

Significance
A major obstacle to developing an effective T-cell–based AIDS vaccine is that the immunization may activate CD4+ T cells, possibly making them more susceptible to infection by HIV. We tested several vaccine candidates and their effects on mucosal CD4+ T cells in a simian model of HIV infection [i.e., Simian immunodeficiency virus (SIV) infection of rhesus macaques]. We find that the immunizations did not protect the animals against infection; however, a lower set-point viral load was observed in the vaccinated animals. Importantly, this study showed that the presence of higher levels of activated CD4+ T cells in mucosal tissues is associated with increased risk of breakthrough SIV infection in vaccinated animals.
Keywords: SIV, HIV, CD4, vaccination
Abstract
An effective T-cell–based AIDS vaccine should induce strong HIV-specific CD8+ T cells in mucosal tissues without increasing the availability of target cells for the virus. Here, we evaluated five immunization strategies that include Human adenovirus-5 (AdHu5), Chimpanzee adenovirus-6 (AdC6) or -7 (AdC7), Vaccinia virus (VV), and DNA given by electroporation (DNA/EP), all expressing Simian immunodeficiency virus group specific antigen/transactivator of transcription (SIVmac239Gag/Tat). Five groups of six rhesus macaques (RMs) each were vaccinated with DNA/EP-AdC6-AdC7, VV-AdC6-AdC7, DNA/-EP-VV-AdC6, DNA/EP-VV-AdC7, or AdHu5-AdHu5-AdHu5 and were challenged repeatedly with low-dose intrarectal SIVmac239. Upon challenge, there were no significant differences among study groups in terms of virus acquisition or viral load after infection. When taken together, the immunization regimens did not protect against SIV acquisition compared with controls but did result in an ∼1.6-log decline in set-point viremia. Although all immunized RMs had detectable SIV-specific CD8+ T cells in blood and rectal mucosa, we found no correlation between the number or function of these SIV-specific CD8+ T cells and protection against SIV acquisition. Interestingly, RMs experiencing breakthrough infection showed significantly higher prechallenge levels of CD4+C-C chemokine receptor type 5 (CCR5)+HLA-DR+ T cells in the rectal biopsies (RB) than animals that remained uninfected. In addition, among the infected RMs, the percentage of CD4+CCR5+Ki-67+ T cells in RBs prechallenge correlated with higher early viremia. Overall, these data suggest that the levels of activated CD4+CCR5+ target T cells in the rectal mucosa may predict the risk of SIV acquisition in RMs vaccinated with vectors that express SIVGag/Tat.
The global spread of HIV infection, which currently affects more than 30 million individuals worldwide, strongly emphasizes the need to develop a safe and effective HIV/AIDS vaccine. Key progress in this direction derived from a number of studies showing that several components of the host antiviral immune response, including CD8+ T-cell–mediated cytotoxic T-lymphocyte (CTL) responses, CD4+ T-cell responses, and neutralizing antibodies, have the potential to prevent or suppress HIV or Simian immunodeficiency virus (SIV) replication effectively in vivo (1–9). However, the development of an AIDS vaccine revealed remarkable scientific challenges that are related to specific aspects of HIV biology. These aspects include (i) the extreme genetic heterogeneity and structural plasticity of the virus; (ii) the ability of the virus to persist as integrated proviral DNA in an immunologically silent form (i.e., latent infection); and (iii) HIV’s preferential targeting of activated memory CD4+ T cells, creates the possibility that any vaccine-induced immune response to HIV will paradoxically favor its transmission (10–12). This last issue has been emphasized by the results two large-scale phase IIb clinical trials testing the efficacy of three candidate AIDS vaccine regimens, the Step trial, the Phambili trial, and the HVTN-505 trial, which independently identify a trend toward a higher frequency of HIV acquisition in vaccinated individuals than in placebo recipients (13–15). Thus the ideal candidate AIDS vaccine should induce strong and persistent antiviral immune responses in mucosal tissues (i.e., HIV-specific neutralizing antibodies and CTL responses) with limited mucosal recruitment of activated CD4+ T cells that can serve as targets for the infection.
Several immunogens currently are being investigated in preclinical and clinical studies as candidate AIDS vaccines, including recombinant DNA and viral vectors expressing HIV and SIV gene products as well as protein-based immunogens [envelope (Env) and virus-like particles, among others] (10, 16). The absence of immunogens that robustly elicit HIV- or SIV-specific broadly neutralizing antibodies has provided impetus for the development of vaccine strategies aimed at eliciting strong antiviral CTL responses (17). A number of these CTL-based immunogens (DNA, adenovirus, Pox viridae, and CMV, among others) have shown promising results in the preclinical nonhuman primate model of rhesus macaques (RMs) in terms of partial protection against acquisition and/or virus replication upon challenge with SIV or the chimeric Simian-human immunodeficient virus (16). However, the immunological correlates of this observed protection remain incompletely understood and may vary substantially based on the specific immunogen. For instance, protection conferred by live-attenuated SIV vaccines appears to be mediated by activated effector memory CD8+ T cells in lymph nodes (18), whereas CMV-based vectors protect against highly pathogenic SIV challenge through the persistent induction of effector CD8+ T cells in mucosal tissues (19). In addition, it remains unclear whether and to what extent different vectors (or vector combinations) can induce the mucosal recruitment of activated CD4+ target T cells and how this event affects virus acquisition and/or replication after challenge.
