

Abstract
Background and Aims
Hepatocellular cancer (HCC) remains a disease of poor prognosis highlighting the relevance of elucidating key molecular aberrations that may be targeted for novel therapies. Wnt signaling activation chiefly due to mutations in CTNNB1 have been identified in a major subset of HCC patients. While several in vitro proof-of-concept studies show the relevance of suppressing Wnt/β-catenin signaling in HCC cells or tumor xenograft models, no study has addressed the impact of β-catenin inhibition in a relevant murine HCC model driven by CTNNB1 mutations.
Methods
We studied the in vivo impact of β-catenin suppression by locked nucleic acid (LNA) antisense treatment after establishing CTNNB1 mutations-driven HCC by Diethylnitrosamine and Phenobarbital (DEN/PB) administration.
Results
The efficacy of LNA-directed against β-catenin versus scrambled sequence on Wnt signaling was demonstrated in vitro in HCC cells and in vivo in normal mice. DEN/PB model led to HCC with CTNNB1 mutations. A complete therapeutic response in the form of abrogation of HCC was observed after ten treatments of tumor-bearing mice with β-catenin LNA every 48 hours as compared to the scrambled control. A decrease in β-catenin activity, cell proliferation and increased cell death was evident after β-catenin suppression. No effect of β-catenin suppression was evident in non-CTNNB1 mutated HCC observed after DEN only administration.
Conclusions
Thus, we provide in vivo proof-of-concept that β-catenin suppression in HCC will be of significant therapeutic benefit provided the tumors display Wnt activation via mechanisms like CTNNB1 mutations.
Keywords: Hepatocellular cancer, Wnt signaling, molecular therapy, oncogene, mutations, targeted therapy
INTRODUCTION
Hepatocellular cancer (HCC) usually occurs in cirrhotic livers associated with chronic liver diseases like hepatitis of viral, alcoholic or metabolic etiology [12]. HCC is a major health burden as reflected by 748,000 new cases diagnosed in 2008 worldwide with corresponding 696,000 deaths [13, 41]. HCC is now the seventh most common cancer in the world, as well as number three in cancer-related deaths. Current effective methods of treating primary HCC are surgical including partial hepatectomy or orthotopic liver transplantation when possible and do improve 5-year disease free survival. Loco-regional therapies are mostly palliative and gradually evolving. The only FDA-approved agent for HCC treatment consists of a multi-tyrosine kinase inhibitor Sorafenib, which has shown some benefit, but does demonstrate effectiveness of targeted therapies in a heterogeneous disease like HCC [20].
Wnt/β-catenin signaling is an evolutionary conserved pathway with many important functions in hepatic development and homeostasis [25]. In the absence of Wnt, cytoplasmic β-catenin is phosphorylated at specific serine and threonine residues in exon-3 by a degradation complex composed of glycogen synthase kinase-3β (GSK-3β), adenomatous polyposis coli (APC), and Axin targeting it for ubiquitin-proteasome degradation. β-Catenin activation occurs upon binding of Wnt to receptor Frizzled (Fzd) and co-receptor low-density lipoprotein receptor related protein (LRP) 5 or 6 via inactivation of degradation complex leading to its nuclear translocation. Here, it can interact with T-cell factor/lymphoid enhancing factor (TCF/LEF) family of transcription factors, to induce target genes such as those encoding for cyclin-D1, c-myc, glutamine synthetase (GS), and others in a tissue- or stage-specific manner.
In around 10–50% of primary HCCs, β-catenin gene (CTNNB1) mutations affecting serine, threonine or adjacent sites in exon-3 and mutations in its degradation components such as AXIN1, are associated with Wnt autonomous nuclear localization and activation of β-catenin, which can have multitude of effects on HCC biology as various downstream targets are induced [7, 19, 23]. This makes β-catenin an attractive therapeutic target in a subset of HCCs. In the current study, we used modified Diethylnitrosamine (DEN) and Phenobarbital (PB) model in which 90% of HCC display activating β-catenin gene mutations [1, 22]. We administer locked nucleic acid antisense (LNA) against β-catenin to show a dramatic therapeutic benefit of β-catenin suppression in this model but not in DEN only treatment where tumors occurred without β-catenin gene mutations. Thus we show an in vivo efficacy of therapeutic inhibition of β-catenin in HCC paving the way for personalized medicine in HCC.
MATERIALS AND METHODS
Animals
All animal experiments were performed under the guidelines of the National Institutes of Health and the Institutional Animal Use and Care Committee at the University of Pittsburgh, School of Medicine.
For examining in vivo efficacy of β-catenin inhibition by LNA around, 3 month-old male C3H/He mice (Jackson Labs) were injected 5 times, every 48 hours (48h), with 15mg/kg of either EZN-3046 (scrambled control) (n=2) and 3 mice with EZN-3892 (directed against β-catenin) obtained under Materials Transfer Agreement from Enzon Pharmaceuticals, New Jersey, USA.
For validation of previously published chemical carcinogenesis model that utilizes CTNNB1 mutations to develop HCC [1, 22], 6-week old C3H/He (Jackson Labs) male mice (n=3) were injected intraperitoneally with 90μg/g DEN (Sigma-Aldrich) followed 3 weeks later by initiation of a diet containing 0.05% PB (LabDiet) (DEN/PB group) for the duration of the experiment (~30 weeks). Additional male mice (n=3) were given DEN at the same dose and time but kept on regular mouse chow without PB (DEN only group) for similar duration. Yet another group of mice (n=3) were started on 0.05% PB (PB only group) at 11 weeks of age for the duration of the experiment (around 30 weeks). Three additional male C3H/He mice without any intervention (control group) and on regular diet were killed when they were around 10 months old. Livers from all these mice were collected for analysis of HCC as described in the forthcoming sections.
For testing therapeutic efficacy of β-catenin suppression on HCC in DEN/PB model, around 7 months after initiation of PB diet, the mice were stratified into two groups. Group 1 received EZN-3046 (n=11) and group 2 received EZN-3892 (n=6). LNA were injected every 48h intraperitoneally at 15mg/kg for a total of 10 times. Livers and serum from mice after treatment were collected for further processing.
For testing any impact of β-catenin knockdown on HCC in chemical carcinogenesis model that is not driven by CTNNB1 mutations, DEN only treated mice at around 7 months received either EZN-3046 (n=6) or EZN-3892 (n=6). LNA were injected every 48h intraperitoneally at 15mg/kg for a total of 6 times. Livers from mice after treatment were collected for further processing.
Cell Culture and Treatment
Hep3B HCC cells stably transfected with serine-33 to tyrosine mutated (S33Y) β-catenin described recently, were grown to approximately 70% confluency and serum-starved for 24h [8, 9, 19, 26]. Cells were then transfected with 800ng of TopFlash plasmid (Millipore), 200ng of Renilla, and 3 μl of lipofectamine 2000 in 100μl Optimem media. After 4–6h, 5μM of EZN-3046 or EZN-3892 was added in media containing 4% FBS. Cells were harvested 48h or 72h later and luciferase activity measured using Dual-Luciferase Reporter Assay System (Promega) and normalized to Renilla levels. Statistical significance was calculated using one-tailed Student t test and p<0.05 was considered significant.
