

Abstract
Splicing of pre-mRNAs is a crucial step in the gene expression pathway. Disruption of splicing has been linked to the pathogenesis of several human diseases and is particularly widespread in cancer. Recently, a number of mutations affecting genes of the core spliceosome machinery have been identified in haematological malignancies, yet the effect of such mutations on RNA splicing is unclear. A better understanding of how mis-splicing contributes to malignancies may provide diagnostic or prognostic information and new drug targets for therapeutic approaches.
Keywords: splicing, cancer, myelodysplastic syndromes, spliceosomal mutations
The splicing reaction
Eukaryotic genes are transcribed as premature messenger RNA (pre-mRNA), consisting of exons interrupted by non-coding introns. Removal of introns and exon joining is a fundamental step of gene expression that occurs primarily co-transcriptionally. Splicing is carried out in the nucleus by the spliceosome, a macromolecular machine consisting of five small nuclear ribonucleoproteins (U1, U2, U4, U5 and U6 snRNAs) and > 150 proteins [1,2]. Four major cis-acting elements contribute to exon definition by the spliceosome (Figure 1). At the 5′-end of introns, a nine-nucleotide sequence with a highly conserved GU dinucleotide at the exon–intron junction marks the 5′-splice site (5′-ss). The 3′-end of the intron contains three consensus sequence motifs: the branch point (BP); the polypyrimidine tract (PPT); and the highly conserved AG dinucleotide at the intron–exon junction. A variety of additional cis-acting regulatory elements within the exon and flanking introns contribute to exon definition. These sequences are bound by non-snRNP splicing factors that enhance or inhibit the assembly of the spliceosome on the adjacent splice site [2].
Figure 1.
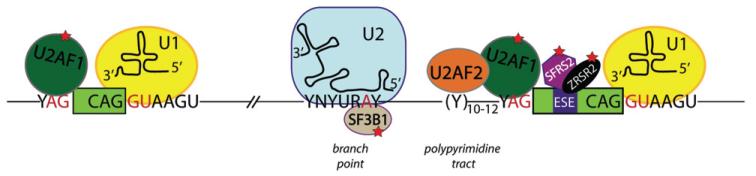
Spliceosome assembly on pre-mRNA and pathogenic mutations. Schematic representation of the consensus sequences on pre-mRNA at the 5′-ss, branch point sequence, polypyrimidine tract and 3′-ss: N, any nucleotide; R, purine (A or G); Y, pyrimidine (C or U). Exons are represented as solid boxes, while introns are depicted as lines. Spliceosome components involved in initial recognition of intron boundaries are shown bound to their target sequence on the pre-mRNA. Red stars mark splicing factors mutated in haematological malignancies
The splicing reaction initiates with U1 snRNA binding to the 5′-splice site. At the 3′-splice site, splicing factor 1 (SF1) binds to the branch point, while the serine/arginine (SR) proteins SRSF2 and ZRSR2 recognize and bind the exon-splicing enhancer (ESE) sequences within the exon. U2AF1 and U2AF2 are recruited to bind the AG dinucleotide and polypyrimidine tract, respectively. Spliceosome assembly continues with replacement of SF1 by U2 snRNP and associated proteins, including SF3B1 (Figure 1). Ultimately, the U4/U6–U5 tri-snRNP engages and the spliceosome becomes catalytically active, driving intron removal and exon ligation.
Abnormal splicing and disease
While exon recognition is guided primarily by the splice site sequences, the splicing process is flexible and auxiliary splicing factors can push the spliceosome to include or skip specific exons, a mechanism known as alternative splicing. More than 90% of human genes generate multiple mRNA isoforms, often encoding divergent protein variants. Thus, alternative splicing allows for a cell- or tissue-specific proteome that can change during development, differentiation and in response to physiological stimuli through expression of individual RNA-binding proteins that regulate the splicing of specific pre-mRNAs. However, with great power comes great responsibility. Mis-regulation of alternative splicing has been linked to several human diseases. For example, splicing can be disrupted by DNA mutations occurring within cis-acting sequences of pre-mRNA, such as the splice sites or the auxiliary elements leading to aberrant splicing and loss of function of the mutated allele [3,4]. Moreover, changes in the intracellular levels, localization and/or activity of trans-acting splicing factors can alter splice site recognition, leading to production of different protein isoforms. Alteration of splicing factor concentration and/or localization is particularly frequent in cancer.