In this study we sought to identify the correlates of immunological protection against low-dose intrarectal SIVmac239 challenges following immunization of RMs with five different regimens aimed at eliciting strong antiviral CTL responses. The regimens used included various combinations (Fig. 1) of the following vectors: Human adenovirus type 5 (AdHu5), Chimpanzee adenovirus type 6 (AdC6) and type 7 (AdC7), Vaccinia virus (VV), and recombinant DNA administered via electroporation (DNA/EP). In an attempt to construct a reductionist experimental system, all vectors used expressed SIVmac239 group specific antigen (Gag) and transactivator of transcription (Tat) but did not contain any Env immunogen. All immunized RMs showed detectable SIV-specific CD8+ T cells in blood and tissues as well as an increase in C-C chemokine receptor type 5 (CCR5)-expressing activated CD4+ T cells. Upon SIV challenge, the vector combinations, taken as a whole, did not protect against virus acquisition but reduced virus replication after SIV infection by ∼1.6 logs. Importantly, we found that SIV infection was associated with higher prechallenge levels of Gag-specific CD8+ T cells in the blood and with higher prechallenge levels of activated CD4+CCR5+ T cells in the rectal mucosa. In addition, prechallenge levels of activated CD4+CCR5+ T cells in the rectal mucosa correlated positively with higher viral load at day 7 after infection. We concluded that the level of activated CD4+CCR5+ T cells in mucosal tissues may be an important correlate of SIV acquisition in the setting of CTL-based candidate AIDS vaccines.
Fig. 1.
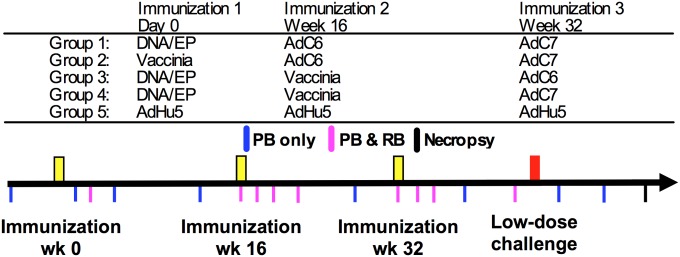
Heterologous prime-boost immunization regimens. Schematic representation of the experimental design that involved five groups of animals given three immunizations 16 wk apart: (1) DNA/EP followed by AdC6 and then AdC7; (2) VV followed by AdC6 and then AdC7; (3) DNA/EP followed by VV and AdC6; (4) DNA/EP followed by VV and AdC7; or (5) three immunizations of AdHu5. Each group included six MamuA*01+ adult Indian RMs. Three to five months following the final immunization all RMs and six additional unvaccinated control animals were challenged with repeated low-dose intrarectal SIVmac239 every week up to 15 times. The times of immunization, challenge, and sample collections are indicated. PB, peripheral blood; RB, rectal biopsy.
Results
Study Design: Immunization and Challenge.
In this study we assessed the immunogenicity and protection from low-dose rectal challenge with SIVmac239 conferred by five combinations of vectors expressing SIVGag and Tat (Fig. 1). The study design is shown in Fig. 1, including immunizations, challenge, and timing of sample collections. Briefly, five groups of six Mamu-A*01+ RMs were vaccinated with three immunizations 16 wk apart as follows: (i) DNA/EP followed by AdC6 and then by AdC7; (ii) VV followed by AdC6 and then by AdC7; (iii) DNA/EP followed by VV and AdC6; (iv) DNA/EP followed by VV and AdC7; or (v) three immunizations of AdHu5 (Materials and Methods). Six unvaccinated controls were also included. Three to five months after the final immunization all RMs were challenged repeatedly intrarectally with a low dose [300 median tissue-culture infective dose (TCID50)] of SIVmac239 that was given every week until infection or for a total of 15 challenges. Animals that tested positive for SIV viremia at a level greater than 1,000 copies/mL of plasma were not challenged further and instead were followed for 6 wk to monitor the early clinical, virological, and immunological course of the infection.
All Tested Immunization Regimens Induced Robust and Polyfunctional SIV-Specific Cellular Immune Responses.
To assess the immunogenicity of the tested vaccination regimens, we first assessed the level of SIV-specific CD8+ T-cell responses by tetramer staining for the well-characterized Mamu-A*01–restricted immunodominant epitopes Gag-CM9 and Tat-SL8. As shown in Fig. 2A and Fig. S1A, tetramer-positive CD8+ T cells were readily identifiable in in whole blood from all groups of immunized RMs, and in general the fraction of CD8+ T cells increased after each vaccination and then contracted within a few weeks. For both Gag-CM9+ and Tat-SL8+ cells we observed a trend toward higher responses in the animals receiving DNA/EP followed by VV and AdC6 and in those receiving DNA/EP followed by VV and AdC7. Conversely, the lower responses after the third immunization were observed in the groups receiving three AdHu5 immunizations and those receiving DNA/EP followed by AdC6 and AdC7. As shown in Fig. 2B and Fig. S1B, we also detected robust levels of Gag-CM9- and Tat-SL8-specific CD8+ T-cell responses by tetramer staining in lymphocytes derived from rectal biopsies (RB) that were collected at several time points during immunization and before SIV challenge. Although Fig. 2 and Fig. S1 A and B show average levels for the SIV-specific CD8+ T-cell responses, we observed substantial variation in the magnitude of these responses among individual animals (Fig. S1 C–F). Of note, the trend toward higher responses following the DNA/EP-VV-AdC6 and DNA/EP-VV-AdC7 regimens and lower responses following three AdHu5 immunizations that was observed in blood also was present in RB, although there were some differences in the ranking of responses at various time points.
Fig. 2.
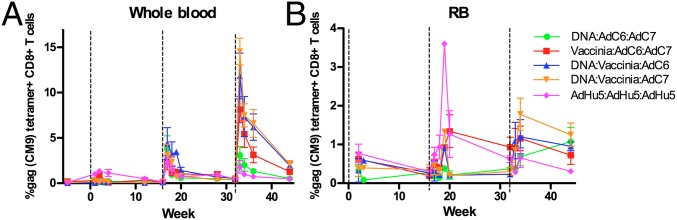
Vaccination regimens result in robust SIV-specific immune responses. Average fraction of CD8+ T cells binding Gag-CM9 Mamu-A*01 tetramers at various time points postimmunization in peripheral blood (A) and rectal mucosa (B). Black dashed lines at 0, 16, and 32 wk indicate immunization time points. Error bars indicate the SEM for each vaccination group. The color scheme is as follows: DNA/EP-AdC6-AdC7, green; VV-AdC6-AdC7, red; DNA/EP-VV-AdC6, blue; DNA/EP-VV-AdC7, orange; and AdHu5-AdHu5-AdHu5, pink.