Whole Cell Lysate Preparation
At time of harvest, mice were anesthetized by isoflurane inhalation and subsequently killed by cervical dislocation. Livers were harvested, washed in PBS, and tissue was flash frozen in liquid nitrogen and stored at −80°C until use. Part of fresh or frozen tissue was homogenized in Radio immunoprecipitation assay (RIPA) buffer with protease/phosphatase inhibitor. Protein concentration was assessed by BCA protein assay (Pierce).
Western Blots (WB)
Around 50μg of whole cell lysate was run on precast 7.5% or 4–14% gradient polyacrylamide gel (Bio-Rad). Gels were transferred onto a polyvinylidene fluoride (PVDF) membrane (Millipore). Membranes were blocked in 5% milk (Labscientific) in Blotto [0.15M NaCl, 0.02M Tris pH 7.5, 0.1% Tween in dH2O] for 1h at room temperature. Primary antibodies were diluted in 5% milk/Blotto and incubated on membranes overnight at 4°C. Primary antibodies used were mouse monoclonal anti-β-catenin (BD Biosociences, 610154; 1:1000), rabbit polyclonal GS (Santa Cruz, SC-9067; 1:200) and rabbit Glyceraldehyde-3-phosphate dehydrogenase or GAPDH antibody (Santa Cruz, SC-25778; 1:800). Membranes were washed in Blotto for 1h at room temperature prior to incubation of membranes with rabbit (1:10,000), mouse (1:25,000), or goat (1:10,000) secondary antibodies (Millipore) for 1h. Membranes were again washed in Blotto for 1h at room temperature prior to expose to either SuperSignal West Pico or Femto Chemiluminescent Substrate (ThermoScientific) for 1–2 minutes at room temperature. The bands reflective of target proteins were viewed by autoradiography.
Histology and Immunohistochemistry (IHC)
Tissue samples were embedded in paraffin and cut into 4μm sections. Tissue sections were de-paraffinized in xylene and hydrated through graded alcohol rinses from 100% to 95% to dH2O, and eventually washed in 1x Phosphate buffered saline (PBS). Slides were next incubated in Eosin stain for 30 seconds followed by 2 washes in 95% ethanol and 2 washes of 100% ethanol. Slides were counterstained in Shandon’s Hematoxylin solution for 1 minute and gradually dehydrated in alcohol and xylene before mounting cover slips with DPX.
For IHC, de-paraffinized sections were microwaved in citrate buffer for antigen retrieval. Endogenous peroxidases were quenched with 3% hydrogen peroxide. Slides were blocked with Super Block (UltraTek) for 10 minutes and additional antigen retrieval was conducted using antigen-unmasking solution (Vector Labs). Primary antibodies used were rabbit anti-β-catenin (Santa Cruz, SC-7199; 1:150) or goat anti-β-catenin (Santa Cruz, SC-1496; 1:150), rabbit anti-GS (Santa Cruz, SC-9067; 1:100) and rabbit anti-cyclin-D1 (Neomarkers, RB-9041; 1:100), diluted in PBS and sections were incubated for 1h at room temperature. Sections were washed in PBS 3x followed by horseradish-peroxidase-conjugated secondary anti-Goat (1:200) or anti-Rabbit (1:200) antibodies (Millipore) added to the slides for 30 minutes at room temperature. The secondary antibody signal was detected with DAB (Vector Labs) and the signal was quenched with dH2O before counterstaining with Shandon solution (Sigma Aldrich). After dehydration in alcohol and xylene, slides were cover-slipped with DPX (Fluka Labs). Negative controls were done without primary antibody. Images were taken on Axioskop 40 (Zeiss) inverted brightfield microscope.
For terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) IHC, slides were stained using the ApopTag peroxidase kit (Intergen Co., Purchase, NY) as per the manufacture’s protocol.
Enzyme-linked immunosorbent assay (ELISA)-based serum Lect2 estimation
We recently reported serum LECT2 as a biomarker of β-catenin gene mutations in murine HCC [26]. Here, we used mouse LECT2 ELISA kit (Medical & Biological Laboratories (MBL) Co, Ltd, Niigata, Japan) to measure serum levels, according to the manufacturer’s protocol.
RESULTS
LNA directed against β-catenin efficaciously inhibits β-catenin activity in vitro and in vivo
To determine in vitro efficacy, Hep3B cells stably expressing S33Y-β-catenin or Snu-398 HCC cells which carry point mutation in CTNNB1, were transfected with TopFlash reporter and treated with 5μM of scrambled (EZN-3046) or β-catenin LNA (EZN-3892) as described in methods. A significant decrease in β-catenin-TCF reporter activity was evident at 48h and 72h after β-catenin-directed LNA treatment when compared to respective scrambled control LNA treatments as shown for S33Y-β-catenin-Hep3B cells (Fig. 1A). Cell lysates from both cell lines were assayed for total β-catenin levels and show a notable knockdown only after 48h EZN-3892 treatment (Fig. 1B).
Figure 1. Locked nucleic acid antisense effectively inhibits β-catenin expression and activity both in vitro and in vivo.
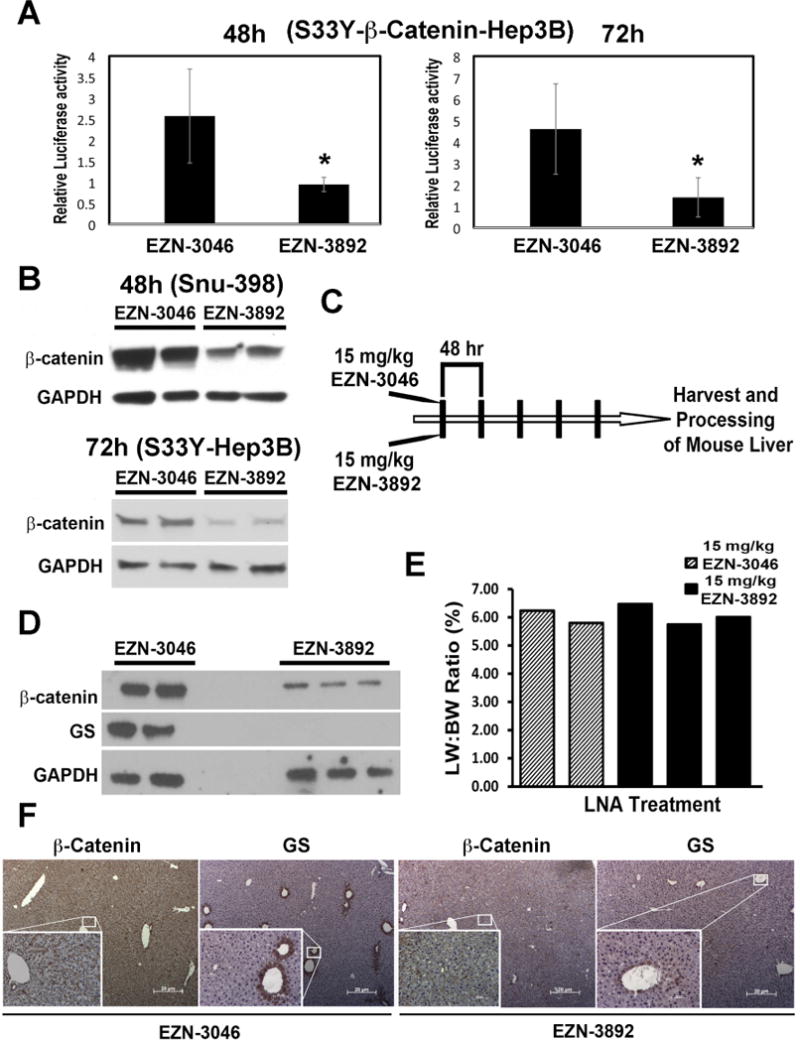
A. Treatment of S33Y-β-catenin-transfected Hep3B cells with 5μM β-catenin LNA (EZN-3892) decreases TopFlash reporter activity significantly (p<0.05) at 48h and 72h as compared to scrambled control LNA (EZN-3046).