Recent studies have also identified mutations affecting core spliceosome proteins as major players for the development of haematopoietic conditions, such as myelodysplastic syndromes (MDS) and chronic lymphocytic leukaemia (CLL) [5,6]. MDS are characterized by impaired haematopoiesis and often develop to acute myeloid leukaemia (AML). Whole-genome and whole-exome sequencing unveiled somatic mutations in the constitutive spliceosomal components SF3B1, U2AF1, SRSF2 and ZRSR2, involved in recognition and binding of the 3′ splice site (Figure 1). Mutational frequencies in spliceosomal genes reach 57% in MDS, suggesting high specificity of splicing pathway mutations for this disease. SF3B1 is the most frequently mutated (15–30% of MDS), increasing to 64–83% in patients with refractory anaemia with ring sideroblasts (RARS). Mutations correlate with MDS phenotypes: SF3B1 mutations occur in early-stage MDS and indicate better prognosis; conversely, U2AF1 and SFRS2 mutations are frequently detected in advanced MDS and correlate with poor prognosis and shorter survival. SF3B1 is also mutated in 10% of CLLs, as well as in other tumour types, both solid and non-solid.
To date, it is unclear whether spliceosomal gene mutations lead to gain or loss of function. Moreover, the specific mechanism(s) by which splicing factor mutations lead to disease remains to be determined. The most straightforward consequence would be failure to remove introns, most likely targeting the affected mRNA to the non-sense-mediated decay (NMD) pathway, leading to down-regulation of gene expression. Indeed, Yoshida et al [5] reported increased intron retention in HeLa cells overexpressing mutant U2AF1 and up-regulation of genes involved in the NMD pathway. NMD was counteracted by co-overexpression of wild-type U2AF1, suggesting that the mutant protein suppresses wild-type protein activity. In contrast, the SF3B1 mutation has been proposed to contribute to disease by deregulating specific target genes rather than generally impairing splicing. RNA-Seq studies of MDS patients showed aberrant splicing of genes involved in several critical cellular pathways, including cell cycle, mitochondrial function and RNA processing. Surprisingly, mutations in pre-mRNA splicing factors do not confer survival advantages. Indeed, in vitro experiments have suggested quite the opposite [5]. Overexpression of mutant U2AF1 in HeLa cells reduces cell proliferation and induces apoptosis. Similar results were obtained by knocking down ZRSR2 in HeLa cells. This contradiction remains unexplained.
Mutations affecting core spliceosomal proteins also impact alternative splicing. Auxiliary splicing factors, such as SRSF2 are known to interact with the basal spliceosome machinery, ie SF3B1 and U2AF1, and modulate alternative splicing of specific genes. As a consequence, mutations affecting these factors could cause alteration in alternative splicing of at least a subset of genes, as has been observed upon knockdown of these factors in culture [7].
(Mis)splicing as a therapeutic target
Several spliceosome inhibitors have been used to treat cancer. These modulators mostly exert their cytotoxicity by altering SF3B1 interactions with pre-mRNA, resulting in abnormal alternative splicing. Since SF3B1 mutations in haematological malignancies are thought to lead to aberrant splicing per se, the effect of these compounds is unpredictable and still needs to be tested in tumours triggered by spliceosomal mutations. Currently, it is thought that cells harbouring mutations in spliceosomal genes would be more sensitive to spliceosome inhibitors. Inhibition of cancer-specific alternative splicing events and/or the splicing factors regulating such events are promising therapeutic approaches. However, extensive clinical and molecular studies are needed to understand in depth the mechanisms and consequences of splicing (mis)regulation in tumours. This understanding will allow a strategic approach to develop therapies aimed at preventing cancer growth and spread.
Suggested reading
- 1.Hoskins AA, Moore MJ. The spliceosome: a flexible, reversible macromolecular machine. Trends Biochem Sci. 2012;37:179–188. doi: 10.1016/j.tibs.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Black DL. Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem. 2003;72:291–336. doi: 10.1146/annurev.biochem.72.121801.161720. [DOI] [PubMed] [Google Scholar]
- 3.Ward AJ, Cooper TA. The pathobiology of splicing. J Pathol. 2010;220:152–163. doi: 10.1002/path.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cartegni L, Chew SL, Krainer AR. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet. 2002;3:285–298. doi: 10.1038/nrg775. [DOI] [PubMed] [Google Scholar]
- 5.Yoshida K, Sanada M, Shiraishi Y, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–69. doi: 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
- 6.Papaemmanuil E, Cazzola M, Boultwood J, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med. 2011;365:1384–1395. doi: 10.1056/NEJMoa1103283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Corey SJ, Minden MD, Barber DL, et al. Myelodysplastic syndromes: the complexity of stem-cell diseases. Nat Rev Cancer. 2007;7:118–129. doi: 10.1038/nrc2047. [DOI] [PubMed] [Google Scholar]