To characterize the development of vaccine-induced SIV-specific T-cell responses functionally, we next stimulated peripheral blood mononuclear cells (PBMCs) from each vaccinated RM with overlapping peptide pools of 15-mers from SIVmac239 Gag and assessed the production of IFN-γ, TNF-α, and IL-2 and the expression of CD107a using multiparametric flow cytometry. As shown in Fig. S2 A and B, all tested vaccination regimens induced detectable Gag-specific CD8+ T-cell responses ranging from 0.0001 to more than 5% of CD8+ T cells, with a substantial representation of cells showing three or four immunological functions both at 1 and 12 wk after the last immunization. However, the level of polyfunctionality was not maintained at 12 wk following the final immunization. Although we found statistical differences among the groups in the strength of the immune response at these two time points, we did not find that these differences predicted protection from infection. The vaccines also induced Gag-specific CD4+ T-cell responses that ranged from 0.0001–1.3% of CD4+ T cells (Fig. S2 C and D). Collectively, these data indicate that expression of SIV antigens from the vectors used (AdHu5, AdC6, AdC7, VV, and DNA/EP) effectively induced robust levels of functional SIV-specific T-cell responses.
Effects of Immunizations on the Levels of Activated CD4+CCR5+ T Cells.
A potential safety risk of any candidate AIDS vaccine regimen is enhancing HIV acquisition by increasing the number of activated CD4+CCR5+ T cells in mucosal tissues that serve as primary targets for the virus. To examine this possibility in our five groups of vaccinated RMs, we measured the total levels of CD4+CCR5+ T cells as well as those of activated and proliferating [i.e., CD4+HLA-DR (DR)+ and CD4+Ki-67+] T cells in the blood and RBs at various time points during the immunization procedure and before SIV challenge. As shown in Fig. 3, there was a variable increase in the level of circulating CD4+CCR5+, CD4+CCR5+Ki-67+, and CD4+CCR5+DR+ T cells that persisted until the last time point before challenge in some of the immunized groups. This trend was more evident for the groups that received VV as part of the immunization regimen (CD4+CCR5+, CD4+CCR5+DR+, and CD4+CCR5+Ki-67+ T cells) and for the AdHu5/AdHu5/AdHu5 regimen (CD4+CCR5+Ki-67+ T cells). We also measured the levels of these CD4+ T-cell subsets in the rectal mucosa. However, the original study design did not include the collection of RB samples before the first immunization. Therefore, we were unable to assess directly the impact of the vaccine regimens used on mucosal CD4+ T-cell activation. As shown in Fig. S3, this analysis did not reveal any major increase in the level of potential SIV target cells in the rectal mucosa between the first time point (i.e., after the first immunization) and the latest time point before challenge. Of note, the highest level of CD4+CCR5+ T cells in the rectal mucosa was observed in RMs receiving three AdHu5-SIV immunizations (Fig. S3A). There were no significant differences among the immunization groups in the levels of CD4+CCR5+ or activated or proliferating CD4+CCR5+ target T cells in the rectal mucosa of the immunized animals 12 wk following the final immunization. (Fig. S3). Differences observed among individual animals may reflect natural animal-to-animal variability in addition to any effect of the vaccination regimens used. Taken together, these data indicate that the immunization regimens used induced an increase of the levels of CD4+CCR5+ target T cells in the peripheral blood of most immunized animals.
Fig. 3.
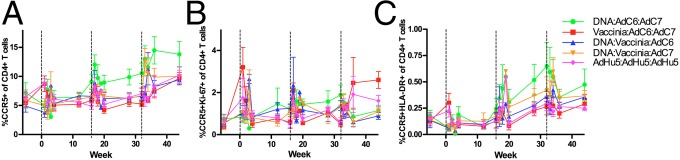
Level of activated and proliferating CCR5+CD4+ T cells after immunization in whole blood. Average levels of CCR5+ (A), Ki-67+CCR5+ (B), and HLA-DR+CCR5+ (C) T cells measured as the percentage of total CD4+ T cells in peripheral blood in vaccinated RMs. Black dashed lines at 0, 16, and 32 wk indicate immunization time points. Error bars represent SEM. The color scheme is as follows: DNA/EP-AdC6-AdC7, green; VV-AdC6-AdC7, red; DNA/EP-VV-AdC6, blue; DNA/EP-VV-AdC7, orange; and AdHu5-AdHu5-AdHu5, pink.
Acquisition of SIV After Repeated Low-Dose Intrarectal Challenge.
In the next phase of this study all five groups of six RMs as well as a group of six unvaccinated controls were challenged weekly up to 15 times with a low dose (300 TCID50) of SIVmac239 given intrarectally. The number of challenges required to acquire the infection (defined as viral load >1,000 copies/mL of plasma) is shown in Fig. 4 A and B. When the six groups of animals were analyzed and presented separately, a trend was observed toward fewer SIV acquisition events in the DNA/EP/VV/AdC7 or DNA/EP/AdC6/AdC7 groups and higher acquisition in the DNA/EP/VV/AdC6 group (Fig. 4A). However, when the entire cohort of vaccinated RMs is examined and compared with unvaccinated controls, no significant protection from mucosal acquisition of SIV was observed (Fig. 4B), with 18 of 29 vaccinated RMs (62%) and four of six control animals (66%) infected after 15 challenges. Of note, one of the RMs from the DNA/EP-VV-AdC7 group showed persistently undetectable viremia after the first positive result (i.e., >1,000 SIV-RNA copies/mL of plasma after challenge no. 8); we excluded this animal from further analysis because of its uncertain infection status. We concluded, based on these results, that none of the SIV immunization regimens used results in significant protection from SIVmac239 transmission, and these findings are consistent with previous preclinical studies of CTL-based immunogens that do not contain an Env antigen (20, 21).