B. Treatment of Snu-398 HCC cells that harbor mutation in CTNNB1, with 5μM EZN-3892 decreases total β-catenin protein at 48h as compared to EZN-3046. Decrease in β-catenin was also observed following 72h treatment of S33Y-β-catenin-Hep3B cells with EZN-3892. GAPDH verifies protein loading.
C. Schematic showing treatment of normal C3H/He adult male mice 5 times with EZN-3892 or EZN-3046.
D. WB from livers of EZN-3892 (n=3) or EZN-3046 (n=2) treated mice show a notable decrease in both β-catenin and GS in the former group. GAPDH verifies protein loading.
E. Comparable liver weight to body weight ratio were evident after 5 injections of EZN-3892 and EZN-3046.
F. A notable decrease in hepatic β-catenin staining as well as GS staining was evident in representative IHC from EZN-3892-treated mice as compared to EZN-3046 group. (Magnificantion-50X; Inset-200X).
In order to assess in vivo efficacy, wild type C3H/He mice were injected 5 times with 15mg/kg of EZN-3046 (n=2) or EZN-3892 treated mice (n=3), every other day (Fig. 1C). At the conclusion of this study, liver weight to body weight ratio showed no significant difference between the two groups (Fig. 1D). However, total β-catenin protein levels by WB were notably less after EZN-3892 treatment (Fig. 1E). Similarly, WB showed a dramatic decrease in GS (Fig. 1E). Furthermore, IHC detected a decrease in β-catenin and pericentral GS expression of EZN-3892-treated group (Fig. 1F). Thus, EZN-3892 effectively suppresses β-catenin expression and activity both in vitro and in vivo.
Validation of chemical carcinogenesis diet selecting for HCC harboring β-catenin gene mutations
To demonstrate the therapeutic efficacy of β-catenin inhibition as a potential treatment strategy for HCC, we next established a previously published model that selects for β-catenin gene mutations, as also described in methods [21]. Upon sacrifice, tumors were visible in DEN only group and DEN/PB group. Microscopic analysis of 4 representative lobes from each animal exhibited notably higher tumor burden in DEN/PB group (Figure 2A). More importantly, while liver tumors in DEN only group never showed nuclear β-catenin, most tumor cells within nodules in DEN/PB group showed nuclear β-catenin (Fig. 2B). These tumors were also strongly positive for GS (Fig. 2C). Quantification of GS-positive nodules as an indicator of CTNNB1 mutations and Wnt activation showed that indeed around 90% of HCC in this model are β-catenin-active as also published elsewhere (Fig. 2D) [22]. The predominant mutations in CTNNB1 observed in this model were S33Y as recently reported [26]. Thus DEN/PB model of HCC is relevant to directly test any therapeutic benefit of β-catenin inhibition.
Figure 2. DEN/PB treatment leads to HCC, which displays β-catenin activation in tumor nodules.
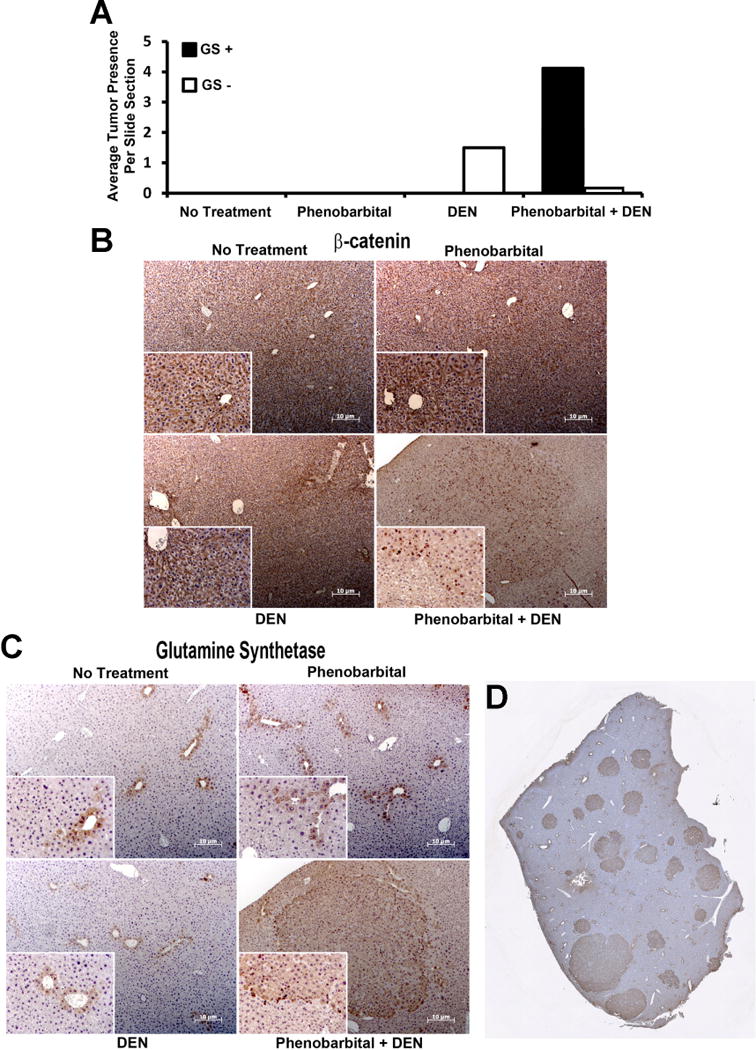
A. Quantification of microscopic nodules after PB, DEN or DEN/PB treatment shows a notable increase in average nodule numbers per section per representative lobe shows a more pronounced disease in DEN/PB model. In addition, while all microscopic nodules in DEN alone were GS-negative, most nodules were GS-positive in DEN/PB model. No tumor nodules were evident in age-matched untreated controls or in PB only treatment.
