette R, Guerois R, Marsolier-Kergoat MC, Peyroche A. 2007. 20S proteasome assembly is orchestrated by two distinct pairs of chaperones in yeast and in mammals. Mol Cell 27:660–674. doi: 10.1016/j.molcel.2007.06.025. [DOI] [PubMed] [Google Scholar]
- 20.Chu-Ping M, Slaughter CA, DeMartino GN. 1992. Purification and characterization of a protein inhibitor of the 20S proteasome (macropain). Biochim Biophys Acta 1119:303–311. doi: 10.1016/0167-4838(92)90218-3. [DOI] [PubMed] [Google Scholar]
- 21.McCutchen-Maloney SL, Matsuda K, Shimbara N, Binns DD, Tanaka K, Slaughter CA, DeMartino GN. 2000. cDNA cloning, expression, and functional characterization of PI31, a proline-rich inhibitor of the proteasome. J Biol Chem 275:18557–18565. doi: 10.1074/jbc.M001697200. [DOI] [PubMed] [Google Scholar]
- 22.Zaiss DM, Standera S, Holzhutter H, Kloetzel P, Sijts AJ. 1999. The proteasome inhibitor PI31 competes with PA28 for binding to 20S proteasomes. FEBS Lett 457:333–338. doi: 10.1016/S0014-5793(99)01072-8. [DOI] [PubMed] [Google Scholar]
- 23.Zaiss DM, Standera S, Kloetzel PM, Sijts AJ. 2002. PI31 is a modulator of proteasome formation and antigen processing. Proc Natl Acad Sci U S A 99:14344–14349. doi: 10.1073/pnas.212257299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bader M, Benjamin S, Wapinski OL, Smith DM, Goldberg AL, Steller H. 2011. A conserved F box regulatory complex controls proteasome activity in Drosophila. Cell 145:371–382. doi: 10.1016/j.cell.2011.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cho-Park PF, Steller H. 2013. Proteasome regulation by ADP-ribosylation. Cell 153:614–627. doi: 10.1016/j.cell.2013.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li X, Thompson D, Kumar B, DeMartino GN. 2014. Molecular and cellular roles of PI31 (PSMF1) protein in regulation of proteasome function. J Biol Chem 289:17392–17405. doi: 10.1074/jbc.M114.561183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hatanaka A, Chen B, Sun J Q, Mano Y, Funakoshi M, Kobayashi H, Ju Y, Mizutani T, Shinmyozu K, Nakayama J, Miyamoto K, Uchida H, Oki M. 2011. Fub1p, a novel protein isolated by boundary screening, binds the proteasome complex. Genes Genet Syst 86:305–314. doi: 10.1266/ggs.86.305. [DOI] [PubMed] [Google Scholar]
- 28.Guthrie C, Fink GR (ed). 2002. Methods in enzymology, vol 350, part B. Guide to yeast genetics and molecular and cell biology. Academic Press, Amsterdam, Netherlands. [Google Scholar]
- 29.Burke D, Dawson D, Stearns T (ed). 2000. Methods in yeast genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 30.Saeki Y, Isono E, Toh EA. 2005. Preparation of ubiquitinated substrates by the PY motif-insertion method for monitoring 26S proteasome activity. Methods Enzymol 399:215–227. doi: 10.1016/S0076-6879(05)99014-9. [DOI] [PubMed] [Google Scholar]
- 31.Perez J, Gomez A, Roncero C. 2010. Upregulation of the PRB1 gene in the Saccharomyces cerevisiae rim101Delta mutant produces proteolytic artefacts that differentially affect some proteins. Yeast 27:575–581. doi: 10.1002/yea.1776. [DOI] [PubMed] [Google Scholar]
- 32.Elsasser S, Schmidt M, Finley D. 2005. Characterization of the proteasome using native gel electrophoresis. Methods Enzymol 398:353–363. doi: 10.1016/S0076-6879(05)98029-4. [DOI] [PubMed] [Google Scholar]
- 33.Kimura Y, Koitabashi S, Kakizuka A, Fujita T. 2001. Initial process of polyglutamine aggregate formation in vivo. Genes Cells 6:887–897. doi: 10.1046/j.1365-2443.2001.00472.x. [DOI] [PubMed] [Google Scholar]
- 34.Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 35.Vagin A, Teplyakov A. 2010. Molecular replacement with MOLREP. Acta Crystallogr D Biol Crystallogr 66:22–25. doi: 10.1107/S0907444909042589. [DOI] [PubMed] [Google Scholar]
- 36.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 37.Murshudov GN, Vagin AA, Dodson EJ. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 38.DeLano WL. 2002. The PyMOL molecular graphics system. DeLano Scientific, San Carlos, CA: http://www.pymol.org. [Google Scholar]
- 39.Chatr-Aryamontri A, Breitkreutz BJ, Heinicke S, Boucher L, Winter A, Stark C, Nixon J, Ramage L, Kolas N, O'Donnell L, Reguly T, Breitkreutz A, Sellam A, Chen D, Chang C, Rust J, Livstone M, Oughtred R, Dolinski K, Tyers M. 2013. The BioGRID interaction database: 2013 update. Nucleic Acids Res 41:D816–D823. doi: 10.1093/nar/gks1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kirk R, Laman H, Knowles PP, Murray-Rust J, Lomonosov M, Meziane EK, McDonald NQ. 2008. Structure of a conserved dimerization domain within the F-box protein Fbxo7 and the PI31 proteasome inhibitor. J Biol Chem 283:22325–22335. doi: 10.1074/jbc.M709900200. [DOI] [PubMed] [Google Scholar]
- 41.Velichutina I, Connerly PL, Arendt CS, Li X, Hochstrasser M. 2004. Plasticity in eucaryotic 20S proteasome ring assembly revealed by a subunit deletion in yeast. EMBO J 23:500–510. doi: 10.1038/sj.emboj.7600059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bajorek M, Finley D, Glickman MH. 2003. Proteasome disassembly and downregulation is correlated with viability during stationary phase. Curr Biol 13:1140–1144. doi: 10.1016/S0960-9822(03)00417-2. [DOI] [PubMed] [Google Scholar]
- 43.Liu CW, Corboy MJ, DeMartino GN, Thomas PJ. 2003. Endoproteolytic activity of the proteasome. Science 299:408–411. doi: 10.1126/science.1079293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fredrickson EK, Rosenbaum JC, Locke MN, Milac TI, Gardner RG. 2011. Exposed hydrophobicity is a key determinant of nuclear quality control degradation. Mol Biol Cell 22:2384–2395. doi: 10.1091/mbc.E11-03-0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sikorski RS, Hieter P. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yashiroda H, Tanaka K. 2004. Hub1 is an essential ubiquitin-like protein without functioning as a typical modifier in fission yeast. Genes Cells 9:1189–1197. doi: 10.1111/j.1365-2443.2004.00807.x. [DOI] [PubMed] [Google Scholar]
- 47.Qadota H, Ishii I, Fujiyama A, Ohya Y, Anraku Y. 1992. RHO gene products, putative small GTP-binding proteins, are important for activation of the CAL1/CDC43 gene product, a protein geranylgeranyltransferase in Saccharomyces cerevisiae. Yeast 8:735–741. doi: 10.1002/yea.320080906. [DOI] [PubMed] [Google Scholar]