Fig. 4.

Viral acquisition and replication after low-dose rectal SIVmac239 challenge. (A) Number of low-dose challenges required for acquisition of SIV infection in all vaccinated RMs and in unvaccinated controls. (B) All immunized animals combined compared with the control animals. (C) SIV plasma viral load (expressed as copies per milliliter of plasma) was measured at days 7, 21, and 42 after SIV infection by real-time PCR. (D) The average viral loads for all infected, immunized RMs combined compared with infected control animals. Error bars represent SEM. The color scheme is as follows: DNA/EP-AdC6-AdC7, green; VV-AdC6-AdC7, red; DNA/EP-VV-AdC6, blue; DNA/EP-VV-AdC7, orange; AdHu5-AdHu5-AdHu5, pink; controls, black; all vaccinated RMs combined, gray. *P < 0.05.
SIVGag/Tat-Vaccinated RMs Show Lower Set-Point Virus Replication After SIV Infection.
Previous studies have shown that the administration of immunogens expressing SIVGag that are designed to elicit a strong antiviral CTL response results in lower levels of virus replication in SIV-infected RMs than in control animals (21–26). To determine whether the immunization regimens used in the current study exhibited the same effect, we monitored the level of SIV viremia at multiple time points after virus acquisition and up to day 42 postinfection, when all animals were killed. As shown in Fig. 4C, in which the six groups of RMs are presented separately, all groups of animals experiencing SIV infection showed a fairly typical trend of virus replication characterized by viremia at day 7 in the range of 104–107 copies/mL of plasma and an early set-point viremia in the range of 102–104 copies/mL of plasma. Importantly, the levels of both early and set-point viremia were similar in all five groups of immunized RMs. Of note, when the entire cohort of immunized RMs is compared with unvaccinated controls, we observed a set-point viremia at day 42 that was ∼1.6 log lower in immunized animals than in controls, and this result is statistically significant (P = 0.0241; t test ) (Fig. 4D). Collectively, these results indicate that CTL-based SIV vaccine regimens that cannot protect against SIV acquisition resulted in a significantly improved control of virus replication after the acute phase of infection (i.e., day 42 postinfection).
SIV Infection Is Associated With Higher Levels of SIVGag-Specific CD8+ T-Cell Responses in Blood and Higher Levels of CD4+CCR5+DR+ T Cells in the Rectal Mucosa Before Challenge.
We next sought to identify correlates of protection from SIV infection in our cohort of immunized RMs during the challenge phase of our experiment. We first investigated the relationship between the vaccine-induced CD8+ T-cell response and the protection (or lack thereof) against SIV acquisition. To this end, we divided the RMs into SIV-infected and uninfected groups at the end of the challenge phase and compared the two groups for the level of SIV-specific CD8+ T-cell responses measured before challenge by either tetramer staining or cytokine production and CD107a up-regulation upon SIVGag peptide stimulation. As shown in Fig. 5 A and B, the levels of total SIVGag-specific CD8+ T cells and the percentage of CD107a+CD8+ T cells (both measured as response to peptide stimulation) were significantly higher in the blood of infected RMs than in animals that remained uninfected throughout the challenge phase (P = 0.0409 and P = 0.0065, respectively). No differences in the level of tetramer-positive CD8+ T cells in the blood or rectal mucosa or in the level of SIV-specific CD4+ T cells were observed in infected and uninfected RMs (Fig. S4 A–E). Next, in our cohort of vaccinated RMs, we examined the relationship between the blood and mucosal levels of CD4+ T cells that may serve as targets for SIV infection (measured as the percentage of total CD4+CCR5+ T cells as well as CD4+CCR5+ T cells expressing the activation/proliferation markers HLA-DR and Ki-67) and the risk of SIV acquisition. No significant differences between SIV-infected and uninfected animals were observed with respect to CD4+CCR5+ T cells and CD4+CCR5+Ki-67+ T cells before challenge (Fig. S4F and Fig. 5C). Interestingly, we observed that SIV-infected RMs had significantly higher levels of CD4+CCR5+DR+ T cells in mucosal tissues than animals that remained uninfected (P = 0.0261) (Fig. 5D). Collectively, these results show that SIV infection in our cohort of SIVGag/Tat-vaccinated RMs is associated with higher levels of SIV-specific CD8+ T-cell responses in blood and increased percentages of CD4+CCR5+DR+ T cells in the rectal mucosa before challenge.
Fig. 5.

SIV infection is associated with higher levels of SIVGag-specific CD8+ T-cell responses in blood and CCR5+DR+CD4+ in rectal mucosa, and a low day 7 viral load is correlated with activated CCR5+CD4+ T cells in rectal mucosa. (A and B) Comparison of the percent of SIVGag peptide-responsive (A) or CD107a-expressing (B) CD8+ T cells in PBMCs before challenge in immunized RMs that remained uninfected (circles) or that acquired SIV infection (squares). Responsive cells are considered to be cytokine producing and/or CD107a expressing after background subtraction. (C and D) Comparison of the percent of CCR5+Ki-67+ (C) and CCR5+HLA-DR+ (D) CD4+ T cells in rectal mucosa before challenge in immunized RMs that remained uninfected (circles) and those that acquired SIV infection (squares). (E and F) The percentage of total CCR5+Ki-67+ (E) and effector memory (EM) CCR5+Ki-67+ (F) CD4+ T cells in rectal mucosa before challenge is plotted against the day 7 viral load in immunized RMs experiencing infection. The Mann–Whitney u test was used to determine differences between uninfected and infected groups. *P < 0.05; **P < 0.01 (Spearman’s correlation).
Early Viremia Correlates Directly with Higher Levels of CD4+CCR5+Ki-67+ T Cells in Rectal Mucosa Before Challenge.