B. Representative IHC for β-catenin from different conditions display mostly membranous β-catenin in all conditions except within tumor nodules in DEN/PB model where clear nuclear localization of β-catenin is evident.
C. Representative IHC for GS from different conditions display mostly pericentral hepatocytes to be GS-positive in all conditions except within tumor nodules in DEN/PB model where entire microscopic nodule is diffusely GS-positive.
D. A representative tiled image from a reconstructed lobe of DEN/PB treated animal displays several microscopic HCC nodules that are strongly GS-positive.
Use of β-catenin LNA in DEN/PB model of HCC leads to notable suppression of β-catenin signaling
To demonstrate efficacy of LNA to suppress β-catenin and its downstream signaling, DEN/PB exposed male C3H/He mice, were stratified into 3 groups at around 7 months after initiation of PB diet. Group one received 15mg/kg of EZN-3046 (n=11), group two received 15mg/kg of EZN-3892 (n=6) and group 3 received no treatment (n=5) as described in methods (Figure 3A).
Figure 3. LNA directed against β-catenin abolishes HCC in DEN/PB model.
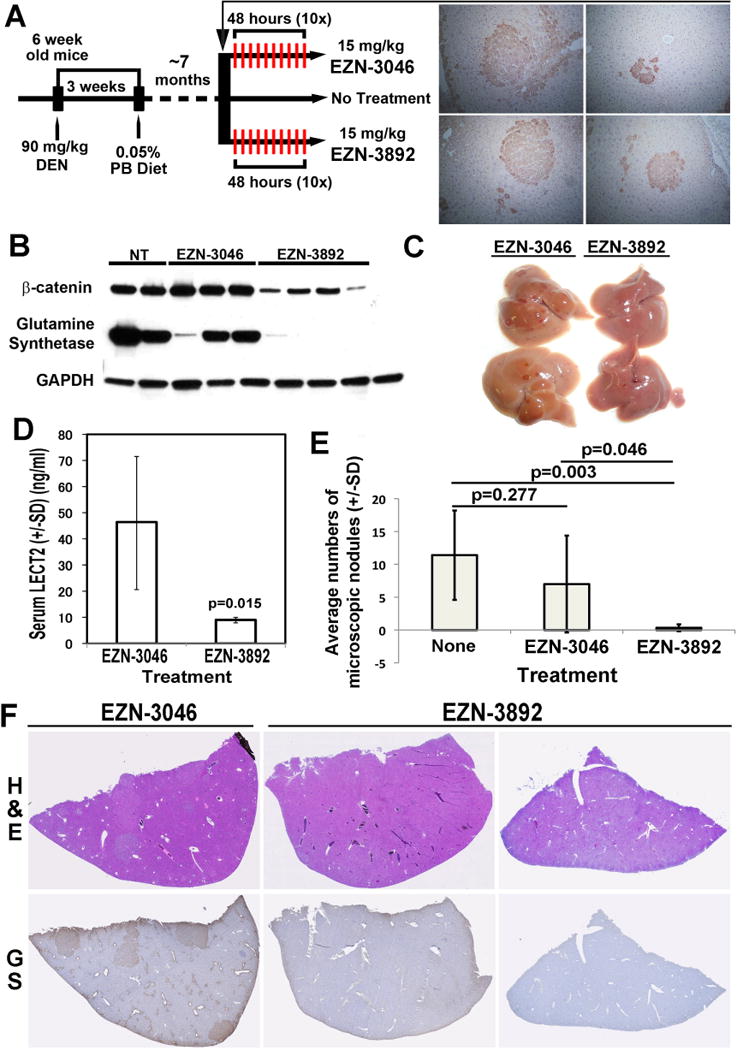
A. Schematic showing DEN/PB protocol and stratification of C3H/He male mice for treatment with EZN-3892, EZN-3046 or no treatment. Representative IHC of liver sections from one mouse at around 7 months after starting PB diet shows existence of 4 GS-positive microscopic foci (total 6 observed in this animal) verifying preexisting disease at the time of initiation of treatment.
B. WB from representative liver samples from mice subjected to DEN/PB protocol and then left either untreated, or treated with EZN-3892 or EZN-3046. A notable decrease in total β-catenin and GS levels are evident in EZN-3892-treated livers as compared to all other groups.
C. Gross pictures of representative livers depict clear loss of gross tumorigenesis after 10 doses of EZN-3892 treatment as compared to EZN-3046.
D. ELISA on serum for Lect2 from mice on DEN/PB protocol treated with EZN-3892 (n=4) showed a significant (p=0.014) decrease as compared to EZN-3046 treated group (n=11). (SD: Standard Deviation).
E. Quantification of microscopic nodules from mice on DEN/PB protocol subjected to EZN-3892 (n=6) shows a significant decrease in average nodule numbers per section per representative lobe as compared to no treatment group (n=11; p=0.003) as well as from EZN-3046-treated group (n=5; p=0.046). No significant differences in microscopic hepatic nodule numbers were evident between untreated and EZN-3046-treated groups (p=0.277). (SD: Standard Deviation).
F. Representative tiled images for liver H&E (upper row) and GS IHC (lower row) depicts presence of several nodules that are GS-positive in EZN-3046-treated animals versus absence of nodules or GS-immunoreactivity in EZN-3892-treated animals.
Following treatment, the livers from mice from all three groups were used to prepare whole cell lysate to test for β-catenin and GS levels by WB analysis. A notable decrease in both β-catenin and GS was observed following the administration of EZN-3892 as compared to the other groups (Fig. 3B). Thus, LNA directed against β-catenin clearly suppresses β-catenin expression and signaling in DEN/PB model of HCC.
Therapeutic efficacy of β-catenin suppression by LNA in DEN/PB model of HCC
The timing of stratification and initiation of therapy at around 7 months was chosen based on previous imaging study that showed existence of MRI-detectable tumors even at 6 months after DEN administration [29]. To verify existence of disease in our study at the time of treatment initiation, a representative animal was sacrificed at this time. Several GS-positive microscopic nodules were observed in the liver (Fig. 3A). After 10 treatments livers from the EZN-3892 group showed a striking abrogation of the disease as compared to EZN-3046 (Fig. 3C).
We next examined serum from EZN-3046 and EZN-3892 treated mice for a recently published biomarker of active β-catenin signaling [26]. Serum was available from 11 mice in EZN-3046 group and 4 mice from EZN-3892 group. As assessed by ELISA, there was a significant decrease in serum LECT2 levels in EZN-3892 versus EZN-3046 treated group, which coincided with a complete response to anti-β-catenin therapy in the former group (Fig. 3D).
Histological analysis from H&E stained sections with all 4 lobes represented on a slide showed a significant decrease in the number of microscopic tumor nodules in EZN-3892 treated group versus both untreated or EZN-3046 treated groups (Fig. 3E). This was more apparent in tiled images of representative liver lobes from each group for H&E and GS IHC, which show a striking absence of microscopic tumor nodules demonstrating a remarkable therapeutic effect of β-catenin suppression in this model (Fig. 3F).
Decreased cell proliferation and enhanced cell death is observed after β-catenin suppression in DEN/PB model
To address mechanism by which β-catenin knockdown led to regression of tumors, we examined β-catenin and cyclin-D1 localization in EZN-3046- and EZN-3892-treated livers. As shown in a representative IHC, several tumor cells within nodules were positive for nuclear β-catenin and cyclin-D1 in EZN-3046 treated group (Fig 4A). As a result, tumor nodules were positive for Ki-67, a marker of cells in S-phase (Fig. 4B). No tumor nodules were evident in the EZN-3892 group and most hepatocytes lacked β-catenin staining other than some at membrane and only isolated hepatocytes showed some remnant cytoplasmic β-catenin (Fig. 4A). In addition, most cells lacked any nuclear cyclin-D1 in this group (Fig. 4A). As a result, hepatocytes were negative for Ki-67, although some non-parenchymal cells, especially endothelial cells were positive for this marker (Fig. 4B). Thus one mechanism by which β-catenin suppression affects tumor growth in vivo is by affecting cell proliferation.
Figure 4. LNA directed against β-catenin decreases hepatic β-catenin, cyclin-D1 and cell proliferation and induces apoptosis in DEN/PB model only.
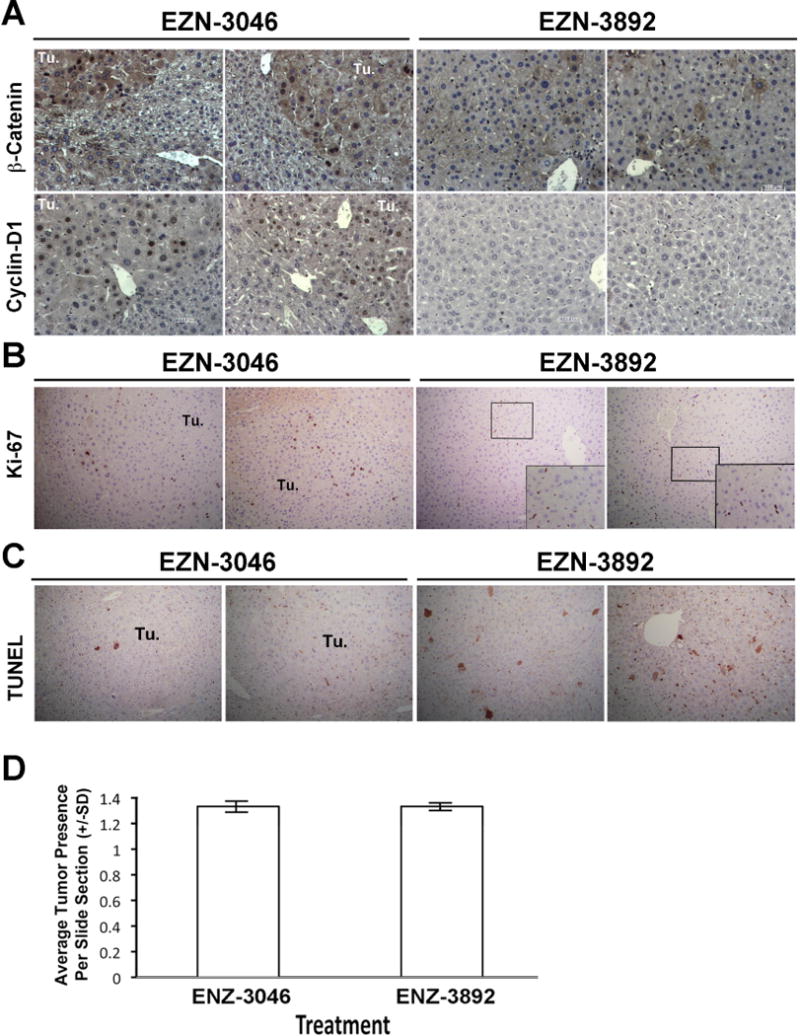
A. Representative IHC shows microscopic tumor nodules (Tu.) in mice on DEN/PB protocol that were treated with EZN-3046 to be strongly nuclear β-catenin-positive (upper row), while also displaying concomitant cyclin-D1-positivity within tumor nodules (lower row). However, following EZN-3892 treatment, no tumor nodules are visible and only occasional hepatocytes were positive for cytoplasmic β-catenin and only rare hepatocyte showed positivity for cyclin-D1. (Magnification: 200×)
B. Representative IHC shows tumor nodules (Tu.) in mice on DEN/PB protocol that were treated with EZN-3046 to be positive for Ki-67, a marker of cells in S-phase. However, following EZN-3892 treatment, only occasional hepatocytes were positive for Ki-67, while some cells with flattened endothelial morphology were Ki-67 positive (inset). (Magnification: 100×; Inset-200x)
C. Representative IHC shows tumor nodules (Tu.) in mice on DEN/PB protocol that were treated with EZN-3046 to be negative for TUNEL. However, following EZN-3892 treatment, some hepatocytes were TUNEL-positive (Magnification: 100×)
D. Quantification of microscopic nodules from mice on DEN only protocol treated with EZN-3892 (n=6) or EZN-3046 show comparable numbers of microscopic nodules per section per representative lobe, showing no decrease in HCC when β-catenin is inhibited in tumors that are not β-catenin-active. (SD: Standard Deviation).
TUNEL staining to address any difference in cell death was also performed. While no or occasional TUNEL-positive cells were evident within tumor nodules in the EZN-3046 group, EZN-3892 treated livers showed increased TUNEL-positivity (Fig. 4D). Thus, impaired cell viability may also be a mechanism of HCC regression after β-catenin suppression.
β-Catenin suppression in non-β-catenin gene mutated HCC does not impact tumorigenesis
To address if β-catenin inhibition could impact tumor growth in HCC that do not harbor CTNNB1 mutation, we administered EZN-3046 or EZN-3892 TO DEN-only injected mice. None of the tumors were positive for nuclear β-catenin- or GS (data not shown) since HCC in this model is due to Ha-ras or B-raf mutations [35]. β-Catenin knockdown by LNA did not affect tumor incidence in this model (Fig. 4D). This demonstrates the therapeutic efficacy of β-catenin suppression only in tumors with active-β-catenin, which in our study were due to CTNNB1 mutations.