We next sought to identify correlates of protection from virus replication in the RMs experiencing SIV infection during the challenge phase of our experiment. We first examined the relationship between the vaccine-induced SIV-specific CD8+ T-cell response before challenge and the level of viremia early in infection on days 7, 21, and 42. We found no significant correlation between viremia and level of SIV-specific CD8+ T cells measured either by tetramer staining or after peptide stimulation (Fig. S5 A–I). Similarly, we found no correlation between viremia and the level of vaccine-induced SIV-specific CD4+ T cells (Fig. S5 J–L). Next, in our cohort of vaccinated RMs, we examined, the relationship between day 7 plasma viremia and the blood or mucosal levels of CD4+ T cells that may serve as targets for SIV infection (i.e., total CD4+CCR5+ T cells, CD4+CCR5+DR+ T cells, and CD4+CCR5+Ki-67+ T cells). Interestingly, we found a significant direct correlation between the level of CD4+CCR5+Ki-67+ T cells in the rectal mucosa before challenge and day 7 plasma viremia (P = 0.0344, r = 0.5005) (Fig. 5E). This correlation was even stronger when CD4+CCR5+Ki-67+ effector-memory T cells were analyzed (P = 0.0196, r = 0.5439) (Fig. 5F). No significant correlation was found between day 7 viremia and the levels of CD4+CCR5+ and CD4+CCR5+DR+ T cells before challenge in either blood (Fig. S6 A and B) or rectal mucosa (Fig. S7 A and B). Nor was there a significant correlation between CD4+CCR5+Ki-67+ T cells in blood before challenge and day 7 viremia (Fig. S6C). We next examined whether the levels of activated and/or CCR5+CD4+ T cells in the RBs correlated with virus replication at later time points after infection (i.e., days 21 and 42 after infection). As shown in Fig. S7, we found a significant direct correlation only between virus replication at day 42 and the percentage of CD4+CCR5+ T cells in the RBs (Fig. S7F). Collectively, these data indicate that the level of target CD4+ T cells (measured as CD4+CCR5+Ki-67+ T cells) in the rectal mucosa before challenge is a correlate of higher day 7 viremia after SIV infection.
Discussion
Despite an enormous effort by the scientific community, a safe and effective AIDS vaccine is not yet available. In absence of immunogens that can induce the production of broadly neutralizing antibodies against HIV reliably, the focus of the AIDS vaccine effort has been on generating vectors expressing HIV or SIV antigens that induce broad and potent virus-specific CD8+ T-cell–mediated CTL responses (“CTL-based vaccines”). Vectors that have been investigated as CTL-based AIDS vaccines in both preclinical and clinical studies include recombinant DNA, various adenovirus types (including AdHu5, AdHu26, AdHu35, AdHu48, and chimpanzee adenoviruses), poxviruses [including Modified vaccinia virus Ankara, canarypox (ALVAC), New York vaccinia virus, and others], CMV, and several others.
In this study we used the well-established preclinical model of SIV immunization and challenges in RMs to identify immunological correlates of protection from virus acquisition and/or virus replication after immunization with five different regimens, including some of the leading vector platforms that currently are being explored in preclinical and clinical studies (AdHu5, AdC6 and AdC7, DNA/EP, and VV). We focused on various aspects of the vector-induced SIV-specific CD8+ T-cell response as potential immunological correlates of protection, and on the levels of target cells (CD4+CCR5+ T cells, in particular those expressing the activation/proliferation markers HLA-DR and Ki-67) in blood and the rectal mucosa as potential correlates of increased risk of virus acquisition. We wish to emphasize that the current study was not designed or powered to compare and rank the immunogenicity or efficacy of these vectors, thus establishing a hierarchy of efficacy, but rather aimed at defining, across all groups of treated RMs, the main correlates of protection or, conversely, of enhanced acquisition.
The main results of this study are that (i) all immunization regimens induced a robust SIV-specific CD8+ T-cell response in blood and rectal mucosa and an increase in the levels of circulating and mucosal target cells; (ii) no significant differences were observed among immunization groups with respect to either virus acquisition or postinfection viral load; (iii) as a whole, immunized animals show no protection from virus infection after challenge but do show significantly lower levels of viral load at day 42 postinfection as compared with control animals; (iv) SIV infection is associated with higher percentages of peptide-responsive, Gag-specific CD8+ T cells in the PBMCs before challenge; (v) the prechallenge levels of CD4+ target T cells in the rectal mucosa correlate with increased risk of SIV acquisition (for CD4+CCR5+DR+ T cells) and higher early virus replication (for CD4+CCR5+Ki-67+ T cells).
A key aspect of this study is the lack of an Env immunogen in the setting of a repeated low-dose intrarectal challenge with SIVmac239. As such, the study was designed to determine the effect of the vaccine-induced cellular immune responses on SIV acquisition (and SIV viremia in the event of infection) in the absence of vaccine-induced Env-specific antibodies. The rationale for this approach resides in the strong cellular immunogenicity of the vectors used (14, 27) and the demonstrations that cellular immune responses can prevent/abort SIVmac infection in the setting of a CMV-based vector platform (28). In this context, it is possible that the significant correlation between risk of infection and activated CD4+ T cells in the rectal mucosa may not be present if the animals are vaccinated with Env-containing immunogens. In terms of protection from virus acquisition, the current set of results also are consistent with our previous work on the effect of AdC6 and AdC7 vectors expressing SIVGag/Tat showing no protection from SIVmac acquisition (24). Interestingly, studies of vector regimens including DNA and AdHu5 expressing an Env immunogen showed protection from virus acquisition when the animals were challenged with SIVsmmE660 but not when the more stringent SIVmac239 was used (29).