DISCUSSION
Activation of Wnt/β-catenin signaling pathway in primary HCC is evident in a significant subset of tumors [24]. Various in vitro proof-of-concept studies have demonstrated that suppression of Wnt signaling can impact tumor cell proliferation and survival. Using siRNA or gamma guanidine-based peptide nucleic acid antisense, we have shown Wnt signaling active HCC cells to succumb to β-catenin knockdown [8, 42]. Additionally, tumor xenograft studies have substantiated Wnt inhibition as a viable therapeutic intervention in HCC [37]. However, to date a study is lacking that demonstrates significance of β-catenin inhibition in a model, which similar to patients innately utilizes activating CTNNB1 mutations to drive HCC. In the current study, we use locked nucleic acid antisense to suppress β-catenin expression in the DEN/PB model of β-catenin-driven HCC [1]. A dramatic impact on HCC was evident in the form of complete response in a model, which utilized activating β-catenin gene mutations to drive tumorigenesis. Thus our study for the first time shows that anti-β-catenin therapies would be highly significant in a select group of tumors that exhibit notable β-catenin activation through mutations in CTNNB1.
DEN/PB model was identified by Dr. Schwarz group to allow for hepatocarcinogenesis through activating CTNNB1 mutations that are commonly observed in a significant subset of HCCs in patients [1, 22, 35]. While the exact mechanism of how this specific protocol selects for β-catenin gene mutations is not know, a possible mechanism can be speculated. DEN at high doses in 6-week-old male leads to DNA damage and adducts possibly through oxidative stress, which may lead to mutations in CTNNB1 along with others [28]. Since Constitutive Androstane Receptor (CAR), a nuclear orphan receptor, was recently identified as a β-catenin target [16], it is likely that introduction of PB, which is a CAR agonist, provides growth advantage to hepatocytes with activating β-catenin gene mutations [3, 17]. Thus tumor nodules in DEN/PB model are predominantly β-catenin mutated. Our study using LNA against β-catenin conclusively demonstrates the druggability and effectiveness of β-catenin inhibition as a therapeutic strategy in the DEN/PB model of HCC. It is relevant to emphasize that β-catenin directed LNA treatment was initiated at 7 months after PB diet was started. At this time the disease is already known to exist as previously reported and was also verified directly in our study [29]. Down-regulation of β-catenin in these existing tumors clearly led to a complete response demonstrating a therapeutic benefit of inhibiting this oncoprotein albeit in only β-catenin mutated HCCs as no effect of β-catenin suppression was observed on non-β-catenin mutated tumors occurring in DEN only treated animals [35]. At the same time, it cannot be ruled out that administration of β-catenin LNA prevented emergence of newer tumor foci and hence may simultaneously have chemoprophylactic role as well. Another recent study using DEN/PB in inducible conditional β-catenin knockout mice also demonstrated that deletion of β-catenin from some tumor nodules affected proliferation and reduced tumor burden [30].
While the precise mechanism of how β-catenin suppression led to involution of tumors is unclear, it is likely through the downregulation of its target genes. Several β-catenin targets play a role in tumor biology, such as cyclin-D1 and c-myc in cell proliferation, survivin in cell viability, GS in tumor cell metabolism, VEGF-A in tumor angiogenesis and EpCAM in cancer stem cell expansion. In current study, we show that β-catenin suppression in vivo led to a complete response. Due to limited LNA, amount of time required to establish HCC in DEN/PB model and numbers of mice required for additional time points, we were unable to do time course to determine temporal effects of β-catenin suppression. However, at the conclusion of the study, there was a notable decrease in cell proliferation and an increase in cell death. This may be due to cumulative effect of β-catenin suppression on expression of targets such as GS, cyclin-D1 and perhaps others [4, 15]. Glutamine is known to play an important role in tumor cell viability and usually glutamine addicted tumors are reprogrammed to derive this amino acid through the tricarboxylic acid cycle [40]. Since β-catenin mutated tumors overexpress GS [22], this could be the major source of glutamine for metabolism and thus β-catenin suppression may deprive such tumors of glutamine to impair tumor cell viability. Indeed glutamine deprivation through specific modalities has revealed glutamine addiction of the β-catenin-mutated tumors [6, 33]. Simultaneously, a direct regulation of cyclin-D1 in liver cells by β-catenin is also likely contributing to abrogation of tumorigenesis upon its suppression [32]. Thus, any suitable modality to inhibit β-catenin expression or activity may have a potential to be investigated for therapeutic benefit.
Could β-catenin be a realistic target in HCC? Since HCCs commonly arise in livers with advanced fibrosis and ongoing regeneration, a target may have antifibrotic function and be redundant in liver regeneration. Indeed β-catenin inhibition has been shown to ameliorate fibrosis and is also redundant in liver regeneration [5, 32]. Also, despite the fact that β-catenin is vital to the adherens junctions, its loss or suppression both in vitro and in vivo have been shown to be compensated by upregulation of plakoglobin [38, 39].
Various groups have utilized different methods to target β-catenin signaling. Our lab has shown R-Etodolac, an enantiomer of a nonsteroidal anti-inflammatory drug S-Etodolac, to inhibit Wnt signaling in HCC cell lines [2]. We have also shown pegylated interferon-α2a to induce export of nuclear β-catenin in HCC [34]. Another group developed a small molecule ICG-001, which reduces β-catenin activity in colon cancer cells [11]. Currently, a second-generation compound of ICG-001, PRI-724, is in multiple Phase I and Phase II trials for pancreatic adenocarcinoma, solid tumors and for myeloid malignancies respectively (NCT01764477) (NCT01302405) (NCT01606579). Recently, we have also utilized structure-function analysis to identify ICG-001 like molecule PMED-1, which suppresses β-catenin activity in HCC cells and in zebrafish [9]. In current study we used LNA to target β-catenin expression.
LNA oligonucleotides were initially used as molecular probes for microarray gene analysis because of their high binding affinity to DNA and RNA [27]. However, LNAs differ from normal DNA or RNA nucleotides, as they have a methylene bridge that “locks” the 2P-oxygen in the ribose backbone to the 4P-carbon and stabilizes the position of the phosphate backbone resulting in superior hybridization [18]. Because LNA can easily hybridize with an intended DNA or RNA target of interest, inhibition of gene expression occurs via activation of RNase H recognition and degradation pathway [36]. In vitro efficacy of LNA in inhibiting gene expression is well recognized with an advantage of transfection by known lipophilic methods [10]. Chemotherapeutic implications were also realized in vivo such as targeting a subunit of RNA polymerase that reduced tumor burden [14]. The ease of LNA synthesis and its mass production makes it an attractive modality for specifically targeting known oncogenes driving cancer progression for personalized medicine [31]. In the current study we show that LNA is a relevant modality to target active Wnt/β-catenin signaling in a subset of HCC cases.
Acknowledgments
FINANCIAL SUPPORT: This study was funded by NIH grants 1R01DK62277, 1R01DK100287 and Endowed Chair for Experimental Pathology to SPSM. This study was also funded by T32
ABBREVIATIONS
- HCC
Hepatocellular cancer
- LNA
Locked nucleic antisense
- DEN/PB
Diethylnitrosamine and Phenobarbital
- GSK-3β
Glycogen synthase kinase-3β
- APC
Adenomatous polyposis coli
- Fzd
Frizzled
- LRP
low-density lipoprotein receptor related protein
- CREB
cAMP response element-binding protein
- CBP
CREB binding protein
- TCF
T-cell factor
- LEF
Lymphoid enhancing factor
- GS
glutamine synthetase
- FBS
Fetal bovine serum
- RIPA
Radio immunoprecipitation assay
- PVDF
polyvinylidene fluoride
- GAPDH
Glyceraldehyde-3-phosphate dehydrogenase
- PBS
Phosphate buffered saline
- TUNEL
Terminal deoxynucleotidyl transferase dUTP nick end labeling
- ELISA
Enzyme linked immunosorbent assay
- IHC
Immunohistochemistry
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST: No conflict of interest related to the study. Dr Yixian Zhang was an employee of Enzon and provided the reagents used in the study and helped with initial study design. He is no longer with Enzon and is employed by leukemia and lymphoma foundation. Dr. Monga is a consultant for Abbvie Pharmaceuticals but there is no conflict of interest with the current study.
AUTHOR CONTRIBUTIONS:
Evan Delgado: Acquisition, analysis and interpretation of data, drafting article
Hirohisa Okabe: Acquisition, analysis, and interpretation of the data
Jacquelyn Olivia Russell: Acquisition and analysis of the data
Tamara Feliciano Alvarado: Acquisition of data
Morgan Preziosi: Acquisition of data
Michael Oertel: Analysis of data
Kari Nichole Nejak-Bowen: Conception and design, analysis, and interpretation of the data
Yixian Zhang: Design of study and providing reagent
Satdarshan P Monga: conception and design, acquisition, analysis, and interpretation of the data, the drafting of the article
References
- 1.Aydinlik H, Nguyen TD, Moennikes O, Buchmann A, Schwarz M. Selective pressure during tumor promotion by phenobarbital leads to clonal outgrowth of beta-catenin-mutated mouse liver tumors. Oncogene. 2001;20(53):7812–7816. doi: 10.1038/sj.onc.1204982. [DOI] [PubMed] [Google Scholar]
- 2.Behari J, Zeng G, Otruba W, Thompson MD, Muller P, Micsenyi A, et al. R-Etodolac decreases beta-catenin levels along with survival and proliferation of hepatoma cells. J Hepatol. 2007;46(5):849–857. doi: 10.1016/j.jhep.2006.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Braeuning A, Heubach Y, Knorpp T, Kowalik MA, Templin M, Columbano A, et al. Gender-specific interplay of signaling through beta-catenin and CAR in the regulation of xenobiotic-induced hepatocyte proliferation. Toxicol Sci. 2011;123(1):113–122. doi: 10.1093/toxsci/kfr166. [DOI] [PubMed] [Google Scholar]
- 4.Cadoret A, Ovejero C, Terris B, Souil E, Levy L, Lamers WH, et al. New targets of beta-catenin signaling in the liver are involved in the glutamine metabolism. Oncogene. 2002;21(54):8293–8301. doi: 10.1038/sj.onc.1206118. [DOI] [PubMed] [Google Scholar]
- 5.Cheng JH, She H, Han YP, Wang J, Xiong S, Asahina K, et al. Wnt antagonism inhibits hepatic stellate cell activation and liver fibrosis. Am J Physiol Gastrointest Liver Physiol. 2008;294(1):G39–49. doi: 10.1152/ajpgi.00263.2007. [DOI] [PubMed] [Google Scholar]
- 6.Chiu M, Tardito S, Pillozzi S, Arcangeli A, Armento A, Uggeri J, et al. Glutamine depletion by crisantaspase hinders the growth of human hepatocellular carcinoma xenografts. Br J Cancer. 2014 doi: 10.1038/bjc.2014.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de La Coste A, Romagnolo B, Billuart P, Renard CA, Buendia MA, Soubrane O, et al. Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc Natl Acad Sci U S A. 1998;95(15):8847–8851. doi: 10.1073/pnas.95.15.8847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delgado E, Bahal R, Yang J, Lee JM, Ly DH, Monga SP. beta-Catenin Knockdown in Liver Tumor Cells by a Cell Permeable Gamma Guanidine-based Peptide Nucleic Acid. Curr Cancer Drug Targets. 2013 doi: 10.2174/15680096113139990081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Delgado ER, Yang J, So J, Leimgruber S, Kahn M, Ishitani T, et al. Identification and characterization of a novel small-molecule inhibitor of beta-catenin signaling. Am J Pathol. 2014;184(7):2111–2122. doi: 10.1016/j.ajpath.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elayadi AN, Braasch DA, Corey DR. Implications of high-affinity hybridization by locked nucleic acid oligomers for inhibition of human telomerase. Biochemistry. 2002;41(31):9973–9981. doi: 10.1021/bi025907j. [DOI] [PubMed] [Google Scholar]
- 11.Emami KH, Nguyen C, Ma H, Kim DH, Jeong KW, Eguchi M, et al. A small molecule inhibitor of beta-catenin/CREB-binding protein transcription [corrected] Proc Natl Acad Sci U S A. 2004;101(34):12682–12687. doi: 10.1073/pnas.0404875101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer. 2006;6(9):674–687. doi: 10.1038/nrc1934. [DOI] [PubMed] [Google Scholar]
- 13.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127(12):2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 14.Fluiter K, ten Asbroek AL, de Wissel MB, Jakobs ME, Wissenbach M, Olsson H, et al. In vivo tumor growth inhibition and biodistribution studies of locked nucleic acid (LNA) antisense oligonucleotides. Nucleic Acids Res. 2003;31(3):953–962. doi: 10.1093/nar/gkg185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gebhardt R, Mecke D. Heterogeneous distribution of glutamine synthetase among rat liver parenchymal cells in situ and in primary culture. EMBO J. 1983;2(4):567–570. doi: 10.1002/j.1460-2075.1983.tb01464.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gougelet A, Torre C, Veber P, Sartor C, Bachelot L, Denechaud PD, et al. T-cell factor 4 and beta-catenin chromatin occupancies pattern zonal liver metabolism in mice. Hepatology. 2014;59(6):2344–2357. doi: 10.1002/hep.26924. [DOI] [PubMed] [Google Scholar]