In this study, we first attempted to determine whether any specific aspects of the SIV-specific CD8+ T-cell response induced by the vectors used was associated with protection from SIV acquisition or from SIV viremia after infection. This extensive analysis did not lead us to identify any clear correlate of CD8+ T-cell–mediated protection. Instead, we identified the level of circulating SIV peptide-responsive CD8+ T cells (and especially those expressing CD107a on the surface) before challenge as a correlate of the risk of virus acquisition. A possible interpretation of this result is that, in this experimental setting, the level of SIV-specific CD8+ T cells is a marker of vector-induced immune activation and/or inflammation at the mucosal level. This latter event can favor virus acquisition by increasing the recruitment of target CD4+ T cells and by inducing other changes in the mucosal microenvironment such as damage to the integrity of the epithelial barrier. In this context, the observed correlation between the levels of activated and proliferating CD4+CCR5+ cells in the rectal mucosa before challenge and SIV acquisition and early viremia, respectively, further emphasize the role of mucosal microenvironmental factors in setting the risk of HIV acquisition and/or early disease progression.
When these findings are examined in the context of the clinical development and testing of candidate HIV/AIDS vaccines, these data underscore the complexity of the potential immunological effects elicited by the vectors used and the intrinsic difficulty in predicting the net results of these various effects in terms of the risk of HIV transmission. The possibility that certain immunization regimens designed to protect against HIV infection and AIDS result in increased risk of virus transmission is not just a theoretical concern, because three recent large-scale clinical trials of candidate AIDS vaccines (the AdHu5-based Step and Phambili trials and the DNA/AdHu5-based HVTN-505 trial) have shown a trend toward higher infection rates in vaccinated individuals than in placebo recipients. Although the exact mechanisms underlying these observations remain poorly defined, none of the data presented so far have ruled out the simple and rather parsimonious hypothesis that these immunization regimens resulted in an increased level of target CD4+CCR5+ T cells in mucosal tissues that are involved in HIV transmission. When taken together with the results of the RV-144 Thai trial, in which an ALVAC- and AIDSVAX-based immunized regimen induced modest but significant protection against HIV acquisition in a large cohort of low-risk individuals (30), our current results indicate that a persistent but balanced antiviral immune response—in which relatively low levels of activated CD4+CCR5+ T cells are present in mucosal tissues—may be crucial to establish a microenvironment that protects against HIV transmission.
In summary, the results of the current preclinical study suggest that the level of target CD4+CCR5+ T cells in mucosal tissues is a key factor favoring virus acquisition in SIV-immunized and -challenged RMs. This potential effect of candidate AIDS vaccines should be evaluated carefully when advancing this type of products to clinical trials in humans.
Materials and Methods
Animals.
Thirty-six healthy, SIV-uninfected, Mamu-A*01+ Indian RMs were used in this study. All animals were housed at the Yerkes National Primate Research Center and were maintained in accordance with National Institutes of Health guidelines (31). These studies were approved by the Emory University Institutional Animal Care and Use Committee.
Vectors.
Ad vectors were derived from the chimpanzee serotypes AdC6 or AdC7. The E1- and E3-deleted Ad vectors expressed a Gag-Tat fusion of SIVmac239. Vectors were generated, rescued, expanded, purified, titrated, and quality controlled as described (32). The DNA vaccines expressing SIVmac239 Gag and Tat were prepared as previously described (33). Recombinant vaccinia viruses (rVV) based on the Copenhagen strain were generated by homologous recombination (SI Materials and Methods).
Vaccination Protocol.
The heterologous prime-boost regimen used in this study consisted of three immunizations performed at day 0 (prime), 16 wk (boost), and 32 wk (boost) as detailed in Fig. 1. Five immunization groups contained six animals each: (i) DNA/EP-AdC6-AdC7; (ii) VV-AdC6-AdC7; (iii) DNA/-EP-VV-AdC6; (iv) DNA/EP-VV-AdC7; and (v) AdHu5-AdHu5-AdHu5. All vectors used expressed codon optimized sequences of SIVmac239 Gag-Tat. Immunizations comprised of the AdHu5, AdC6, or AdC7 vector expressing SIVGag/Tat were injected intramuscularly (i.m.) at the dose of 1011 virus particles per RM. VV expressing SIVGag/Tat 107 pfu was injected s.c. Animals were vaccinated with 0.5 mg of each plasmid mixed together and formulated in water, delivered i.m. with in vivo electroporation. DNA was delivered to a single site in the quadriceps followed by in vivo electroporation with the adaptive current CELLECTRA device (Inovio Pharmaceuticals) with three pulses at 0.5-A constant current, a 52-ms pulse length, and 1-s rest between pulses (34). Six additional unvaccinated animals were used as controls. Further details of this protocol and subsequent samplings can be found in SI Materials and Methods.
Viral Challenge.
Three to five months after the final immunization, all vaccinated RMs and the control animals were challenged intrarectally every week with a repeated low dose (300 TCID50) of SIVmac239 that was provided by Koen Van Rampay at the California National Primate Research Center, Davis, CA. Animals were considered to be infected if they had 1,000 copies/mL 7 d after the 15th challenge. One animal from the DNA/EP-VV-AdC7 group was thought to be infected at challenge no. 8 but did not show any viral load greater than 1,000 copies/mL at any later time point. This animal was removed from the analysis because its infectious status remained uncertain.
Tissue Collection and Processing.
PBMCs were isolated by gradient centrifugation. Procedures for lymph node biopsy, RBs, and isolation of lymphocytes from the obtained samples were performed as previously described (24, 35).
Immunophenotyping and Flow Cytometry.
Multicolor flow cytometric analysis was performed on mononuclear cells isolated from blood, lymph nodes, and mucosal tissues (RB) according to standard procedures using human monoclonal antibodies that were found to cross-react with RMs. Further information detailing antibodies is given in SI Materials and Methods. Flow cytometric acquisition and analysis of samples was performed on at least 100,000 lymphocytes on an LSRII flow cytometer driven by the DiVa software package (BD Biosciences). Analysis of the acquired data was performed using FlowJo software (Tree Star, Inc.).
Tetramer Staining.