- 17.Kawamoto T, Sueyoshi T, Zelko I, Moore R, Washburn K, Negishi M. Phenobarbital-responsive nuclear translocation of the receptor CAR in induction of the CYP2B gene. Mol Cell Biol. 1999;19(9):6318–6322. doi: 10.1128/mcb.19.9.6318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kumar R, Singh SK, Koshkin AA, Rajwanshi VK, Meldgaard M, Wengel J. The first analogues of LNA (locked nucleic acids): phosphorothioate-LNA and 2′-thio-LNA. Bioorg Med Chem Lett. 1998;8(16):2219–2222. doi: 10.1016/s0960-894x(98)00366-7. [DOI] [PubMed] [Google Scholar]
- 19.Lee JM, Yang J, Newell P, Singh S, Parwani A, Friedman SL, et al. beta-Catenin signaling in hepatocellular cancer: Implications in inflammation, fibrosis, and proliferation. Cancer Lett. 2014;343(1):90–97. doi: 10.1016/j.canlet.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 21.Loeppen S, Koehle C, Buchmann A, Schwarz M. A beta-catenin-dependent pathway regulates expression of cytochrome P450 isoforms in mouse liver tumors. Carcinogenesis. 2005;26(1):239–248. doi: 10.1093/carcin/bgh298. [DOI] [PubMed] [Google Scholar]
- 22.Loeppen S, Schneider D, Gaunitz F, Gebhardt R, Kurek R, Buchmann A, et al. Overexpression of glutamine synthetase is associated with beta-catenin-mutations in mouse liver tumors during promotion of hepatocarcinogenesis by phenobarbital. Cancer Res. 2002;62(20):5685–5688. [PubMed] [Google Scholar]
- 23.Nault JC, Mallet M, Pilati C, Calderaro J, Bioulac-Sage P, Laurent C, et al. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat Commun. 2013;4:2218. doi: 10.1038/ncomms3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nault JC, Zucman-Rossi J. Genetics of hepatocellular carcinoma: the next generation. J Hepatol. 2014;60(1):224–226. doi: 10.1016/j.jhep.2013.08.019. [DOI] [PubMed] [Google Scholar]
- 25.Nejak-Bowen KN, Monga SP. Beta-catenin signaling, liver regeneration and hepatocellular cancer: sorting the good from the bad. Semin Cancer Biol. 2011;21(1):44–58. doi: 10.1016/j.semcancer.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Okabe H, Delgado E, Lee JM, Yang J, Kinoshita H, Hayashi H, et al. Role of leukocyte cell-derived chemotaxin 2 as a biomarker in hepatocellular carcinoma. PLoS One. 2014;9(6):e98817. doi: 10.1371/journal.pone.0098817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Petersen M, Bondensgaard K, Wengel J, Jacobsen JP. Locked nucleic acid (LNA) recognition of RNA: NMR solution structures of LNA:RNA hybrids. J Am Chem Soc. 2002;124(21):5974–5982. doi: 10.1021/ja012288d. [DOI] [PubMed] [Google Scholar]
- 28.Santos NP, Pereira IC, Pires MJ, Lopes C, Andrade R, Oliveira MM, et al. Histology, bioenergetics and oxidative stress in mouse liver exposed to N-diethylnitrosamine. In Vivo. 2012;26(6):921–929. [PubMed] [Google Scholar]
- 29.Schmid A, Rignall B, Pichler BJ, Schwarz M. Quantitative analysis of the growth kinetics of chemically induced mouse liver tumors by magnetic resonance imaging. Toxicol Sci. 2012;126(1):52–59. doi: 10.1093/toxsci/kfs018. [DOI] [PubMed] [Google Scholar]
- 30.Singh Y, Port J, Schwarz M, Braeuning A. Genetic ablation of beta-catenin inhibits the proliferative phenotype of mouse liver adenomas. Br J Cancer. 2014;111(1):132–138. doi: 10.1038/bjc.2014.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stenvang J, Silahtaroglu AN, Lindow M, Elmen J, Kauppinen S. The utility of LNA in microRNA-based cancer diagnostics and therapeutics. Semin Cancer Biol. 2008;18(2):89–102. doi: 10.1016/j.semcancer.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 32.Tan X, Behari J, Cieply B, Michalopoulos GK, Monga SP. Conditional deletion of beta-catenin reveals its role in liver growth and regeneration. Gastroenterology. 2006;131(5):1561–1572. doi: 10.1053/j.gastro.2006.08.042. [DOI] [PubMed] [Google Scholar]
- 33.Tardito S, Chiu M, Uggeri J, Zerbini A, Da Ros F, Dall’Asta V, et al. L-Asparaginase and inhibitors of glutamine synthetase disclose glutamine addiction of beta-catenin-mutated human hepatocellular carcinoma cells. Curr Cancer Drug Targets. 2011;11(8):929–943. doi: 10.2174/156800911797264725. [DOI] [PubMed] [Google Scholar]
- 34.Thompson MD, Dar MJ, Monga SP. Pegylated interferon alpha targets Wnt signaling by inducing nuclear export of beta-catenin. J Hepatol. 2011;54(3):506–512. doi: 10.1016/j.jhep.2010.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Unterberger EB, Eichner J, Wrzodek C, Lempiainen H, Luisier R, Terranova R, et al. Ha-ras and beta-catenin oncoproteins orchestrate metabolic programs in mouse liver tumors. Int J Cancer. 2014;135(7):1574–1585. doi: 10.1002/ijc.28798. [DOI] [PubMed] [Google Scholar]
- 36.Wahlestedt C, Salmi P, Good L, Kela J, Johnsson T, Hokfelt T, et al. Potent and nontoxic antisense oligonucleotides containing locked nucleic acids. Proc Natl Acad Sci U S A. 2000;97(10):5633–5638. doi: 10.1073/pnas.97.10.5633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wei W, Chua MS, Grepper S, So S. Small molecule antagonists of Tcf4/beta-catenin complex inhibit the growth of HCC cells in vitro and in vivo. Int J Cancer. 2010;126(10):2426–2436. doi: 10.1002/ijc.24810. [DOI] [PubMed] [Google Scholar]
- 38.Wickline ED, Awuah PK, Behari J, Ross M, Stolz DB, Monga SP. Hepatocyte gamma-catenin compensates for conditionally deleted beta-catenin at adherens junctions. J Hepatol. 2011;55(6):1256–1262. doi: 10.1016/j.jhep.2011.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wickline ED, Du Y, Stolz DB, Kahn M, Monga SP. gamma-Catenin at adherens junctions: mechanism and biologic implications in hepatocellular cancer after beta-catenin knockdown. Neoplasia. 2013;15(4):421–434. doi: 10.1593/neo.122098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci. 2010;35(8):427–433. doi: 10.1016/j.tibs.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang JD, Roberts LR. Hepatocellular carcinoma: A global view. Nat Rev Gastroenterol Hepatol. 2010;7(8):448–458. doi: 10.1038/nrgastro.2010.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zeng G, Apte U, Cieply B, Singh S, Monga SP. siRNA-mediated beta-catenin knockdown in human hepatoma cells results in decreased growth and survival. Neoplasia. 2007;9(11):951–959. doi: 10.1593/neo.07469. [DOI] [PMC free article] [PubMed] [Google Scholar]