MHC class I tetramers were prepared and conjugated to Streptavidin APC fluorophore (Molecular Probes) as previously described (24, 36). The level of SIV-specific CD8+ T cells was assessed using soluble tetrameric Mamu-A*01 MHC class I tetramers specific for SIVmac239 immunodominant epitopes Gag181–189 CM9 (CTPYDINQM) and Tat28–35 SL8 (STPESANL). Lymphocytes isolated from blood and tissues were incubated with conjugated tetramer, along with surface antibody conjugates, and analyzed for tetramer and surface-marker expression using an LSRII Flow Cytometer (BD Biosciences) equipped with FACS DiVa software.
Multifunctional Assessment of SIV-Specific T-Cell Responses.
The function of SIV-specific CD8+ T cells was assessed by flow cytometry after stimulation with peptide pools of 15-mers (overlapping by 11 amino acids) spanning the SIVmac239 Gag proteins as described in refs. 3 and 24. Peptides were prepared from peptide stocks obtained from the National Institutes of Health AIDS Research and Reference Reagent Program (Bethesda, MD), reconstituted in DMSO, and pooled. All peptides were used at a final concentration of 2 µg/mL of each peptide. Further information detailing antibodies is given in SI Materials and Methods. The unstimulated control was used as a background, and the percentage of responding cells is depicted after background subtraction.
Plasma Viral Load Determination.
The quantitative real-time RT-PCR assay to determine SIVmac239 viral load was performed as previously described (24, 37). The sensitivity of the assay is 50 copies/mL of plasma.
Statistical Analysis.
Measurements among all treatment groups were compared using one-way ANOVA with Bonferroni multiple comparisons adjustments. An exact χ2 was used when comparing differences in acquisition of infection among groups. Mann–Whitney u tests were performed to compare differences between uninfected versus infected groups or immunized groups versus controls. All tests were conducted as two-tailed tests with a type one-error rate of 5%. We used both k-sample Kruskal–Wallis tests and zero-inflated negative binomial models to compare the polyfunctionality among vaccination groups. Statistical analyses were conducted using Prism Software (GraphPad) and the open-source software R (www.cran.r-project.org).
Supplementary Material
Acknowledgments
We thank the Emory Center for AIDS Research Virology Core for technical support. This work was supported by National Institutes of Health (NIH) Grant HIVRAD P01 A080082 (to H.C.J.E. and G.S.) and by NIH/National Center for Research Resources Grant P51RR000165 and currently is supported by the Office of Research Infrastructure Programs Grant OD P51OD011132 to the Yerkes National Primate Research Center. N.Y.S., J. Yan, and M.P.M. were supported in part by NIH/National Institute of Allergy and Infection Diseases/Division of AIDS Grant HHSN272200800063C for HIV vaccine development. D.B.W. has grant funding, participates in industry collaborations, has received speaking honoraria, and fees for consulting.
Footnotes
Conflict of interest statement: J. Yan, M.P.M., and N.Y.S. are employees of Inovio Pharmaceuticals. D.B.W. has received speaking honoraria and direct payments or stock or stock options as fees for consulting (including service on scientific review committees and advisory boards) with Pfizer, Medimmune, Inovio, Merck, VGXI, OncoSec, Roche, Ferring, Novartis, Aldevron, and possibly others. Licensing of technology from his laboratory has created over 150 jobs in the biotech/pharma sector.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1407466112/-/DCSupplemental.
References
- 1.Koup RA, et al. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J Virol. 1994;68(7):4650–4655. doi: 10.1128/jvi.68.7.4650-4655.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Letvin NL. Correlates of immune protection and the development of a human immunodeficiency virus vaccine. Immunity. 2007;27(3):366–369. doi: 10.1016/j.immuni.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 3.Betts MR, et al. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood. 2006;107(12):4781–4789. doi: 10.1182/blood-2005-12-4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borrow P, Lewicki H, Hahn BH, Shaw GM, Oldstone MB. Virus-specific CD8+ cytotoxic T-lymphocyte activity associated with control of viremia in primary human immunodeficiency virus type 1 infection. J Virol. 1994;68(9):6103–6110. doi: 10.1128/jvi.68.9.6103-6110.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goulder PJ, Watkins DI. Impact of MHC class I diversity on immune control of immunodeficiency virus replication. Nat Rev Immunol. 2008;8(8):619–630. doi: 10.1038/nri2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schmitz JE, et al. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science. 1999;283(5403):857–860. doi: 10.1126/science.283.5403.857. [DOI] [PubMed] [Google Scholar]
- 7.Matano T, et al. Administration of an anti-CD8 monoclonal antibody interferes with the clearance of chimeric simian/human immunodeficiency virus during primary infections of rhesus macaques. J Virol. 1998;72(1):164–169. doi: 10.1128/jvi.72.1.164-169.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jin X, et al. Dramatic rise in plasma viremia after CD8(+) T cell depletion in simian immunodeficiency virus-infected macaques. J Exp Med. 1999;189(6):991–998. doi: 10.1084/jem.189.6.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klatt NR, et al. CD8+ lymphocytes control viral replication in SIVmac239-infected rhesus macaques without decreasing the lifespan of productively infected cells. PLoS Pathog. 2010;6(1):e1000747. doi: 10.1371/journal.ppat.1000747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burton DR, et al. A Blueprint for HIV Vaccine Discovery. Cell Host Microbe. 2012;12(4):396–407. doi: 10.1016/j.chom.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garber DA, Silvestri G, Feinberg MB. Prospects for an AIDS vaccine: Three big questions, no easy answers. Lancet Infect Dis. 2004;4(7):397–413. doi: 10.1016/S1473-3099(04)01056-4. [DOI] [PubMed] [Google Scholar]
- 12.Qureshi H, et al. Low-dose penile SIVmac251 exposure of rhesus macaques infected with adenovirus type 5 (Ad5) and then immunized with a replication-defective Ad5-based SIV gag/pol/nef vaccine recapitulates the results of the phase IIb step trial of a similar HIV-1 vaccine. J Virol. 2012;86(4):2239–2250. doi: 10.1128/JVI.06175-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buchbinder SP, et al. Step Study Protocol Team Efficacy assessment of a cell-mediated immunity HIV-1 vaccine (the Step Study): A double-blind, randomised, placebo-controlled, test-of-concept trial. Lancet. 2008;372(9653):1881–1893. doi: 10.1016/S0140-6736(08)61591-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hammer SM, et al. Efficacy trial of a DNA/rAd5 HIV-1 preventive vaccine. N Engl J Med. 2013;369(22):2083–2092. doi: 10.1056/NEJMoa1310566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gray GE, et al. HVTN 503/Phambili study team Safety and efficacy of the HVTN 503/Phambili study of a clade-B-based HIV-1 vaccine in South Africa: A double-blind, randomised, placebo-controlled test-of-concept phase 2b study. Lancet Infect Dis. 2011;11(7):507–515. doi: 10.1016/S1473-3099(11)70098-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Picker LJ, Hansen SG, Lifson JD. New paradigms for HIV/AIDS vaccine development. Annu Rev Med. 2012;63:95–111. doi: 10.1146/annurev-med-042010-085643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haynes BF, McElrath MJ. Progress in HIV-1 vaccine development. Curr Opin HIV AIDS. 2013;8(4):326–332. doi: 10.1097/COH.0b013e328361d178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fukazawa Y, et al. Lymph node T cell responses predict the efficacy of live attenuated SIV vaccines. Nat Med. 2012;18(11):1673–1681. doi: 10.1038/nm.2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hansen SG, et al. Effector memory T cell responses are associated with protection of rhesus monkeys from mucosal simian immunodeficiency virus challenge. Nat Med. 2009;15(3):293–299. doi: 10.1038/nm.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vogel TU, et al. Multispecific vaccine-induced mucosal cytotoxic T lymphocytes reduce acute-phase viral replication but fail in long-term control of simian immunodeficiency virus SIVmac239. J Virol. 2003;77(24):13348–13360. doi: 10.1128/JVI.77.24.13348-13360.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Engram JC, et al. Vaccine-induced, simian immunodeficiency virus-specific CD8+ T cells reduce virus replication but do not protect from simian immunodeficiency virus disease progression. J Immunol. 2009;183(1):706–717. doi: 10.4049/jimmunol.0803746. [DOI] [PubMed] [Google Scholar]
- 22.Barouch DH, et al. Protective efficacy of a single immunization of a chimeric adenovirus vector-based vaccine against simian immunodeficiency virus challenge in rhesus monkeys. J Virol. 2009;83(18):9584–9590. doi: 10.1128/JVI.00821-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Casimiro DR, et al. Efficacy of multivalent adenovirus-based vaccine against simian immunodeficiency virus challenge. J Virol. 2010;84(6):2996–3003. doi: 10.1128/JVI.00969-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cervasi B, et al. Immunological and virological analyses of rhesus macaques immunized with chimpanzee adenoviruses expressing SIVgag/tat and challenged intra-rectally with repeated low-dose of SIVmac. J Virol. 2013;87(17):9420–9430. doi: 10.1128/JVI.01456-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Casimiro DR, et al. Attenuation of simian immunodeficiency virus SIVmac239 infection by prophylactic immunization with dna and recombinant adenoviral vaccine vectors expressing Gag. J Virol. 2005;79(24):15547–15555. doi: 10.1128/JVI.79.24.15547-15555.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilson NA, et al. Vaccine-induced cellular immune responses reduce plasma viral concentrations after repeated low-dose challenge with pathogenic simian immunodeficiency virus SIVmac239. J Virol. 2006;80(12):5875–5885. doi: 10.1128/JVI.00171-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tatsis N, et al. Chimpanzee-origin adenovirus vectors as vaccine carriers. Gene Ther. 2006;13(5):421–429. doi: 10.1038/sj.gt.3302675. [DOI] [PubMed] [Google Scholar]
- 28.Hansen SG, et al. Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature. 2011;473(7348):523–527. doi: 10.1038/nature10003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Letvin NL, et al. Immune and Genetic Correlates of Vaccine Protection Against Mucosal Infection by SIV in Monkeys. Sci Transl Med. 2011;3(81):81ra36. doi: 10.1126/scitranslmed.3002351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rerks-Ngarm S, et al. MOPH-TAVEG Investigators Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N Engl J Med. 2009;361(23):2209–2220. doi: 10.1056/NEJMoa0908492. [DOI] [PubMed] [Google Scholar]
- 31.Committee on Care and Use of Laboratory Animals 1996. Guide for the Care and Use of Laboratory Animals (Natl Inst Health, Bethesda), DHHS Publ No (NIH) 85-23.
- 32.Zhou D, et al. An efficient method of directly cloning chimpanzee adenovirus as a vaccine vector. Nat Protoc. 2010;5(11):1775–1785. doi: 10.1038/nprot.2010.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yan J, et al. Novel SIVmac DNA vaccines encoding Env, Pol and Gag consensus proteins elicit strong cellular immune responses in cynomolgus macaques. Vaccine. 2009;27(25-26):3260–3266. doi: 10.1016/j.vaccine.2009.01.065. [DOI] [PubMed] [Google Scholar]
- 34.Laddy DJ, et al. Heterosubtypic protection against pathogenic human and avian influenza viruses via in vivo electroporation of synthetic consensus DNA antigens. PLoS ONE. 2008;3(6):e2517. doi: 10.1371/journal.pone.0002517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gordon SN, et al. Severe depletion of mucosal CD4+ T cells in AIDS-free simian immunodeficiency virus-infected sooty mangabeys. J Immunol. 2007;179(5):3026–3034. doi: 10.4049/jimmunol.179.5.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Altman JD, et al. Phenotypic analysis of antigen-specific T lymphocytes. Science. 1996;274(5284):94–96. doi: 10.1126/science.274.5284.94. [DOI] [PubMed] [Google Scholar]
- 37.Silvestri G, et al. Nonpathogenic SIV infection of sooty mangabeys is characterized by limited bystander immunopathology despite chronic high-level viremia. Immunity. 2003;18(3):441–452. doi: 10.1016/s1074-7613(03)00060-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.